Introduction
Retinitis pigmentosa (RP) is a rare, inherited
disease, which is caused by the progressive loss of rod and cone
photoreceptors. The clinical hallmarks of RP include sensitivity to
dim light, abnormal visual function and characteristic bone spicule
deposits of pigment in the retina (1). RP has been associated with mutations
in >45 genes, a number of which affect proteins expressed
exclusively in rod photoreceptor cells, including the α and β
subunits of the rod cyclic guanosine monophosphate (cGMP)
phosphodiesterase, the rod cGMP-gated channel and rhodopsin
(2). Rhodopsin is a photon
receptor and light-sensitive visual pigment, which initiates
phototransduction. Autosomal dominant retinitis pigmentosa (ADRP)
is associated with mutations in rhodopsin (2). At present, >140 RP-associated
rhodopsin mutations have been identified (3). The human rhodopsin gene encodes a
348-amino acid protein that has seven transmembrane domains, with a
luminal N terminus and a cytoplasmic C terminus (4). Dominant rhodopsin mutations are
divided into class I, II and III (5,6).
T17M rhodopsin is a class II rhodopsin mutant and it lacks the
capacity to form the normal rhodopsin chromophore with
11-cis-retinal due to a defect in thermal stability folding
(7). Patients carrying the T17M
rhodopsin mutant have altitudinal visual field defects, associated
with less impaired rod and cone functions in the inferior than in
the superior field (8,9). Expression of the human T17M mutant
rhodopsin transgene in mice has been associated with photoreceptor
apoptosis in response to moderate exposure to light. However, this
phenotype was not observed in non-transgenic littermates or in mice
expressing the human P28H mutant rhodopsin transgene. This finding
suggests that the T17M mutation abolishes glycosylation at the
Asn-15 site of rhodopsin and that this elimination of glycosylation
is associated with an increased sensitivity to light-induced damage
(10).
The present study aimed to investigate the
differences in the localization and degradation of RP-associated
T17M mutant rhodopsin compared with wild-type (WT) rhodopsin,
elucidate the role of endoplasmic reticulum (ER) stress and
identify potential candidates for therapy.
Materials and methods
Cell culture, plasmid constructs and
transfection
Human embryonic kidney 293 (HEK-293) and ARPE-19
retinal pigmented epithelium cells were obtained from American Type
Culture Collection (Rockville, MD, USA) and maintained according to
the providers instructions. HEK-293 and ARPE-19 cells were cultured
in Dulbecco’s modified Eagle’s medium (DMEM) and DMEM/F-12,
respectively, supplemented with 10% fetal bovine serum and 50 μg/ml
penicillin-streptomycin at 37°C and 5% CO2. The coding
sequence of the rhodopsin gene was amplified from a human cDNA
library (Invitrogen Life Technologies, Carlsbad, CA, USA) using LA
Taq DNA polymerase (Takara Bio Inc., Shiga, Japan). The
fragment was ligated into the mammalian expression vector
pcDNA™3.1/myc HisA(+) (Invitrogen Life Technologies) and
transformed into competent Escherichia coli DH5α cells
according to the manufacturer’s instructions. The integrity of all
inserts was determined using direct sequence analysis and
transfections were performed using Lipofectamine® 2000
(Invitrogen Life Technologies) according to the manufacturer’s
instructions. In brief, cells were cultured in 12-well plates in
serum-free DMEM supplemented with 1.6 μg DNA and 4 μl Lipofectamine
2000 for 4 h. The transfection media was then replaced and
incubated with fresh standard HEK-293 media.
Immunofluorescence microscopy
Cells expressing WT and T17M rhodopsin were cultured
on glass coverslips for 24–48 h. Cells were then fixed using 4%
paraformaldehyde for 10 min and permeabilized using 0.1% Triton
X-100 in phosphate-buffered saline (PBS) for 10 min at 25°C.
Subsequent to blocking with 3% bovine serum albumin for 30 min,
cells were incubated with mouse anti-myc (dilution, 1:300) and
rabbit anti-protein disulfide isomerase (PDI) (dilution, 1:300)
antibodies (Sigma-Aldrich, St. Louis, MO, USA). Cells were then
incubated with Alexa Fluor® 488-labeled goat anti-mouse
IgG and Alexa Fluor® 549-labeled goat anti-rabbit IgG
antibodies (dilution, 1:400; Invitrogen Life Technologies). Cell
nuclei were counter-stained using DAPI (Invitrogen Life
Technologies). Images were captured using a Leica microscope (Leica
Microsystems, Wetzlar, Germany) with appropriate excitation and
emission filter pairs.
Western blot analysis
For the degradation experiment, cells were treated
with or without 10 μM proteasome inhibitor MG132 for 12 h (Sigma
Aldrich). For the turnover rate experiment, ARPE-19 cells were
treated with 50 μg/ml cycloheximide for 0, 2, 4 or 6 h. (Sigma
Aldrich). Subsequently, the cells were lysed in 2X SDS sample
buffer (63 mM Tris HCl, 10% glycerol and 2% SDS). The supernatant
was collected and the protein concentration was determined using a
Pierce™ protein assay kit (Pierce Chemical Company, Rockford, IL,
USA). Protein extracts (30 μg) were separated using SDS-PAGE and
were then transferred to polyvinylidene fluoride membranes
(Millipore Corporation, Billerica, MA, USA). Membranes were
incubated for 1 h in blocking solution (5% dry milk in 0.1% Triton
X-100/PBS), followed by incubation with mouse anti-myc (dilution,
1:5,000; Sigma-Aldrich), mouse anti-binding immunoglobulin protein
(BiP; dilution, 1:1,000; Sigma-Aldrich) and mouse anti-actin
(dilution, 1:10,000; Sigma-Aldrich) antibodies in blocking
solution. Subsequent to being washed with 0.1% Triton X-100/PBS
buffer, membranes were incubated with goat anti-mouse IgG antibody
conjugated to horseradish peroxidase (dilution, 1:10,000;
Invitrogen Life Technologies) for 1 h and visualized using an
enhanced chemiluminescence kit according to the manufacturer’s
instructions (GE Healthcare, Little Chalfont, UK).
Cell death detection
Following a pre-coat with 50 μg/ml poly-L-lysine,
HEK293 cells were transfected with myc-tagged WT or T17M mutant
vectors. A total of 20 h after transfection, cells were treated
with dimethylsulfoxide (DMSO; Sigma-Aldrich), 2 mg/ml tunicamycin
for 8 h, or 100 μM ROS scavengers butylated hydroxyanisole (BHA;
Sigma-Aldrich) and 1 mM N-acetylcysteine (NAC;
Sigma-Aldrich) for 12 h, respectively. Cell death was then
quantified by counting the cells that exhibited highly condensed
and/or fragmented DNA subsequent to staining with Hoechst 33342
nuclear dye. At least 1,000 cells were counted per well.
ROS detection
Cells were digested with 0.125% trypsin, pelleted
and suspended in medium containing 20 μM
dichloro-dihydro-fluorescein diacetate (Sigma-Aldrich). After
incubation for 30 min, cells were centrifuged at 2000 × g for 10
min, resuspended in fresh medium and subjected to
fluorescence-activated cell sorting analysis using the MoFLo™ XDP
cell sorter (Beckman Coulter, Inc., Brea, CA, USA). Data were
analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA)
in order to assess the mean fluorescence intensity. Data are
presented relative to the control cells.
Statistical analysis
Data represent quantification of three independent
experiments. Results are presented as the mean ± standard
deviation. One way analysis of variance followed by Tukey or
Dunnett’s tests were performed using GraphPad Prism 5 software
(GraphPad Software, San Diego California USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
RP-associated T17M mutant rhodopsin is
translocated to the ER
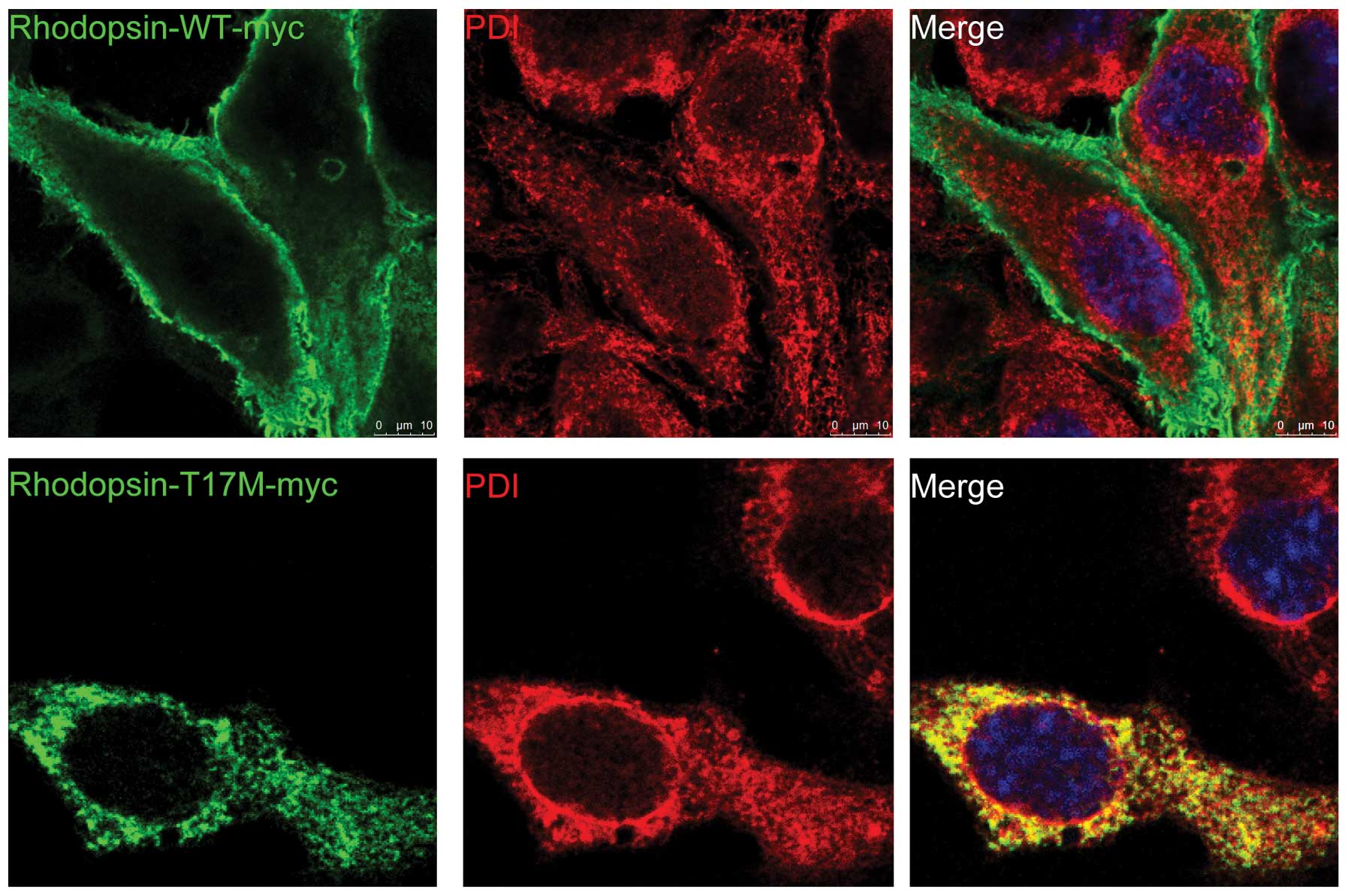
In order to investigate whether WT and T17M
rhodopsin have a similar localization, myc-tagged WT and T17M
proteins were heterologously expressed in ARPE-19 cells and the
proteins were visualized using immunofluorescence microscopy.
Following transient transfection, double immunofluorescence
staining revealed that the T17M rhodopsin mutant protein was
localized in the ER, as evidenced by the strong co-localization
with the ER marker PDI (Fig. 1).
By contrast, WT rhodopsin was observed to be localized at the cell
membrane and did not co-localize with PDI (Fig. 1).
T17M mutant rhodopsin is unstable and
degraded by proteasomes
It was examined whether accumulation of the T17M
protein in HEK293 cells was sensitive to proteasome inhibition,
which is a characteristic of ER-associated protein degradation
(ERAD) substrates. Cells were treated with or without the
proteasome inhibitor MG132 (10 μM for 12 h) and the accumulation of
transiently expressed myc-tagged WT and T17M rhodopsin proteins
were compared in HEK293 cells. The levels of WT and T17M rhodopsin
were observed to markedly increase following MG132 treatment.
However, the aggregation of rhodopsin was observed to be higher in
the T17M rhodopsin-transfected cells than in the WT
rhodopsin-transfected cells following MG132 treatment (Fig. 2).
The turnover rates of the myc-tagged rhodopsin
proteins were assessed in ARPE-19 cells, which were treated with 50
μg/ml cycloheximide for various time-periods (Fig. 3A). T17M rhodopsin exhibited a more
rapid turnover than WT rhodopsin, with a half-life of 2 h compared
with 6 h, respectively (Fig.
3B).
T17M mutant rhodopsin activates ER-stress
and sensitizes cells to ER stress-induced cell death
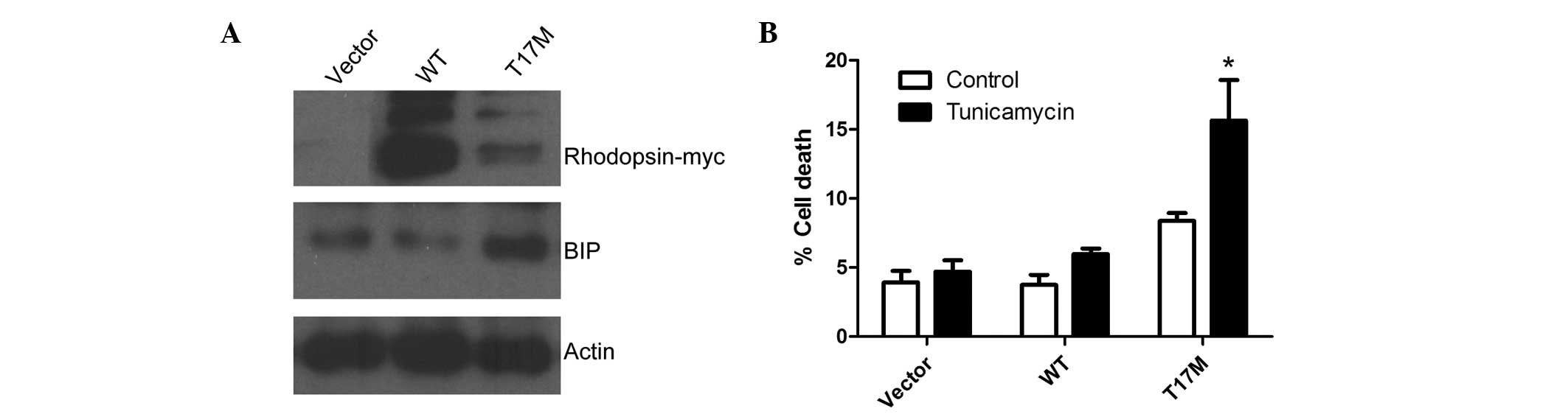
To determine whether the accumulation of T17M in the
ER activates ER stress, the expression of the resident ER chaperone
BiP, which is an unfolded protein response marker, was assessed in
WT and T17M rhodopsin-transfected cells. BiP was observed to be
upregulated in cells transfected with rhodopsin T17M constructs,
indicating that the ER stress response was activated (Fig. 4A). Prolonged ER stress caused by
the accumulation of unfolded proteins in the ER leads to cell
death; therefore, WT and misfolded rhodopsin proteins were
overexpressed in HEK293 cells and the effect on cell death was
assessed. HEK293 cells were transfected with myc-tagged WT and T17M
rhodopsin constructs and empty vectors. Cell death was then
quantified in the absence or presence of 2 mg/ml tunicamycin, which
is an ER stress inducer. Cells transfected with the T17M rhodopsin
mutant were found to be more sensitive to ER stress-induced cell
death than those transfected with WT rhodopsin or empty vectors.
Moreover, an increase in cell death was observed in the cells
exogenously expressing T17M rhodopsin (Fig. 4B).
ROS scavengers protect against T17M
rhodopsin-induced cell death
To determine whether ROS production contributes to
T17M rhodopsin-induced cell death, cells expressing T17M rhodopsin
were treated with the ROS scavengers BHA (100 μM) and NAC (1 mM).
Subsequent to 12 h of treatment, cell death was assessed using
Hoechst 33342 staining. ROS levels were observed to be
significantly increased in the cells transfected with T17M
rhodopsin compared with those transfected with the empty vector or
WT rhodopsin (Fig. 5A).
Furthermore, cell death was found to be significantly decreased in
the cells treated with BHA and NAC compared with those treated with
DMSO (Fig. 5B). These findings
suggest that T17M rhodopsin induces cell death, at least in part,
through a ROS-mediated pathway.
Discussion
In the present study, human T17M mutant rhodopsin
was found to be abnormally localized in the ER. Furthermore,
misfolded T17M mutant rhodopsin was more rapidly degraded by
proteasomes than WT rhodopsin. Overexpression of T17M rhodopsin was
found to increase cytotoxicity and predispose cells to ER
stress-induced cell death. Moreover, the cell death induced by T17M
rhodopsin overexpression was observed to be decreased with ROS
scavenger treatment.
Rhodopsin is a G-protein-coupled receptor that
mediates light-induced signal transduction in rod photoreceptor
cells (11) and is glycosylated
and transported to the plasma membrane (12). Heterozygous rhodopsin mutations
which cause amino acid substitutions have been reported to cause
human ADRP (6). It has also been
reported that transgenic animals expressing a mutant rhodopsin gene
demonstrate retinal degeneration (7,13,14).
Mutant rhodopsin phenotypes are divided into three classes: class
I, II and III. Class I mutants are capable of forming normal
rhodopsin chromophores with 11-cis-retinal and are trafficked to
the plasma membrane. However, their transducing activation is
inefficient upon illumination. Class III mutants are only capable
of forming a few rhodopsin chromophores, due to their low
expression levels. Moreover, class III mutants that accumulate in
the ER exhibit high mannose glycosylation (5). Class II mutants accumulate in the ER
and are incapable of forming the chromophore due to the inability
to bind with 11-cis-retinal (7).
The rhodopsin T17M mutation is an example of a class II mutant and
has a defect in protein folding (15). The substitution of threonine to
methionine at position 17 in the T17M rhodopsin mutant, has been
proposed to affect protein folding due to the proximity of the
substitution to the N2–N15 glycosylation site (16). The present study confirmed that the
rhodopsin T17M mutant, which is involved in RP, was localized in
the ER and was incapable of plasma membrane trafficking. This
retention of the misfolded protein in the ER supports the
pathogenicity of RP mutations at a cellular level.
The pathogenesis of neurodegeneration is often
associated with disturbances in protein folding, aggregation and
degradation (17). Accurate
protein folding and processing are required to maintain cellular
homeostasis (18). Protein
misfolding not only affects protein function, but leads to the
formation of potentially toxic aggregates (19). Cells have complex systems in order
to eliminate unwanted and potentially toxic proteins. Incorrectly
folded rhodopsin is detected by specialized ER sensors and is
either subject to additional folding cycles or degradation. In T17M
rhodopsin transgene mice, no significant difference was observed in
ERAD gene expression compared with the control; however, the
autophagy degradation pathway was observed to be upregulated
(20). In the present study,
proteasomal inhibition and analysis of protein turnover revealed
that the T17M mutant protein has an accelerated rate of degradation
compared with the WT protein. Furthermore, the proteasome was found
to be responsible for this increased protein turnover. It has been
reported that some ER-associated degradation is
proteasome-independent (21).
Therefore, the mechanism underlying the degradation of the T17M
mutant requires further investigation.
The accumulation of misfolded proteins in the ER
induces ER stress and has been implicated in human diseases,
including neurodegenerative diseases (22,23).
In the present study, it was hypothesized that ER stress induced by
T17M rhodopsin may have a role in the development of RP due to
three reasons. Firstly, the T17M rhodopsin mutant was observed to
be localized in the ER. Secondly, the presence of the T17M mutant
in the ER was found to increase cell death following exposure to
the ER stressor tunicamycin, suggesting that the misfolded T17M
rhodopsin protein negatively impacts ER homeostasis, thereby
increasing the susceptibility of cells to ER stress-induced cell
death. Of note, ER stress-induced retinal pathology in rhodopsin
P23H-expressing rats has been observed to be reduced by BiP protein
(13). Therefore, increased
expression of BiP or other chaperones may reduce T17M-induced ER
stress. The rhodopsin T17M mutant has been reported to exhibit a
reversible folding capacity (15)
and is transported partially to the rod outer segments in
transgenic Xenopus photoreceptor neurons (16). Thus, small pharmacological molecule
chaperones that correct folding-deficient rhodopsin may be
potential therapeutic options. Thirdly, activation of the ER stress
response has been reported to lead to changes in intracellular
Ca2+ concentration (24), as well as the release of ROS from
protein folding chaperones involved in disulphide bond formation
(25). Chaperones, including BiP
require energy to function. ER stress therefore increases the
demand on ATP synthesis by the mitochondria. Thus, it is possible
that ER stress may induce other downstream effects and may cause a
cascade of damage, leading to cell death (26,27).
Moreover, in the present study, ROS scavengers were found to reduce
T17M rhodopsin-induced cell death. This study identifies a
mechanism of RP pathogenesis induced by rhodopsin T17M mutant and
provides a potential treatment strategy of the disease.
Acknowledgements
This paper was supported by the grant from the
National Natural Science Foundation of China (No. 81170844).
References
|
1
|
Dryja TP, McGee TL, Hahn LB, et al:
Mutations within the rhodopsin gene in patients with autosomal
dominant retinitis pigmentosa. N Engl J Med. 323:1302–1307. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mendes HF, van der Spuy J, Chapple JP and
Cheetham ME: Mechanisms of cell death in rhodopsin retinitis
pigmentosa: implications for therapy. Trends Mol Med. 11:177–185.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nathans J and Hogness DS: Isolation and
nucleotide sequence of the gene encoding human rhodopsin. Proc Natl
Acad Sci USA. 81:4851–4855. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaushal S and Khorana HG: Structure and
function in rhodopsin 7. Point mutations associated with autosomal
dominant retinitis pigmentosa. Biochemistry. 33:6121–6128. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sung CH, Davenport CM and Nathans J:
Rhodopsin mutations responsible for autosomal dominant retinitis
pigmentosa. Clustering of functional classes along the polypeptide
chain. J Biol Chem. 268:26645–26649. 1993.
|
|
7
|
Li T, Sandberg MA, Pawlyk BS, et al:
Effect of vitamin A supplementation on rhodopsin mutants
threonine-17 --> methionine and proline-347 --> serine in
transgenic mice and in cell cultures. Proc Natl Acad Sci USA.
95:11933–11938. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jacobson SG, Kemp CM, Sung CH and Nathans
J: Retinal function and rhodopsin levels in autosomal dominant
retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol.
112:256–271. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sung CH, Davenport CM, Hennessey JC, et
al: Rhodopsin mutations in autosomal dominant retinitis pigmentosa.
Proc Natl Acad Sci USA. 88:6481–6485. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
White DA, Fritz JJ, Hauswirth WW, Kaushal
S and Lewin AS: Increased sensitivity to light-induced damage in a
mouse model of autosomal dominant retinal disease. Invest
Ophthalmol Vis Sci. 48:1942–1951. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ridge KD, Abdulaev NG, Sousa M and
Palczewski K: Phototransduction: crystal clear. Trends Biochem Sci.
28:479–487. 2003. View Article : Google Scholar
|
|
12
|
Chapple JP, Grayson C, Hardcastle AJ,
Saliba RS, van der Spuy J and Cheetham ME: Unfolding retinal
dystrophies: a role for molecular chaperones? Trends Mol Med.
7:414–421. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gorbatyuk MS, Knox T, LaVail MM, et al:
Restoration of visual function in P23H rhodopsin transgenic rats by
gene delivery of BiP/Grp78. Proc Natl Acad Sci USA. 107:5961–5966.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krebs MP, White DA and Kaushal S: Biphasic
photoreceptor degeneration induced by light in a T17M rhodopsin
mouse model of cone bystander damage. Invest Ophthalmol Vis Sci.
50:2956–2965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krebs MP, Holden DC, Joshi P, Clark CL
III, Lee AH and Kaushal S: Molecular mechanisms of rhodopsin
retinitis pigmentosa and the efficacy of pharmacological rescue. J
Mol Biol. 395:1063–1078. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tam BM and Moritz OL: The role of
rhodopsin glycosylation in protein folding, trafficking, and
light-sensitive retinal degeneration. J Neurosci. 29:15145–15154.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Selkoe DJ: Cell biology of protein
misfolding: The examples of Alzheimer’s and Parkinson’s diseases.
Nat Cell Biol. 6:1054–1061. 2004.
|
|
18
|
Wickner S, Maurizi MR and Gottesman S:
Posttranslational quality control: folding, refolding, and
degrading proteins. Science. 286:1888–1893. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chaudhuri TK and Paul S:
Protein-misfolding diseases and chaperone-based therapeutic
approaches. FEBS J. 273:1331–1349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kunte MM, Choudhury S, Manheim JF, et al:
ER stress is involved in T17M rhodopsin-induced retinal
degeneration. Invest Ophthalmol Vis Sci. 53:3792–3800. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Finger A, Knop M and Wolf DH: Analysis of
two mutated vacuolar proteins reveals a degradation pathway in the
endoplasmic reticulum or a related compartment of yeast. Eur J
Biochem. 218:565–574. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao L and Ackerman SL: Endoplasmic
reticulum stress in health and disease. Curr Opin Cell Biol.
18:444–452. 2006. View Article : Google Scholar
|
|
23
|
Griciuc A, Aron L and Ueffing M: ER stress
in retinal degeneration: a target for rational therapy? Trends Mol
Med. 17:442–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scorrano L, Oakes SA, Opferman JT, et al:
BAX and BAK regulation of endoplasmic reticulum Ca2+: a
control point for apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haynes CM, Titus EA and Cooper AA:
Degradation of misfolded proteins prevents ER-derived oxidative
stress and cell death. Mol Cell. 15:767–776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stefani IC, Wright D, Polizzi KM and
Kontoravdi C: The role of ER stress-induced apoptosis in
neurodegeneration. Curr Alzheimer Res. 9:373–387. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayashi T, Saito A, Okuno S, Ferrand-Drake
M, Dodd RL and Chan PH: Damage to the endoplasmic reticulum and
activation of apoptotic machinery by oxidative stress in ischemic
neurons. J Cereb Blood Flow Metab. 25:41–53. 2005. View Article : Google Scholar : PubMed/NCBI
|