Introduction
Bufalin is a traditional Chinese medicine and it is
the major digoxin-like immunoreactive component of Chan Su, which
is obtained from the skin and parotid venom glands of toads
(1). It is a cardioactive C-24
steroid, with the molecular formula
C24H34O4 and a relative molecular
weight of 386.5. Bufalin exhibits a variety of biological
activities, such as cardiotonic, anesthetic, blood pressure
stimulatory, respiratory and antineoplastic effects (2). In terms of its antitumor activity,
bufalin has been demonstrated to inhibit the growth of gastric,
colon, breast, prostate, gynecological, hepatocellular and bladder
tumors (3). It is also able to
induce strong differentiation and apoptosis of myeloid leukemia
cells, such as HL60, U937 and K562 cells. The mechanisms by which
bufalin induces leukemia cell apoptosis are complex and involve a
diverse range of cell signals (4–7). It
is capable of inhibiting sodium-potassium-ATPase, and then
activating apoptotic pathways (8,9). It
is also able to activate the mitogen-activated protein kinase
(MAPK) signaling pathway (9);
downregulate the expression of the proto-oncogene c-myc and the
anti-apoptotic protein Bcl-2 (10); and reduce the quantity, activity
and mRNA levels of topoisomerase II (11). However, the precise pathways by
which bufalin induces apoptosis remain unclear, and an improved
understanding of such pathways is an essential goal of further
study. To determine the global molecular mechanisms underlying
bufalin-induced leukemia cell apoptosis, the present study analyzed
the effects of bufalin on the gene expression of K562 cells using a
Microarray assay. Based on this experiment, the expression of the
BMI-1 pathway components in K562 cell apoptosis was also
analyzed.
BMI-1 is a member of the polycomb-group gene family,
that is known to be essential in supporting the self-renewal of
stem cells through their epigenetic transcriptional regulation
(12–15). BMI-1 is located on chromosome
10p13, a region involved in chromosomal translocations in infant
leukemia (16). BMI-1 is a
transcriptional repressor, the most widely known of the INK4a/ARF
locus, that encodes the cell cycle regulators and tumor suppressors
p16INK4a and p14ARF (17,18).
Downregulation of p16INK4a and p14ARF
inhibits cell apoptosis that is induced by p53 and retinoblastoma
protein (pRB). It has become clear that BMI-1 also functions in
protecting against oxidative stress. In the absence of BMI-1,
reactive oxygen species (ROS) accumulate, which are associated with
the activation of DNA damage response pathways and increased
apoptosis (19). Therefore,
overexpression of BMI-1 results in cell immortalization and
tumorigenesis, whereas downregulation in tumor (stem) cells results
in impaired self-renewal and proliferation (20,21).
It has been demonstrated that BMI-1 is often overexpressed in
various types of leukemia and its depletion in those leukemia cells
leads to cell proliferation arrest, differentiation and apoptosis
(22). Lessard and Sauvageau
(20) demonstrated that the
proliferative potential of acute myeloid leukemia (AML) stem and
progenitor cells that lacked BMI-1 was compromised, as the cells
eventually underwent proliferation arrest and exhibited signs of
differentiation and apoptosis. In addition, the absence of BMI-1
led to transplant failure of the leukemia, and these proliferative
defects were completely rescued by BMI-1. These findings suggest
that the BMI-1 gene is a potential target for therapeutic
intervention in leukemia. Zhu et al (22) demonstrated that small interfering
RNA (siRNA)-mediated silencing of BMI-1 in U937 cells led to
reduced cell growth and proliferation, and increased cell
apoptosis. Meng et al (23)
also demonstrated that K562 cells transfected with an antisense
BMI-1 plasmid grew significantly slower than controls. Their
colony-forming ability decreased significantly, and their
p16INK4a expression levels were upregulated more than
that of the controls.
In the present study, the effect of bufalin on BMI-1
expression in K562 cells was investigated, and the expression of
p16INK4a and p14ARF during the
bufalin-induced apoptosis was also determined.
Materials and methods
Materials
Bufalin (10.0 mg/vial) was purchased from
Sigma-Aldrich (St. Louis, MO, USA) and was dissolved in anhydrous
alcohol to make 0.01 mol/l stock solution. The solution was stored
at −20°C and diluted in RPMI-1640 when used. K562 cells were
obtained from the central laboratory of The First Affiliated
Hospital of Xi’an Jiaotong University (Xi’an, China). Bone marrow
mononuclear cells were acquired from two female healthy
iron-deficiency anemia volunteers (ages, 38 and 29 years) as normal
controls. The study was approved by the Ethics Committee of Shaanxi
Provincial People’s Hospital and written informed consent was
obtained from the volunteers.
Cell culture
The K562 human erythrocyte leukemia cell line was
grown in suspension culture in RPMI-1640 supplemented with 10%
(v/v) fetal bovine serum (HyClone, Logan, UT, USA), at 37°C in a
humidified atmosphere with 5% CO2. K562 cells at an
exponential growth stage were employed in all of the
experiments.
MTT assay
Cells were seeded in 96-well plates at a density of
105 cells/ml in 100 μl of medium and cultured
overnight. Each experiment was performed in triplicate.
Subsequently, the cells in the wells were treated with fresh medium
containing different concentrations of bufalin diluted from the
stock solution. The resulting concentrations of bufalin were 0.025,
0.05, 0.1, 0.5, 1.0 and 2.0 μmol/l. The culture solution was
applied to the wells as a blank control. The cells that were not
treated with bufalin served as negative controls. Cells were
cultured for 48 or 72 h. At the indicated times, MTT solution [5
mg/ml in 20 μl phosphate-buffered saline (PBS)] was added to
each well and incubated for 4 h. Following removal of the medium,
200 μl dimethylsulfoxide was added to each well to dissolve
the formazan crystals. The absorbance at 490 nm was determined
using a microplate reader (Beijing Pulangxin Technology Co., Ltd.,
Beijing, China). The inhibition rate of cell proliferation was
calculated as follows: Inhibition rate (%) = A490 (control) − A490
(test) / A490 (control) − A490 (blank) × 100.
Flow cytometric analysis
To evaluate the effect of bufalin on K562 cell early
apoptosis and to determine a suitable concentration of bufalin for
microarray experiments, an Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit (BD
Immunocytometry Systems, Franklin Lakes, NJ, USA) for flow
cytometry was used to examine the apoptosis. K562 cells were
treated with 0.025, 0.05, 0.5 or 1.0 μmol/l bufalin for 48
h. The cells were washed twice with cold PBS and then stained with
Annexin V-FITC and PI according to the manufacturer’s instructions.
Samples were analyzed using a FACSCalibur flow cytometer (Beckman
Coulter, Inc., Brea, CA, USA) following staining.
RNA isolation and microarray
analysis
RNA extraction and purification
Total RNA was extracted using TRIzol reagent (cat.
no. 15596-018; Life technologies, Carlsbad, CA, USA) following the
manufacturer’s instructions, and checked for an RNA integrity
number to inspect RNA integration by an Agilent Bioanalyzer 2100
(Agilent technologies, Santa Clara, CA, USA). Qualified total RNA
was further purified using an RNeasy mini kit (cat. no. 74106;
Qiagen GmBH, Hilden, Germany) and RNase-Free DNase set (cat. no.
79254; Qiagen GmBH).
RNA amplification and labeling
Total RNA was amplified and labeled using a Low
Input Quick Amp Labeling kit, one-color (cat. no. 5190-2305;
Agilent Technologies), according to the manufacturer’s
instructions. Labeled cRNA were purified using the RNeasy mini kit
(cat. no. 74106; Qiagen GmBH).
Hybridization
Each slide was hybridized with 1.65 μg
Cy3-labeled cRNA using a Gene Expression Hybridization kit (cat.
no. 5188–5242; Agilent Technologies) in a hybridization oven (cat.
no. G2545A; Agilent Technologies), according to the manufacturer’s
instructions. Following 17 h of hybridization, slides were washed
in staining dishes (cat. no. 121; Thermo Shandon, Waltham, MA, USA)
with the Gene Expression Wash Buffer kit (cat. no. 5188–5327;
Agilent Technologies), following the manufacturer’s
instructions.
Data acquisition
Slides were scanned with an Agilent Microarray
Scanner (cat. no. G2565CA; Agilent Technologies) with the following
default settings: Dye channel, green; scan resolution, 5 μm;
PMT, 100%, 10%. Additionally, 16-bit Feature Extraction software
10.7 (Agilent Technologies) was used, and raw data were normalized
by a quantile algorithm (GeneSpring software 11.0; Agilent
Technologies).
qPCR
Total RNA was isolated and purified as previously
described. Reverse transcription of the total RNA was conducted
using the Cloned AMV First-Strand cDNA Synthesis kit (Invitrogen
Life Technologies, Carlsbad, CA, USA). qPCR was performed using
SYBR-Green chemistry in an ABI StepOnePlus system (Applied
Biosystems, New York, NY, USA). GAPDH served as a housekeeping
gene. PCR primer sequences were as follows: Forward:
5′-TGGGTGTGAACCATGAGAAGT-3′ and reverse:
5′-TGAGTCCTTCCACGATACCAA-3′ for human GAPDH; forward:
5′-CTTGGCTCGCATTCATTTTCT-3′ and reverse:
5′-CTCAGTGATCTTGATTCTCGTTGT-3′ for BMI-1; forward:
5′-GACAAAGAAAACGCCACAAATC-3′ and reverse:
5′-CTGAAACTGAATCCTGATCCAAC-3′ for MDM2; forward:
5′-GTCAGGACCTTCGTAGCATTG-3′ and reverse:
5′-CTCAGGGCACAGGAAAACATC-3′ for E2F; forward:
5′-GAGGGCTTCCTGGACACG-3′ and reverse: 5′-TCTTTCAATCGGGGATGTCTG- 3′
for p16INK4a; forward: 5′-TGTGGAGTTGGACTGAATGCT-3′ and
reverse: 5′-TGACAAGGTCTTCTCTTCATAGGTT-3′ for p14ARF; and
forward: 5′-GGAAGAAATACAGCCTGACGG-3′ and reverse:
5′-AGGAGGTTCCCGTAGGTCAT-3′ for BCR/ABL.
Triplicates were made for each sample. For each
sample, an amplification plot and corresponding dissociation curves
were examined. Relative quantification analysis was performed using
the comparative CT method (2−ΔΔCT). The formula was as
follows: 2−ΔΔCT = 2-[(CT gene of interest − CT
internal control) sample A − (CT gene of interest − CT internal
control) sample B)]
Statistical analysis
The data are expressed as the mean ± standard
deviation from experiments performed in triplicate. Student’s
t-test was used to identify statistically significant differences
between the experimental and control groups. The statistical
analyses were performed using SPSS software, version 13.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of bufalin on the growth of K562
cells
To investigate the effects of bufalin on K562 cells
and to determine a suitable concentration for microarray
experiments, the effect of various doses of bufalin on the
viability of K562 cells was tested using an MTT assay and flow
cytometry.
MTT results
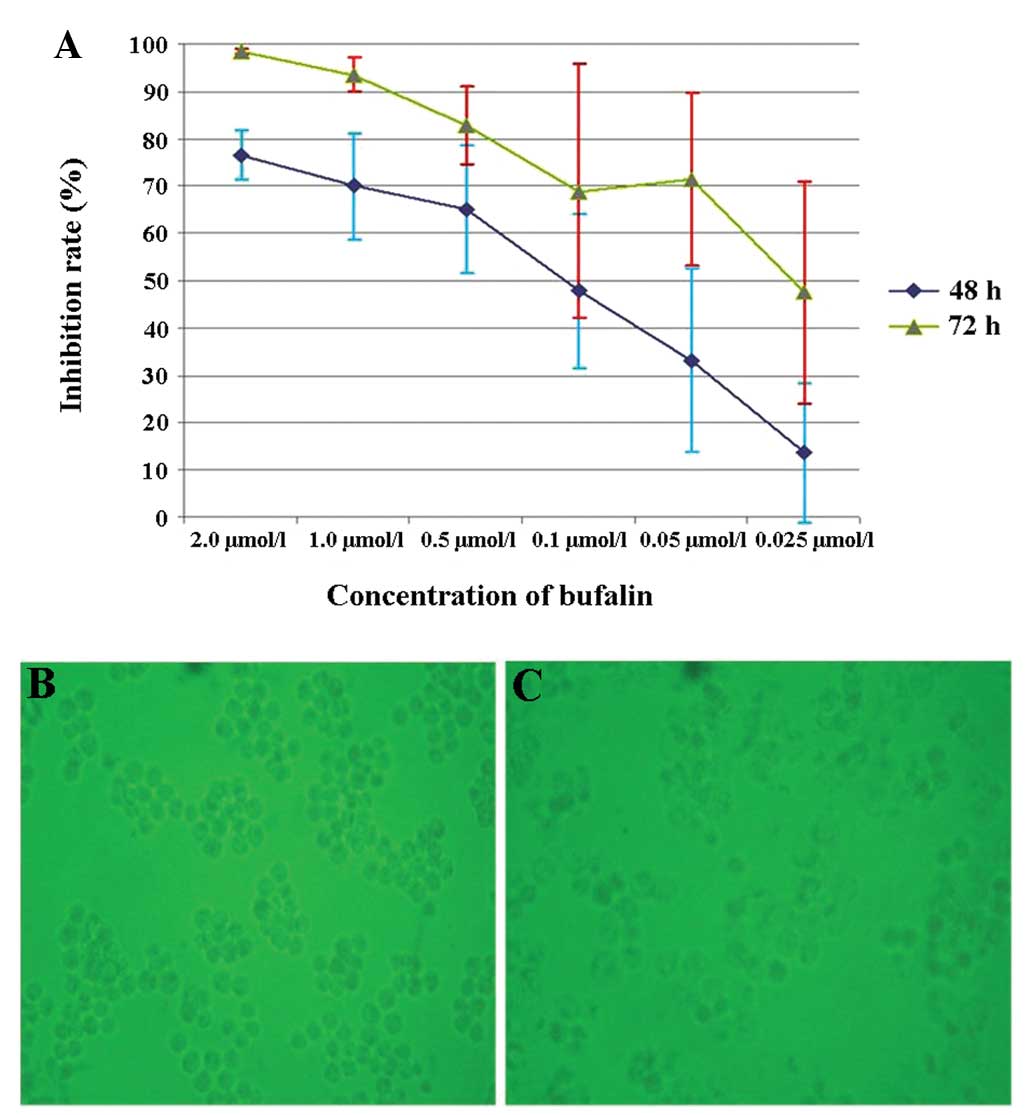
The cells were treated for 48 and 72 h as shown in
Fig. 1A, and cell growth was
inhibited by bufalin in a dose- and time-dependent manner (Fig. 1A–C). The calculated IC50
values (50% inhibitory concentration) were 0.153 and 0.028
μmol/l for cells treated for 48 and 72 h, respectively. The
difference in the apoptosis rate between bufalin treatment at a
concentration of 0.025 and 0.05 μmol/l at 48 h was
significant (P<0.05).
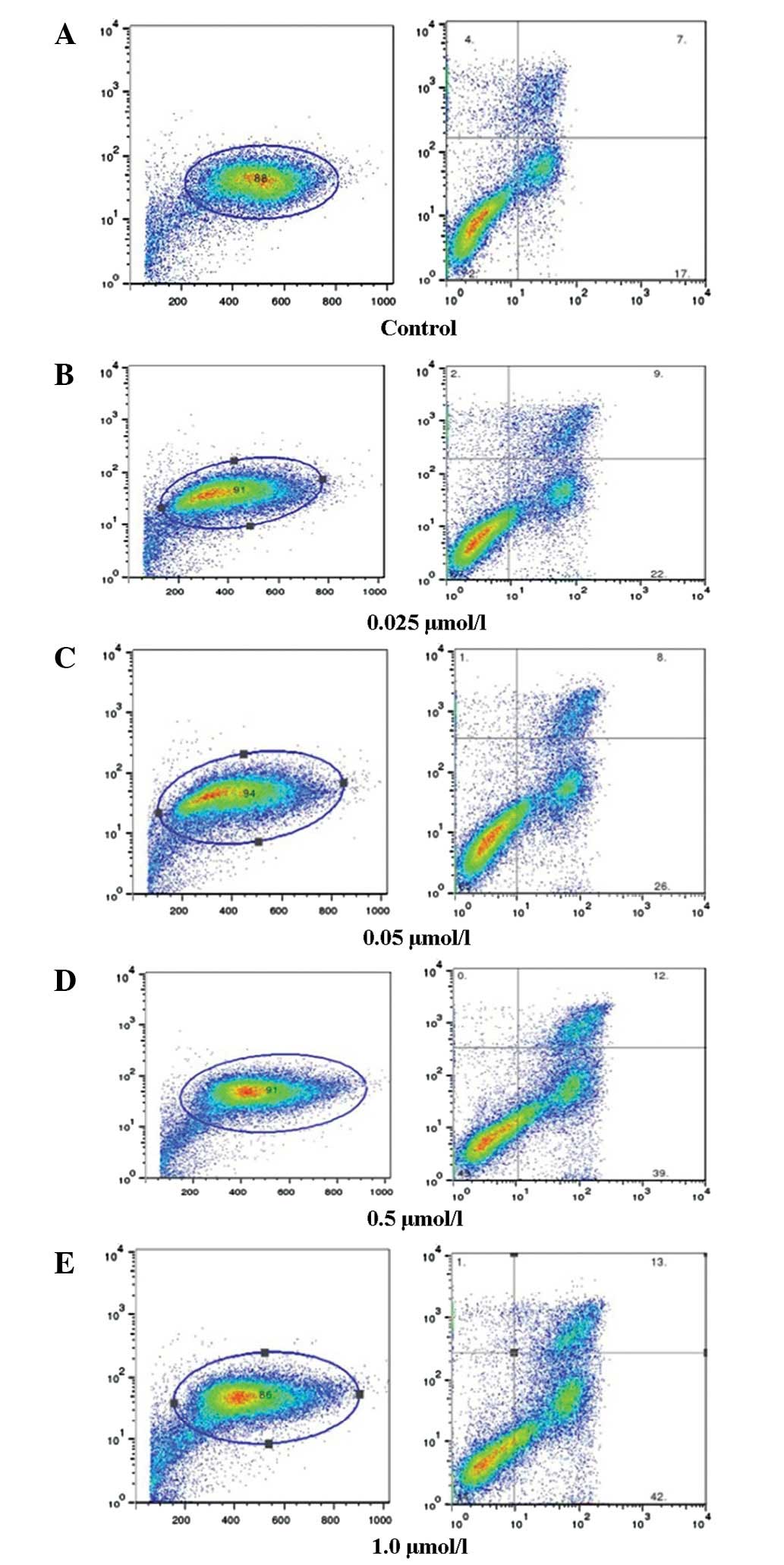
Flow cytometric analysis
K562 cells were treated with 0.025, 0.05, 0.5 or 1.0
μmol/l bufalin for 48 h. Flow cytometric analysis showed
that bufalin induced the apoptosis of cells in a dose-dependent
manner (Fig. 2A–E).
Microarray analysis of bufalin-induced
K562 cell apoptosis
As there is no standard concentration for the use of
bufalin in cell culture for microarray experiments, several
different concentrations were used to determine the effect of
bufalin on the growth of K562 cells at various time points. Chen
et al (5) used
IC50 values as their standard concentrations for
microarray analysis of bufalin-induced HL-60 apoptosis. In the
present study, based on the results of the MTT and flow cytometry
assays, a concentration of 0.5 μmol/l for 48 h was selected
for the microarray experiments. Cells that were not treated with
bufalin served as the control. Triplicates were made for the
samples treated with bufalin. Gene functional annotation analysis
and gene network analysis were performed using the Database for
Annotation, Visualization, and Integrated Discovery (DAVID;
http://david.abcc.ncifcrf.gov/). All
data underwent log transformation. A ratio of ≥1 was regarded as
upregulated and a ratio of ≤1 was considered as downregulated. The
results showed that bufalin induced significant changes in the gene
expression of K562 cells; 4296 genes were differentially expressed,
2185 were upregulated and 2111 were downregulated. As the focus of
the study was on enriched functional categories rather than on
individual genes, DAVID Annotation Cluster was employed to analyze
the molecular mechanisms of bufalin-induced apoptosis. Among the
genes with high fold-change values, the most upregulated genes were
associated with transcription regulation (Table I), while the most downregulated
genes were associated with the non-coding RNA (ncRNA) metabolic
process and DNA repair (Table
II). Moreover, a significant signaling pathway, the MAPK
pathway, was identified, which had been verified as a pathway in
bufalin-induced apoptosis (9). Of
the genes overexpressed by bufalin in K562 cells, BMI-1, E2F and
MDM2 were selected for qPCR analysis and the results were in
agreement with the microarray data (Table III).
 | Table IMost upregulated functional
annotation clusters of bufalin-induced apoptosis of K562 cells. |
Table I
Most upregulated functional
annotation clusters of bufalin-induced apoptosis of K562 cells.
| Category | Term | Count | % | P-value |
|---|
| GOTERM_BP_FAT | GO:0006350;
transcription | 258 | 19.06874 | 1.94 E-21 |
| GOTERM_BP_FAT | GO:0045449;
regulation of transcription | 297 | 21.95122 | 3.82 E-20 |
| GOTERM_BP_FAT | GO:0006355;
regulation of transcription, DNA-dependent | 204 | 15.07761 | 2.26 E-13 |
| GOTERM_BP_FAT | GO:0051252;
regulation of RNA metabolic process | 206 | 15.22542 | 5.77 E-13 |
| Table IIMost downregulated functional
annotation clusters of bufalin-induced K562 cell apoptosis. |
Table II
Most downregulated functional
annotation clusters of bufalin-induced K562 cell apoptosis.
| A, Annotation
cluster 1a |
|---|
|
|---|
| Category | Term | Count | % | P-value |
|---|
| GOTERM_BP_FAT | GO:0034660; ncRNA
metabolic process | 50 | 2.9036 | 6.25 E-11 |
| GOTERM_BP_FAT | GO:0034470; ncRNA
processing | 44 | 2.555168 | 7.16 E-11 |
| GOTERM_BP_FAT | GO:0006399; tRNA
metabolic process | 26 | 1.509872 | 3.38 E-06 |
| GOTERM_BP_FAT | GO:0008033; tRNA
processing | 20 | 1.16144 | 3.94 E-06 |
| GOTERM_BP_FAT | GO:0042254;
ribosome biogenesis | 26 | 1.509872 | 6.34 E-06 |
| GOTERM_BP_FAT | GO:0022613;
ribonucleoprotein complex biogenesis | 33 | 1.916376 | 8.37 E-06 |
| GOTERM_BP_FAT | GO:0006364; rRNA
processing | 21 | 1.219512 | 2.12 E-05 |
| GOTERM_BP_FAT | GO:0016072; rRNA
metabolic process | 21 | 1.219512 | 4.07 E-05 |
|
| B, Annotation
cluster 2b |
|
| Category | Term | Count | % | P-value |
|
| GOTERM_BP_FAT | GO:0006259; DNA
metabolic process | 60 | 3.484321 | 0.001126 |
| GOTERM_BP_FAT | GO:0006281; DNA
repair | 38 | 2.206736 | 0.001353 |
| GOTERM_BP_FAT | GO:0006974;
response to DNA damage stimulus | 45 | 2.61324 | 0.003657 |
| GOTERM_BP_FAT | GO:0033554;
cellular response to stress | 53 | 3.077816 | 0.121174 |
| Table IIIResults of qPCR confirmation of
differentially-expressed genes compared with microarray data. |
Table III
Results of qPCR confirmation of
differentially-expressed genes compared with microarray data.
| Fold
upregulation |
|---|
|
|
|---|
| Gene | qPCR | Microarray |
|---|
| BMI-1 | 1.63 | 1.11 |
| E2F-1 | 4.89 | 1.45 |
| MDM2 | 1.04 | 1.00 |
Effect of bufalin on the BMI-1,
p16INK4a, E2F, p14ARF, MDM2 and Brc/Abl
expression
K562 cells were treated with 0.5 or 0.05
μmol/l bufalin for 3, 6, 18, 24 or 48 h, respectively, and
then qPCR was performed. The results indicated that BMI-1, E2F and
MDM2 expression levels were upregulated in the K562 cells treated
with 0.5 μmol/l bufalin for 48 h, which was in agreement
with the results of the microarray experiment (Table III).
The expression of BMI-1 was decreased relative to
the baseline level at 18 and 24 h following treatment with a
concentration of 0.5 μmol/l bufalin, and then increased
1.63-fold 48 h after treatment. The minimum expression of the gene
was at 24 h after bufalin treatment. In addition, the expression of
BMI-1 target genes p16INK4a and p14ARF, and
the effect of bufalin on BCR/ABL expression was investigated. The
results indicated that the expression of p16INK4a began
to increase 18 h following bufalin treatment, and increased
107.60-fold 48 h after bufalin treatment. The rate of increase of
p14ARF expression was slower than that of
p16INK4a, and p14ARF expression initially
decreased and returned to the orignial level at 48 h after
treatment (Fig. 3A). The
expression of BCR/ABL decreased gradually in a time-dependent
manner, and had decreased 6.73-fold 48 h after bufalin treatment
(Fig. 3B).
The effect of bufalin treatment at a concentration
of 0.05 μmol/l on K562 cells was the same as that of 0.5
μmol/l, except that the minimum expression of BMI-1 was at
18 h following treatment. p16INK4a expression levels
began to increase 6 h following bufalin treatment and increased
30.18-fold 48 h after treatment. p14ARF expression began
to increase 24 h following bufalin treatment, and increased
1.26-fold 48 h after the treatment.
In conclusion, p16INK4a expression began
to increase as BMI-1 expression decreased, and the increase in
p14ARF expression lagged behind that of
p16INK4a. The expression of BCR/ABL does not appear to
be correlated with the upregulation or downregulation of BMI-1.
Bufalin-induced apoptosis may be mediated by downregulating the
expression of BMI-1, and accordingly upregulating the expression of
p16INK4a and p14ARF, and downregulating the
expression of BCR/ABL in K562 cells.
In addition, BMI-1 expression was observed in K562
cells treated with bufalin for 24 h. Bone marrow mononuclear cells
from healthy iron-deficiency anemia volunteers served as the normal
control. BMI-1 expression levels in the untreated control cells
were higher than those in the normal control cells, and they were
downregulated in the drug-treated cells (Fig. 4).
Discussion
The potential antitumor activity of natural products
used in traditional Chinese medicines has been noted (24). Cinobufacini (Huachansu), a Chinese
medicine prepared from dried toad skin, has been widely used for
the treatment of various types of cancer in China (25). Bufalin, which is the main active
component of cinobufacini, has attracted much attention from
researchers since the 1990’s (2).
Experiments have demonstrated that it is capable of inducing
apoptosis in a variety of types of tumor cell, including leukemia
cells such as U937, HL-60, ML1 and THP1 (5,6,9–11,26).
The mechanisms by which bufalin induces apoptosis are complex and
involve a diverse range of cell signals (2,3).
Data suggest that activation of the MAPK signaling pathway is one
of the most important signal transduction pathways for the
induction of apoptosis by bufalin (2). Bufalin is also able to induce
apoptosis of prostate cancer cells via p53- and Fas-mediated
apoptotic pathways (27). In
addition, bufalin induces lung cancer A549 cell apoptosis via
inhibition of the PI3K/Akt signaling pathway (28). Recently, it has been indicated that
bufalin-induced apoptosis is associated with the disruption of the
mitochondrial membrane potential, cytochrome c release, ROS
generation and caspase activation (29).
In the present study, it was demonstrated that
bufalin is able to induce K562 cell apoptosis in a time- and
dose-dependent manner, and the microarray analysis results suggest
that bufalin is able to induce significant changes in the gene
expression of K562 cells. The most upregulated functional
annotation cluster of bufalin-induced apoptosis of K562 cells was
associated with transcription regulation. For example, BMI-1, AFF4,
DDIT3, ING3, E2F1, E2F8, DENND4A, ATF, CCNL1, CCNL2, CCTI, CCT2,
CDK9, JUN, CDKN1B and certain zinc finger proteins were upregulated
in K562 cells treated with bufalin. DDIT3, also known as
CCAAT/enhancer binding protein ζ, has been demonstrated to be
involved in the regulation of cellular growth and differentiation
(30). The levels of DDIT3
transcription have been demonstrated to be downregulated in myeloid
malignancies (31), and
overexpression of DDIT3 transcripts has been shown to induce
increased apoptosis of myeloid cells and block cell progression
from the G1 to S phase (32,33).
ING3, a member of the ING family, has been reported to be involved
in p53-mediated transcription modulation, cell cycle control and
the induction of apoptosis. It was demonstrated that the expression
levels of ING3 are decreased in melanoma and head and neck squamous
cell carcinoma, and predict a poor prognosis (34). DENND4A, also known as C-myc
promoter-binding protein MBP-1, acts as a general transcriptional
repressor (35). It binds to c-myc
promoter sequences and transcriptionally represses the c-myc gene
(36–38). It has been demonstrated that
overexpression of the MBP-1 gene induces apoptosis in several types
of cancer cell line (39). The
family of zinc finger proteins is composed of regulatory proteins
that participate in a number of molecular and cellular pathways,
and is considered to be one of the most profuse regulatory protein
families in eukaryotic cells. Their functions are extremely diverse
and include DNA recognition, RNA packaging, transcriptional
activation, regulation of apoptosis, protein folding and assembly,
and lipid binding (40–41).
The present study demonstrated that bufalin
downregulated certain genes participating in ncRNA metabolic
processes, such as DGCR8, FTSJ3, MINA, NSUN2, U2AF1, CPSF3, GEMIN4,
LCMT2 and PRMT5. DGCR8 is an RNA-binding protein that assists the
RNase III enzyme Drosha in the processing of microRNAs (miRNAs); it
is required for the processing of pri-miRNAs to pre-miRNAs. Using
RNA from wild-type embryonic stem cells as a reference, Wang et
al (42) observed a global
loss of miRNAs in Dgcr8 knockout cells but identified normal
expression in heterozygous Dgcr8 cells, by using miRNA microarray
analysis. As a result, bufalin is able to interfere with the
processing of miRNAs via downregulating certain genes, such as
DGCR8.
FTSJ3 is a human NIP7-interacting protein. NIP7 is
one of the numerous trans-acting factors required for eukaryotic
ribosome biogenesis, which interacts with nascent pre-ribosomal
particles and dissociates as they complete maturation and are
exported to the cytoplasm. Conditional knockdown revealed that
depletion of FTSJ3 affects cell proliferation and causes pre-rRNA
processing defects (43). Mina53
is involved in cell division (44)
and is directly involved in ribosome biogenesis, most likely during
the assembly process of pre-ribosomal particles (45). It may be important in cell growth
and survival. The inhibition of Mina53 expression results in the
suppression of cell proliferation in certain cell lines (46). PRMT5 is a type II PRMT that
catalyzes the symmetrical dimethylation of arginine residues within
target proteins (47). PRMT5 is
highly conserved among yeast, animals and higher plants, and has
been implicated in diverse cellular and biological processes
including transcriptional regulation, RNA metabolism, ribosome
biogenesis, Golgi apparatus structural maintenance and cell cycle
progression (48). Overexpression
of PRMT5 promotes tumor cell growth and is associated with poor
disease prognosis (49).
Bufalin also downregulated certain genes associated
with DNA repair, such as RUVBL1, Cdc7, ERCC5, Grx2, MSH6, MUTYH,
NEIL1, NTHL1, PRMT6, tyrosyl-DNA phosphodiesterase I (Tdp1) and
MYC. RUVBL1 is a member of the AAA+ (ATPase associated with diverse
cellular activities) family of proteins that is associated with
several chromatin-remodeling complexes and is important in
transcriptional regulation, the DNA damage response, telomerase
activity, snoRNP assembly, cellular transformation and cancer
metastasis (50–52). RUVBL1 is overexpressed in a number
of different types of cancer and interacts with major oncogenic
factors, such as by regulating the function of β-catenin and c-Myc
(50–51). RUVBL1 blocks p53-mediated apoptosis
by repressing the expression of p53 and its target genes, and
siRUVBL1 is also able to induce the apoptotic death of HCT116
(p53−/−) cells, suggesting that RUVBL1 is able to suppress
apoptosis that is mediated by p53 and other molecules (53). Cdc7 is a conserved serine-threonine
kinase, as well as an essential replication regulator (54). Knockdown of Cdc7 has been
demonstrated to cause the death of cancer cells, but not normal
cells, via p53-dependent pathways which are able to arrest the cell
cycle, presumably in the G1 phase (55,56).
It has also been reported that Cdc7 knockdown induced p38-dependent
cell death in HeLa cells (57).
Cdc7 depletion is able to induce DNA damage in cancer cells
(58), and activated Chk2 is
capable of stabilizing the FoxM1 transcription factor, which
induces the expression of cyclin B1 (59). Thus, anticancer drugs, such as
bufalin, that facilitate Cdc7 depletion are able to induce cancer
cell death via numerous pathways.
Grx2 belongs to the oxidoreductase family and it has
been reported that Grx2 protects cells against a variety of
oxidative insults (60–62). Conversely, knockdown of Grx2
sensitizes HeLa cells to anticancer drugs (63). A study indicated that Grx2 also has
an anti-apoptotic function. Grx2 was demonstrated to protect cells
against H2O2-induced apoptosis via its
peroxidase and dethiolase activities; in particular, Grx2 prevents
complex I inactivation and preserves mitochondrial function
(64).
The DNA mismatch repair (MMR) system is one of a
number of DNA repair mechanisms. MSH6 is an MMR-related protein
that belongs to the MutS family. MSH proteins recognize errors in
the genome sequence during replication, and prevent the duplication
of the damaged strand and repair single strand breaks (65). Bufalin also downregulates certain
genes involved in the base excision repair and nucleotide excision
repair pathways of oxidization-induced DNA damage, such as MUTYH,
NEIL1 and NTHL1. Bufalin is capable of inducing apoptosis via a
ROS-dependent mitochondrial death pathway (29) and as a result, bufalin induces
apoptosis by inducing ROS production and inhibiting repair of DNA
damage induced by oxidization.
Tdp1 is a cellular enzyme that repairs the
irreversible topoisomerase I (Top1)-DNA complexes and confers
chemotherapeutic resistance to Top1 inhibitors. Inhibiting Tdp1
provides an attractive approach to potentiating clinically used
Top1 inhibitors (66,67). Bufalin is able to reduce the
quantity, activity and mRNA levels of topoisomerase II (11). Thus, bufalin is a Top II inhibitor
and a sensitizer that increases the cytotoxic effects of Top I
inhibitors.
The qPCR analysis of the present study demonstrated
that bufalin is able to downregulate BMI-1 expression levels in
K562 cells after 3 h. The expression levels in the K562 cells were
higher than in the normal controls and decreased following
treatment with bufalin. BMI-1 is required for maintenance and
self-renewal of normal and leukemia stem and progenitor cells.
Leukemia stem and progenitor cells lacking BMI-1 eventually undergo
proliferation arrest and exhibit signs of differentiation and
apoptosis, leading to transplant failure of the leukemia (20). Rizo et al (68) suggested that repression of BMI-1 in
cord blood CD34+ cells impaired their long-term
expansion and progenitor-forming capacity, in cytokine-driven
liquid cultures and in bone marrow stromal co-cultures. In
addition, the long-term culture-initiating cell frequencies were
markedly decreased upon knockdown of BMI-1, indicating an impaired
maintenance of stem and progenitor cells. The reduced progenitor
and stem cell frequencies were associated with increased expression
levels of p14ARF and p16INK4A and enhanced
apoptosis (68). It has been
verified that BMI-1 is overexpressed in hematological malignancies
and is correlated with a poor prognosis (22,69).
The findings of the present study also demonstrated that BMI-1 is
overexpressed in K562 cells, and that the increase in
p16INK4a and p14ARF expression levels is
associated with the downregulation of BMI-1 expression levels. We
propose that bufalin may induce K562 cell apoptosis via
downregulating BMI-1 expression and accordingly upregulating the
expression of p16INK4a and p14ARF.
To date, it has not been demonstrated that
expression of BMI-1 alone is sufficient to induce leukemia.
However, various studies suggest that BMI-1 may be an important
collaborating factor in the transformation process. For example,
BMI-1 is able to cooperate with H-RAS to induce aggressive breast
cancer with brain metastases (70,71).
Using qPCR, Merkerova et al (72) demonstrated significantly increased
levels of BMI-1 transcripts in chronic myelogenous leukemia (CML)
cells. Using siRNA silencing, the impact of BMI-1 inhibition on the
growth and cell cycle of BCR/ABL-positive CML cell lines was
measured, and it was demonstrated that BMI-1 inhibition did not
affect the proliferation rate or the cell cycle of these cells. As
it is well-known that the growth deregulation in CML cells is
caused by the BCR/ABL oncogene, BMI-1 may have a secondary role in
the proliferation of these cells (73). Merkerova et al (72) also reduced BCR/ABL expression by
siRNA in K562 cells to determine if BMI-1 upregulation was
dependent on BCR/ABL expression, and demonstrated that the BMI-1
transcript levels did not change after BCR/ABL inhibition, which
suggests that the increased expression levels of BMI-1 are not
directly associated with BCR/ABL. In the present study, the
expression of BCR/ABL did not appear to be correlated with the
upregulation or downregulation of BMI-1 expression levels. Bufalin
may induce K562 cell apoptosis via downregulating BCR/ABL
expression levels, and this pathway may occur independently of the
BMI-1 pathway.
It has become clear that BMI-1 also functions in
protecting against oxidative stress. In the absence of BMI-1, ROS
accumulate and are associated with the activation of DNA damage
response pathways and increased apoptosis. BMI-1-mediated control
over ROS levels may occur via impaired mitochondrial functions,
independent of the INK4a/ARF pathway (19). BMI-1 may be required to protect
hematopoietic stem/progenitor cells from apoptosis induced by
oxidative stress conditions (68).
As mentioned previously, bufalin is able to induce apoptosis via a
ROS-dependent mitochondrial death pathway and inhibit repair of DNA
damage induced by oxidization (29). The association between bufalin,
BMI-1 and DNA damage repair genes requires further
investigation.
The signaling pathways that regulate BMI-1 have not
been extensively studied. SALL4, an oncogene that is expressed in
AML, is capable of upregulating BMI-1 expression by directly
binding to its promoter (74).
Furthermore, BMI-1 expression appears to be under the control of
miRNAs, including miRNA-128. miRNA-128 causes a marked decrease in
the expression levels of BMI-1 by direct regulation of the BMI-1
mRNA 3-untranslated region, through a single miRNA-128 binding site
(75,76). miRNA-128a increases intracellular
ROS levels by targeting BMI-1 and inhibits medulloblastoma cancer
cell growth by promoting senescence (77). However, BMI-1 is not expressed in a
cell cycle-regulated manner in various types of primary and
transformed cells and is not affected by overexpression of
p16INK4a (19). In the
present study, BMI-1 expression levels increased 48 h following
bufalin treatment. Although the mechanism requires in-depth
research, we propose certain potential possibilities: i) Bufalin
may upregulate genes upstream of BMI-1, such as SALL4; ii) bufalin
may downregulate mRNAs and eliminate their suppression of BMI-1,
for example, bufalin is capable of interfering with the processing
of miRNA via downregulating DGCR8; and iii) transcription factor
E2F-1 may directly regulate BMI-1, which represses the INK4A gene,
thereby promoting phosphorylation of the pRb and activation of E2F,
as has been demonstrated previously (78). The present study showed that
bufalin is able to upregulate E2F-1 expression and this may be a
reason why BMI-1 increased 48 h after drug treatment. The phenomena
of BMI-1 expression levels increasing at later time also suggests
that BMI-1 alone cannot induce apoptosis of K562 cells.
References
|
1
|
Krenn L and Kopp B: Bufadienolides from
animal and plant sources. Phytochemistry. 48:1–29. 1998.PubMed/NCBI
|
|
2
|
Qi F, Li A, Inagaki Y, Kokudo N, Tamura S,
Nakata M and Tang W: Antitumor activity of extracts and compounds
from the skin of the toad Bufo bufo gargarizans Cantor. Int
Immunopharmacol. 11:342–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takai N, Kira N, Ishii T, Yoshida T,
Nishida M, Nishida Y, Nasu K and Narahara H: Bufalin, a traditional
oriental medicine, induces apoptosis in human cancer cells. Asian
Pac J Cancer Prev. 13:399–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang L, Nakaya K, Yoshida T and Kuroiwa
Y: Induction by bufalin of differentiation of human leukemia cells
HL60, U937, and ML1 toward macrophage/monocyte-like cells and its
potent synergistic effect on the differentiation of human leukemia
cells in combination with other inducers. Cancer Res. 52:4634–4641.
1992.
|
|
5
|
Chen A, Yu J, Zhang L, Sun Y, Zhang Y, Guo
H, Zhou Y, Mitchelson K and Cheng J: Microarray and biochemical
analysis of bufalin-induced apoptosis of HL-60 cells. Biotechnol
Lett. 31:487–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang C, Chen A, Guo M and Yu J: Membrane
dielectric responses of bufalin-induced apoptosis in HL-60 cells
detected by an electrorotation chip. Biotechnol Lett. 29:1307–1313.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Numazawa S, Shinoki MA, Ito H, Yoshida T
and Kuroiwa Y: Involvement of Na+, K(+)-ATPase inhibition in K562
cell differentiation induced by bufalin. J Cell Physiol.
160:113–210. 1994.
|
|
8
|
Kawazoe N, Aiuchi T, Masuda Y, Nakajo S
and Nakaya K: Induction of apoptosis by bufalin in human tumor
cells is associated with a change of intracellular concentration of
Na+ ions. J Biochem. 126:278–286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watabe M, Masuda Y, Nakajo S, Yoshida T,
Kuroiwa Y and Nakaya K: The cooperative interaction of two
different signaling pathways in response to bufalin induces
apoptosis in human leukemia U937 cells. J Biol Chem.
271:14067–14072. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Masuda Y, Kawazoe N, Nakajo S, Yoshida T,
Kuroiwa Y and Nakaya K: Bufalin induces apoptosis and influences
the expression of apoptosis-related genes in human leukemia cells.
Leuk Res. 19:549–556. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hashimoto S, Jing Y, Kawazoe N, Masuda Y,
Nakajo S, Yoshidaz T, et al: Bufalin reduces the level of
topoisomerase II in human leukemia cells and affects the
cytotoxicity of anticancer drugs. Leuk Res. 21:875–883. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park IK, Qian D, Kiel M, et al: Bmi-1 is
required for maintenance of adult self-renewing haematopoietic stem
cells. Nature. 423:302–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dick JE: Stem cells: self-renewal writ in
blood. Nature. 423:231–233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lessard J and Sauvageau G: Bmi-1
determines the proliferative capacity of normal and leukaemic stem
cells. Nature. 423:255–260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iwama A, Oguro H, Negishi M, et al:
Enhanced self-renewal of hematopoietic stem cells mediated by the
polycomb gene product Bmi-1. Immunity. 21:843–851. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alkema MJ, Wiegant J, Raap AK, Berns A and
van Lohuizen M: Characterization and chromosomal localization of
the human proto-oncogene BMI-1. Hum Mol Genet. 2:1597–1603. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and polycomb-group gene bmi-1
regulates cell proliferation and senescence through the Ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dhawan S, Tschen SI and Bhushan A: Bmi-1
regulates the Ink4a/Arf locus to control pancreatic beta-cell
proliferation. Genes Dev. 23:906–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schuringa JJ and Vellenga E: Role of the
polycomb group gene BMI1 in normal and leukemic hematopoietic stem
and progenitor cells. Curr Opin Hematol. 17:294–299. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lessard J and Sauvageau G: Bmi-1
determines the proliferative capacity of normal and leukaemic stem
cells. Nature. 423:255–260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Valk-Lingbeek ME, Bruggeman SW and van
Lohuizen M: Stem cells and cancer; the polycomb connection. Cell.
118:409–418. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu W, Huang L, Xu X, Qian H and Xu W:
Anti-proliferation effect of BMI-1 in U937 cells with siRNA. Int J
Mol Med. 25:889–895. 2010.PubMed/NCBI
|
|
23
|
Meng XX, Liu WH, Liu DD, Zhao XY and Su
BL: Construction of antisense Bmi-1 expression plasmid and its
inhibitory effect on K562 cells proliferation. Chin Med J (Engl).
118:1346–1350. 2005.PubMed/NCBI
|
|
24
|
Mehta RG, Murillo G, Naithani R and Peng
X: Cancer chemoprevention by natural products: how far have we
come? Pharm Res. 27:950–961. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge
Y, Newman RA, Cohen L, Liu L, Thornton B, Chang DZ, Liao Z and
Kurzrock R: Pilot study of huachansu in patients with
hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic
cancer. Cancer. 115:5309–5318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kawazoe N, Aiuchi T, Masuda Y, Nakajo S
and Nakaya K: Induction of apoptosis by bufalin in human tumor
cells is associated with a change of intracellular concentration of
Na+ ions. J Biochem. 126:278–286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu CH, Kan SF, Pu HF, Jea Chien E and Wang
PS: Apoptotic signaling in bufalin- and cinobufagin-treated
androgen-dependent and -independent human prostate cancer cells.
Cancer Sci. 99:2467–2476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu Z, Sun H, Ma G, Wang Z, Li E and Liu Y
and Liu Y: Bufalin induces lung cancer cell apoptosis via the
inhibition of PI3K/Akt pathway. Int J Mol Sci. 13:2025–2035. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun L, Chen T, Wang X, Chen Y and Wei X:
Bufalin induces reactive oxygen species dependent bax translocation
and apoptosis in ASTC-a-1 cells. Evid Based Complement Alternat
Med. 2011:2490902011.PubMed/NCBI
|
|
30
|
Wang YL, Qian J, Lin J, Yao DM, Qian Z,
Zhu ZH and Li JY: Methylation status of DDIT3 gene in chronic
myeloid leukemia. J Exp Clin Cancer Res. 29:542010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qian J, Chen Z, Lin J, Wang W and Cen J:
Decreased expression of CCAAT/enhancer binding protein zeta
(C/EBPzeta) in patients with different myeloid diseases. Leuk Res.
29:1435–1441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Friedman AD: GADD153/CHOP, a DNA
damage-inducible protein, reduced CAAT/enhancer binding protein
activities and increased apoptosis in 32D c13 myeloid cells. Cancer
Res. 56:3250–3256. 1996.PubMed/NCBI
|
|
33
|
Matsumoto M, Minami M, Takeda K, Sakao Y
and Akira S: Ectopic expression of CHOP (GADD153) induces apoptosis
in M1 myeloblastic leukemia cells. FEBS Lett. 395:143–147. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu M, Chen F, Wang Q, Wang K, Pan Q and
Zhang X: Downregulation of inhibitor of growth 3 is correlated with
tumorigenesis and progression of hepatocellular carcinoma. Oncol
Lett. 4:47–52. 2012.PubMed/NCBI
|
|
35
|
Ghosh AK, Steele R and Ray RB: Functional
domains of c-myc promoter binding protein 1 involved in
transcriptional repression and cell growth regulation. Mol Cell
Biol. 19:2880–2886. 1999.PubMed/NCBI
|
|
36
|
Ray R and Miller DM: Cloning and
characterization of a human c-myc promoter-binding protein. Mol
Cell Biol. 11:2154–2161. 1996.PubMed/NCBI
|
|
37
|
Chaudhary D and Miller DM: The c-myc
promoter binding protein (MBP-1) and TBP bind simultaneously in the
minor groove of the c-myc P2 promoter. Biochemistry. 34:3438–3445.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ray RB: Induction of cell death in murine
fibroblasts by a c-myc promoter binding protein. Cell Growth
Differ. 6:1089–1096. 1995.PubMed/NCBI
|
|
39
|
Ghosh AK, Steele R, Ryerse J and Ray RB:
Tumor-suppressive effects of MBP-1 in non-small cell lung cancer
cells. Cancer Res. 66:11907–11912. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim MK, Jeon BN, Koh DI, Park SY, Yun CO
and Hur MW: Regulation of the cyclin-dependent kinase inhibitor 1A
gene (CDKNIA) by the repressor BOZF1 through inhibition of p53
acetylation and transcription factor Sp1 binding. J Biol Chem.
288:7053–7064. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Laity JH, Lee BM and Wright PE: Zinc
finger proteins: new insights into structural and functional
diversity. Curr Opin Struct Biol. 11:39–46. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Medvid R, Melton C, Jaenisch R and
Blelloch R: DGCR8 is essential for microRNA biogenesis and
silencing of embryonic stem cell self-renewal. Nat Genet.
39:380–385. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morello LG, Coltri PP, Quaresma AJ,
Simabuco FM, Silva TC, Singh G, Nickerson JA, Oliveira CC, Moore MJ
and Zanchin NI: The human nucleolar protein FTSJ3 associates with
NIP7 and functions in pre-rRNA processing. PLoS One. 6:e291742011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsuneoka M, Koda Y, Soejima M, Teye K and
Kimura H: A novel myc target gene, mina53, that is involved in cell
proliferation. J Biol Chem. 277:35450–35459. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Eilbracht J, Kneissel S, Hofmann A and
Schmidt-Zachmann MS: Protein NO52-a constitutive nucleolar
component sharing high sequence homologies to protein NO66. Eur J
Cell Biol. 84:279–294. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tan XP, Zhang Q, Dong WG, Lei XW and Yang
ZR: Upregulated expression of Mina53 in cholangiocarcinoma and its
clinical significance. Oncol Lett. 3:1037–1041. 2012.PubMed/NCBI
|
|
47
|
Bedford MT and Clarke SG: Protein arginine
methylation in mammals: who, what, and why. Mol Cell. 33:1–13.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gu Z, Li Y, Lee P, Liu T, Wan C and Wang
Z: Protein arginine methyltransferase 5 functions in opposite ways
in the cytoplasm and nucleus of prostate cancer cells. PLoS One.
7:e440332012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bao X, Zhao S, Liu T, Liu Y, Liu Y and
Yang X: Overexpression of PRMT5 promotes tumor cell growth and is
associated with poor disease prognosis in epithelial ovarian
cancer. J Histochem Cytochem. 61:206–217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gallant P: Control of transcription by
Pontin and Reptin. Trends Cell Biol. 17:187–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huber O, Ménard L, Haurie V, Nicou A,
Taras D and Rosenbaum J: Pontin and reptin, two related ATPases
with multiple roles in cancer. Cancer Res. 68:6873–6876. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jha S and Dutta A: RVB1/RVB2: running
rings around molecular biology. Mol Cell. 34:521–533. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Taniue K, Oda T, Hayashi T, Okuno M and
Akiyama T: A member of the ETS family, EHF, and the ATPase RUVBL1
inhibit p53-mediated apoptosis. EMBO Rep. 12:682–689. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ito S, Ishii A, Kakusho N, Taniyama C,
Yamazaki S, Fukatsu R, Sakaue-Sawano A, Miyawaki A and Masai H:
Mechanism of cancer cell death induced by depletion of an essential
replication regulator. PLoS One. 7:e363722012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Montagnoli A, Tenca P, Sola F, Carpani D,
Brotherton D, Albanese C and Santocanale C: Cdc7 inhibition reveals
a p53-dependent replication checkpoint that is defective in cancer
cells. Cancer Res. 64:7110–7116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tudzarova S, Trotter MW, Wollenschlaeger
A, Mulvey C, Godovac-Zimmermann J, Williams GH and Stoeber K:
Molecular architecture of the DNA replication origin activation
checkpoint. EMBO J. 29:3381–3394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Im JS and Lee JK: ATR-dependent activation
of p38 MAP kinase is responsible for apoptotic cell death in cells
depleted of Cdc7. J Biol Chem. 283:25171–25177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim JM, Kakusho N, Yamada M, Kanoh Y,
Takemoto N and Masai H: Cdc7 kinase mediates Claspin
phosphorylation in DNA replication checkpoint. Oncogene.
27:3475–3482. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tan Y, Raychaudhuri P and Costa RH: Chk2
mediates stabilization of the FoxM1 transcription factor to
stimulate expression of DNA repair genes. Mol Cell Biol.
27:1007–1016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Daily D, Vlamis-Gardikas A, Offen D,
Mittelman L, Melamed E, Holmgren A and Barzilai A: Glutaredoxin
protects cerebellar granule neurons from dopamine-induced apoptosis
by dual activation of the ras-phosphoinositide 3-kinase and jun
n-terminal kinase pathways. J Biol Chem. 276:21618–21626. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Enoksson M, Fernandes AP, Prast S, Lillig
CH, Holmgren A and Orrenius S: Overexpression of glutaredoxin 2
attenuates apoptosis by preventing cytochrome c release. Biochem
Biophys Res Commun. 327:774–779. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Diotte NM, Xiong Y, Gao J, Chua BH and Ho
YS: Attenuation of doxorubicin-induced cardiac injury by
mitochondrial glutaredoxin 2. Biochim Biophys Acta. 1793:427–438.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lillig CH, Lonn ME, Enoksson M, Fernandes
AP and Holmgren A: Short interfering RNA-mediated silencing of
glutaredoxin 2 increases the sensitivity of HeLa cells toward
doxorubicin and phenylarsine oxide. Proc Natl Acad Sci USA.
101:13227–13232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu H, Xing K and Lou MF: Glutaredoxin 2
prevents H(2)O(2)-induced cell apoptosis by protecting complex I
activity in the mitochondria. Biochim Biophys Acta. 1797:1705–1715.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Conde-Pérezprina JC, León-Galván MÁ and
Konigsberg M: DNA mismatch repair system: repercussions in cellular
homeostasis and relationship with aging. Oxid Med Cell Longev.
2012:7284302012.PubMed/NCBI
|
|
66
|
Sirivolu VR, Vernekar SK, Marchand C,
Naumova A, Chergui A, Renaud A, Stephen AG, Chen F, Sham YY,
Pommier Y and Wang Z: 5-Arylidenethioxothiazolidinones as
inhibitors of tyrosyl-DNA phosphodiesterase I. J Med Chem.
55:8671–8684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Alagoz M, Gilbert DC, El-Khamisy S and
Chalmers AJ: DNA repair and resistance to topoisomerase I
inhibitors: mechanisms, biomarkers and therapeutic targets. Curr
Med Chem. 19:3874–3885. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rizo A, Olthof S, Han L, Vellenga E, de
Haan G and Schuringa JJ: Repression of BMI1 in normal and leukemic
human CD34(+) cells impairs self-renewal and induces apoptosis.
Blood. 114:1498–1505. 2009.PubMed/NCBI
|
|
69
|
Bhattacharyya J, Mihara K, Yasunaga S,
Tanaka H, Hoshi M, Takihara Y and Kimura A: BMI-1 expression is
enhanced through transcriptional and posttranscriptional regulation
during the progression of chronic myeloid leukemia. Ann Hematol.
88:333–340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Datta S, Hoenerhoff MJ, Bommi P, Sainger
R, Guo WJ, Dimri M, Band H, Band V, Green JE and Dimri GP: Bmi-1
cooperates with H-Ras to transform human mammary epithelial cells
via dysregulation of multiple growth-regulatory pathways. Cancer
Res. 67:10286–10295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hoenerhoff MJ, Chu I, Barkan D, Liu ZY,
Datta S, Dimri GP and Green JE: BMI1 cooperates with H-RAS to
induce an aggressive breast cancer phenotype with brain metastases.
Oncogene. 28:3022–3032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Merkerova M, Bruchova H, Kracmarova A,
Klamova H and Brdicka R: Bmi-1 over-expression plays a secondary
role in chronic myeloid leukemia transformation. Leuk Lymphoma.
48:793–801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Danisz K and Blasiak J: Role of
anti-apoptotic pathways activated by BCR/ABL in the resistance of
chronic myeloid leukemia cells to tyrosine kinase inhibitors. Acta
Biochim Pol. 60:503–514. 2013.PubMed/NCBI
|
|
74
|
Yang J, Chai L, Liu F, Fink LM, Lin P,
Silberstein LE, Amin HM, Ward DC and Ma Y: Bmi-1 is a target gene
for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci
USA. 104:10494–10499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Godlewski J, Nowicki MO, Bronisz A,
Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca
EA and Lawler S: Targeting of the Bmi-1 oncogene/stem cell renewal
factor by microRNA-128 inhibits glioma proliferation and
self-renewal. Cancer Res. 68:9125–9130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wu XM, Liu X, Bu YQ, Sengupta J, Cui HJ,
Yi FP, Liu T, Yuan CF, Shi YY and Song FZ: RNAi-mediated silencing
of the Bmi-1 gene causes growth inhibition and enhances
doxorubicin-induced apoptosis in MCF-7 cells. Genet Mol Biol.
32:697–703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Venkataraman S, Alimova I, Fan R, Harris
P, Foreman N and Vibhakar R: MicroRNA 128a increases intracellular
ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer
cell growth by promoting senescence. PLoS One. 5:e107482010.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nowak K, Kerl K, Fehr D, Kramps C, Gessner
C, Killmer K, Samans B, Berwanger B, Christiansen H and Lutz W:
BMI1 is a target gene of E2F-1 and is strongly expressed in primary
neuroblastomas. Nucleic Acids Res. 34:1745–1754. 2006. View Article : Google Scholar : PubMed/NCBI
|