Parkinson’s disease (PD) is a common
neurodegenerative disorder characterized by the gradually
progressive and selective loss of dopaminergic neurons and by
intracellular inclusions known as Lewy bodies (LBs) expressed in
the surviving neurons of the substantia nigra (SN) (1). The progressive loss of dopaminergic
neurons is a complex process, and multiple pathological events,
including oxidative stress, mitochondrial dysfunction, protein
aggregation and neuroinflammation, are indicated in PD pathogenesis
(2,3). Substantial evidence indicates that
mitochondrial dysfunction induced by diverse stress conditions
plays a crucial role in the pathogenesis of PD (4,5). A
central event in the mitochondrial cell death pathway is the
formation of a mitochondrial permeability transition pore (mPTP).
The production of reactive oxygen species (ROS) triggered by
complex I inhibition are believed to be key inducers of mPTP
formation (6). Complex I
deficiency has been shown to contribute to the dopaminergic cell
death in idiopathic PD patients (7,8). The
administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP) and rotenone, well-known inhibitors of complex I, induce PD
syndrome characterized by the loss of SN neurons in animal models
(9–11), supporting the involvement of
mitochondrial dysfunction in the pathogenesis of PD. Decreased
complex I activity in the mitochondrial respiratory chain leads to
excessive ROS production, which contributes to the oxidative damage
of cellular macromolecules and the activation of mPTP, ultimately
leading to cell death (12,13).
Neuroinflammation has increasingly been recognized as a
pathological contributor to neurodegenerative diseases (14–16),
and particularly as a key promoter to the chronic loss of nigral
dopaminergic neurons in PD (17).
Postmortem studies revealed activated microglia and accumulation of
inflammatory mediators expressed in the SN of PD patients and
animal models (16,18,19).
Inhibition of the inflammatory response promotes dopaminergic
neuron survival in various PD models (20–22),
confirming the indicated action of the inflammatory response in
neurodegenerative diseases. Several studies have revealed that the
inhibition of GSK-3β reduces dopaminergic neuron injury induced by
MPTP toxicity, indicating the association of GSK-3β with the
pathogenesis of PD (23,24). GSK-3β is a central point in a
number of signaling pathways in the pathogenesis of this
neurodegenerative disease, affecting multiple pathological events
involved in dopaminergic neuron degeneration, thus providing a
potential target in the therapeutic management by blocking the
pathogenic pathways involved in PD pathogenesis.
GSK-3 is a serine/threonine (Ser/Thr) protein kinase
expressed in the cytosol, nucleus and mitochondria of all
eukaryotic cells. There are two major GSK-3 protein isoforms (GSK3α
and GSK3β) encoded by two highly homologous genes, gsk-3α and
gsk-3β (25). This enzyme was
originally identified as a regulator of glycogen synthase (26). However, GSK-3 has been recognized
as a pleiotropic enzyme, affecting numerous biological functions
including gene expression and cellular processes such as cell
proliferation, differentiation and apoptosis (27,28).
GSK-3 phosphorylates and regulates >50 substrates, which allows
this enzyme to modulate a wide range of biological functions
(27,28). Dysregulation of GSK-3 is implicated
in diverse diseases, including diabetes, ischemia/reperfusion
injury, bipolar disorder, cancer and neurodegenerative disease
(27,29–32).
GSK-3β is a point of convergence for multiple signaling pathways
and thus plays a crucial role in regulating the pathogenesis of
diverse diseases. Its activity and functions are controlled by
phosphorylation at specific sites. Phosphorylation at Ser9 of
GSK-3β markedly inhibits its activity (33), whereas phosphorylation at Tyr216
increases its activity. The inactivation of GSK-3β is mainly
targeted by the Akt signaling pathway by the phosphorylation of
Ser9 of this enzyme. GSK-3β is mainly localized in the cytosol, but
lower amounts are expressed in the nucleus and mitochondria
(34–37), and its regulatory role in the
mitochondrial cell death pathway has been elicited by a variety of
stress conditions shown in neuronal cells (38–43).
GSK-3β facilitates numerous apoptotic conditions involved in PD
pathogenesis, including mitochondrial dysfunction, oxidative
stress, protein aggregation and the inflammatory response, by
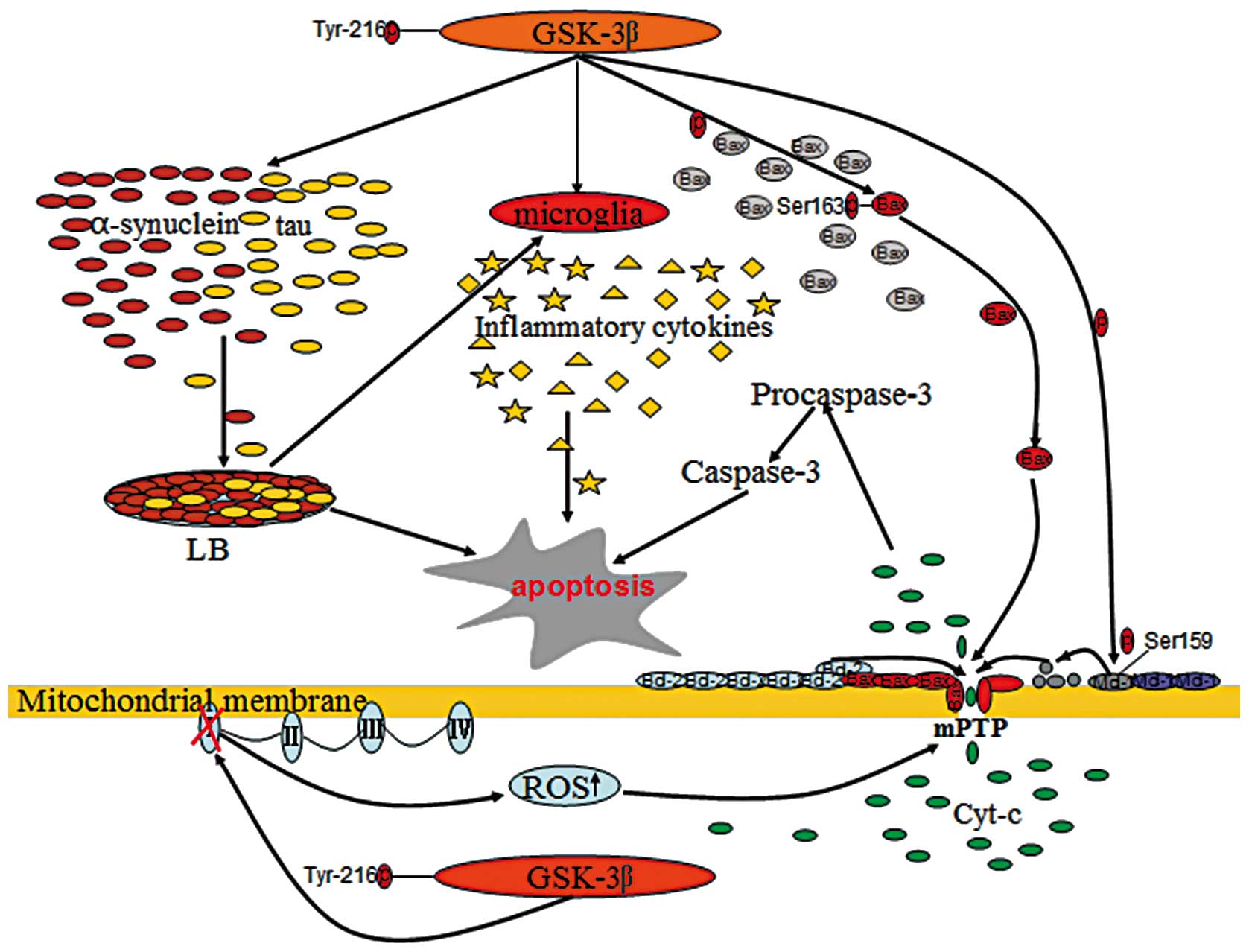
modulating diverse signaling pathways (Fig. 1) (23,24,42).
Inhibition of GSK-3β is indicated in the suppression of a number of
pathogenic events in PD, thus promoting dopaminergic neuronal
survival (23,24,44,45).
In eukaryotic cells, mitochondria are key organelles
providing essential energy for cell metabolism through adenosine
triphosphate (ATP) generation. Complex I is a protein component of
the electron transport chain located in the inner part of the
mitochondrial membrane and functioning as the effective enzyme of
the oxidative phosphorylation system responsible for the generation
of cellular ATP. Mitochondrial complex I is the main site of ROS
formation as it transfers single electrons to oxygen, thus
generating O2− and subsequently
H2O2 (46,47).
Inhibition of complex I leads to a decrease in ATP levels and
excessive production of ROS, the central events of mitochondrial
dysfunction that have been indicated in PD pathogenesis (48). The first evidence for the
involvement of complex I inhibition in PD was the recognition that
MPTP caused a severe and irreversible parkinsonian syndrome in drug
abusers (49). MPTP is a
lipophilic molecule and can rapidly cross the blood-brain barrier.
Once it crosses the barrier, it is oxidized in the brain to its
toxic metabolite 1-methyl-4-phenylpyridinium (MPP+) by
type B monoamine oxidase (50).
MPP+ is then taken up by dopaminergic neurons via a
dopamine transporter and accumulates in the mitochondria where it
causes excessive ROS formation by inhibiting respiration complex I
(51), finally leading to
dopaminergic neuron death. Mitochondrial complex I inhibition has
also been reported in the SN, platelets and skeletal muscle of
idiopathic PD patients (7,8,52).
Complex I is generally known to be the primary source of
mitochondrial ROS (53–55). Inhibition of mitochondrial complex
I elicited by neurotoxins MPP+ and rotenone,
well-established dopaminergic cell death inducers in PD, have been
shown to increase the production of ROS (5,56).
GSK-3β has been shown to be located into the mitochondria, where it
is highly activated compared with the cytosolic form (36). Although the significance of the
presence of GSK-3β in the mitochondria remains poorly understood,
its involvement in mitochondrial dysfunction has been reported
(57,58). GSK-3β can regulate cell survival
and apoptosis by controlling mitochondrial complex I activity and
ROS production (43). GSK-3β
regulates oxidative phosphorylation by inhibiting NADH (complex I),
which is the main site of ROS formation, whereas this enzyme is
implicated in homeostatic redox equilibrium (43). Previous studies have shown that
mitochondrial toxins, including rotenone and MPTP treatments,
increase GSK3β activity. Inhibition of GSK-3β protects dopaminergic
neurons from the toxicity of rotenone and MPTP, indicating the
involvement of GSK-3β in the complex I inhibition-induced cell
death pathway in PD (23). Studies
in the MPTP model of PD also demonstrate that mitochondrial GSK-3β
significantly promotes ROS production by further inhibiting complex
I, and that this can be reversed by GSK-3β inhibitors (43). Similar studies have indicated that
GSK-3β inhibition promotes mitochondrial biogenesis and prevents
ROS production during ischemic cerebral damage (59). Although the mechanism underlying
the contribution of GSK-3β to the mitochondrial complex I
inhibition remains unclear, these reports clearly indicate that
GSK-3β inhibition contributes to cell survival induced by
mitochondrial complex I inhibition and ROS formation.
Mitochondria are integrated in diverse signaling
pathways linked to multiple cell processes, including apoptosis.
The major property for mitochondria is the maintenance of its
membrane potential and the low-conductance state of the
mitochondrial permeability transition pore (mPTP) in living cells.
mPTP activation is a central event in mitochondria-mediated
intrinsic cell apoptosis, which has been implicated in the
pathogenesis of PD and several other neurodegenerative disorders
(60–62). The mPTP pathway of cell death is
mediated by the disruption of the mitochondrial membrane and the
release of apoptogenic molecules, which can be regulated by GSK-3β
signaling pathways through modulating the opening of the mPTP
(63,64). Studies have shown that GSK-3β
inactivation protects cardiac cells from ischemia/reperfusion
injury through the inhibition of the mPTP opening, indicating its
regulatory role in the mitochondrial cell death pathway (65–68).
It has also been shown in cell and animal models of PD that GSK-3β
inhibition can protect dopaminergic neurons from MPTP toxicity
(23,24,42,69).
This contribution of GSK-3β to cell death and survival appears to
correlate with its ability to control the mitochondrial
localization and activation of a number of proteins, particularly
B-cell lymphoma 2 (Bcl-2) family proteins, including Bax, Bcl-2 and
Mcl-1, considered as central players in mPTP formation (70,71).
Generally, Bax is a cytosolic protein that can be translocated to
the mitochondrial membrane in response to apoptotic stimuli
(72). Once located in the
mitochondrial membrane, this protein increases the mitochondrial
membrane permeabilization by sequestering Bcl-2 and by
oligomerization within the mitochondrial membrane, leading to the
release of pro-apoptotic molecules into the cytoplasm (73,74).
By contrast, Bcl-2 and Mcl-1 are anti-apoptotic members that
preserve mitochondrial membrane integrity, thereby preventing the
release of apoptogenic molecules and cell apoptosis (75). GSK-3β activation promotes
mitochondria-mediated apoptosis by the upregulation of Bax
expression levels (58,76). Treatment with lithium, a
pharmacological inhibitor of GSK-3β, could suppress the
pro-apoptotic pathway by decreasing the expression levels of Bax,
but promote anti-apoptotic signaling through increasing Bcl-2
expression (77–79). In addition, GSK-3β can facilitate
the mitochondrial localization of Bax by directly phosphorylating
Ser163 of this protein (70). In
PD, models reveal that the inhibition of GSK-3β protects
dopaminergic cells against neurotoxin-induced damage through
attenuating the translocation of Bax to the mitochondria (80–82).
Additionally, GSK-3β phosphorylates Mcl-1 on Ser159, resulting in
the destabilization of this protein and the blockage of the
Mcl-1-dependent integrity of the mitochondrial membrane (71). Overall, GSK-3β may be vital in the
regulation of cell death and survival through the modulation of the
mitochondrial apoptotic cell death pathway.
Protein aggregation and inclusion body formation in
selected areas of the neuronal system are pathological hallmarks of
neurodegenerative diseases, including PD, in which intracellular
inclusions known as LBs are expressed in surviving SN neurons. LBs
are composed mainly of the α-synuclein protein, a presynaptic
neuronal protein abundantly expressed in the nervous system
(83–85). The regulatory role of α-synuclein
in the production of dopamine through the interaction with tyrosine
hydroxylase has been shown in cultured cells (86,87).
TH is the rate-limiting enzyme responsible for the conversion of
tyrosine to L-3,4-dihydroxyphenylalanine in the dopamine synthesis
pathway (86,88). Overexpression of α-synuclein
inhibits TH activity and decreases dopamine biosynthesis, while
suppression of α-synuclein expression levels promotes TH activity
and consequently increases dopamine production (86,87,89).
The toxicity of α-synuclein overexpression and accumulation to the
neurons has been established in in vivo and in vitro
models (90–93). α-synuclein protein overexpression
and aggregation exacerbate the impairment of mitochondrial
functions by augmenting oxidative stress (94–97).
This protein overexpression can also directly activate microglia
via a classical activation pathway, leading to the increase of the
inflammatory response by the production and release of
proinflammatory mediators (98–100). The actions of α-synuclein in
promoting oxidative stress and the inflammatory response may be the
underlying mechanism responsible for the toxicity of its
overexpression and accumulation to dopaminergic neurons in PD.
α-synuclein is a substrate for GSK-3β phosphorylation. GSK-3β
inhibition decreases α-synuclein protein expression and prevents
cell death in a cellular model of PD, indicating that inhibition of
GSK-3β activity may be neuroprotective to dopaminergic neurons by
attenuating the toxicity of α-synuclein overexpression (101). τ protein was originally
discovered as a key component of intracellular neurofibrillary
tangles within the brain of AD patients, however, this protein is
also expressed highly in LBs and in the striatum of PD brains,
indicating that it contributes to the pathogenesis of PD (102,103). Blockage of τ phosphorylation with
special inhibitors prevents the dopaminergic neuronal death of PD
models (101). GSK-3β is a main
kinase affecting τ function through interfering with τ
phosphorylation. Activation of GSK-3β increases τ phosphorylation
(104–106), which can be reversed by GSK-3β
inhibitors or upstream Akt inhibitors (107,108). Additionally, GSK-3β may also
facilitate the aggregation of τ protein and neurodegeneration
(109,110). Animal models indicate that the
inhibition of GSK-3β promotes neuron survival by reducing τ-induced
toxicity (111–113). These findings provide a potential
target in the therapeutic management of PD by blocking the
pathogenic pathway of protein overexpression and aggregation.
The inflammatory response, including a host of
cytokines has been shown to be implicated in neuronal degeneration
in PD and other neurodegenerative diseases (15,114). The activation of microglia and
the upregulation of proinflammatory cytokines are key characters of
brain inflammation. Microglia are resident immunocompetent cells in
the brain and become activated in response to infection and damage
(115). The release of
proinflammatory and neurotoxic mediators from activated microglia
contributes to progressive neuron damage in neurodenerative
conditions (116,117). Studies have shown that microglia
are activated regionally in the SN of PD patients and animal models
(16,18,19,118), and that the levels of a number of
inflammatory cytokines, including tumor necrosis factor-α (TNF-α),
interleukin (IL)-1β, IL-2 and IL-6, are also upregulated in PD
(119–122), indicating the involvement of the
inflammatory response in PD pathogenesis. The contribution of
inflammation-derived oxidative stress and cytokine-dependent
toxicity to the nigrostriatal dopaminergic neuron death has also
been reported in PD models (117,123,124). Additionally, suppression of the
inflammatory response leads to the protection of dopaminergic
neurons against neurotoxin-induced cell damage (22,125), which further supports the
indication that the inflammatory mechanism is involved in
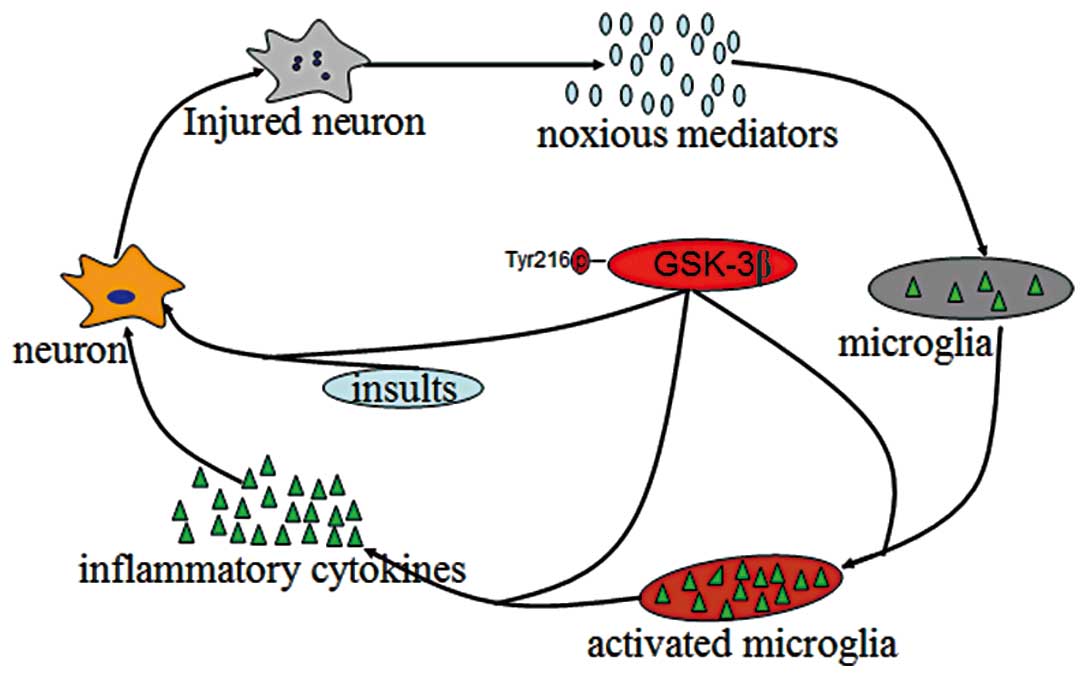
neurodegenerative disease. Microglia can be activated by injured
neurons through generating a spectrum of noxious endogenous
mediators. Once activated, microglia produce and release multiple
proinflammatory factors. This production of proinflammatory factors
in turn exacerbates neuron damage by oxidative stress and cytokine
toxicity (14,19), leading to further release of
noxious endogenous mediators from injured neurons and an
everlasting inflammatory response. This positive feedback between
activated microglia and damaged neurons contributes to an
uncontrolled, prolonged inflammatory process, which is believed to
be, at least in part, responsible for the progressive loss of
dopaminergic neurons in PD (17,114). Thereby, inhibition of the
inflammatory response caused by microglia activation may be
beneficial in neurodegenerative conditions. GSK-3β is a point of
convergence of a wide range of signaling pathways, and has been
recognized as a key regulator of inflammation (126,127). Activation of GSK-3β promotes
inflammatory responses by activating microglia and increasing the
production of inflammatory cytokines (45,128–129). The signals of GSK-3β can also
promote various insult-induced neuronal injuries and the noxious
generation of endogenous mediators. Thus, GSK-3β plays a central
role in the maintenance of the vicious cycle between activated
microglia and damaged neurons responsible for the progressive loss
of dopaminergic cell loss in PD (Fig.
2). Inhibition of GSK-3β attenuates the microglia response to
inflammatory stimuli and reduces cytokine production, thereby
providing protection from inflammation-induced toxicity (45,126). However, the direct substrates of
GSK-3β that are involved in inflammation-induced neuron damage
remain unclear. TNF-α may be a key downstream signal transducer
that is indicated in the proinflammatory effect of GSK-3β in
activated microglia-mediated neuroinflammation (130). Within the brain, TNF-α is a
mainly proinflammatory cytokine that is released by activated
microglia in response to various insults or injury. This production
of TNF-α triggers the uncontrolled inflammatory response by further
activating microglia (131),
which can be blocked by GSK-3β inhibition through modulation of
nuclear factor κB and mixed lineage kinase 3/c-Jun N-terminal
kinase 3 signaling cascades (130). These findings indicate that
GSK-3β activity is critical for neuronal death in response to the
neuroinflammation elicited by the microglial activation.
Attenuation of the microglia-mediated inflammatory response
targeted by GSK-3β inhibition to prevent dopaminergic neuron
degeneration in PD requires further investigation.
The pathogenesis of PD is a complex process, and
multiple pathological events, including oxidative stress,
mitochondrial dysfunction, protein aggregation and
neuroinflammation, are considered to mediate and drive the gradual
loss of dopaminergic neurons in PD (8,132–134). Understanding the intracellular
signaling processes that regulate the events involved in the
pathogenesis of PD is critical for developing novel therapeutics
for PD treatment. GSK-3β is a multifaceted enzyme that has been
indicated to be involved in the pathogenesis of neurodegenerative
diseases, including PD, by modulating multiple signaling pathways
(101,135–137). GSK-3β inhibition protects
dopaminergic neurons from various stress-induced injuries in the
cell culture and animal models of PD (23,42).
The cellular and molecular mechanisms of the protective effects of
GSK-3β inhibition on dopaminergic neurons in pathogenic conditions
require further elucidation, and may provide a potential efficient
target for treating PD by blocking the pathogenic pathway.
|
1
|
Forno LS: Neuropathology of Parkinson’s
disease. J Neuropathol Exp Neurol. 55:259–272. 1996.
|
|
2
|
McNaught KS and Olanow CW: Protein
aggregation in the pathogenesis of familial and sporadic
Parkinson’s disease. Neurobiol Aging. 27:530–545. 2006.
|
|
3
|
Martinez-Vicente M, Talloczy Z, Kaushik S,
et al: Dopamine-modified alpha-synuclein blocks chaperone-mediated
autophagy. J Clin Invest. 118:777–788. 2008.PubMed/NCBI
|
|
4
|
Keeney PM, Xie J, Capaldi RA and Bennett
JP Jr: Parkinson’s disease brain mitochondrial complex I has
oxidatively damaged subunits and is functionally impaired and
misassembled. J Neurosci. 26:5256–5264. 2006.
|
|
5
|
Li DW, Li GR, Lu Y, et al: alpha-lipoic
acid protects dopaminergic neurons against MPP+-induced apoptosis
by attenuating reactive oxygen species formation. Int J Mol Med.
32:108–114. 2013.PubMed/NCBI
|
|
6
|
Rasola A and Bernardi P: Mitochondrial
permeability transition in Ca(2+)-dependent apoptosis
and necrosis. Cell Calcium. 50:222–233. 2011. View Article : Google Scholar
|
|
7
|
Parker WD Jr, Boyson SJ and Parks JK:
Abnormalities of the electron transport chain in idiopathic
Parkinson’s disease. Ann Neurol. 26:719–723. 1989.
|
|
8
|
Schapira AH, Cooper JM, Dexter D, Jenner
P, Clark JB and Marsden CD: Mitochondrial complex I deficiency in
Parkinson’s disease. Lancet. 1:12691989.
|
|
9
|
Betarbet R, Sherer TB, MacKenzie G,
Garcia-Osuna M, Panov AV and Greenamyre JT: Chronic systemic
pesticide exposure reproduces features of Parkinson’s disease. Nat
Neurosci. 3:1301–1306. 2000.
|
|
10
|
Cannon JR, Tapias V, Na HM, Honick AS,
Drolet RE and Greenamyre JT: A highly reproducible rotenone model
of Parkinson’s disease. Neurobiol Dis. 34:279–290. 2009.PubMed/NCBI
|
|
11
|
Hantraye P, Brouillet E, Ferrante R, et
al: Inhibition of neuronal nitric oxide synthase prevents
MPTP-induced parkinsonism in baboons. Nat Med. 2:1017–1021. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shang T, Kotamraju S, Kalivendi SV,
Hillard CJ and Kalyanaraman B: 1-Methyl-4-phenylpyridinium-induced
apoptosis in cerebellar granule neurons is mediated by transferrin
receptor iron-dependent depletion of tetrahydrobiopterin and
neuronal nitric-oxide synthase-derived superoxide. J Biol Chem.
279:19099–19112. 2004. View Article : Google Scholar
|
|
13
|
Hartley A, Stone JM, Heron C, Cooper JM
and Schapira AH: Complex I inhibitors induce dose-dependent
apoptosis in PC12 cells: relevance to Parkinson’s disease. J
Neurochem. 63:1987–1990. 1994.PubMed/NCBI
|
|
14
|
Kreutzberg GW: Microglia: a sensor for
pathological events in the CNS. Trends Neurosci. 19:312–318. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Monahan AJ, Warren M and Carvey PM:
Neuroinflammation and peripheral immune infiltration in Parkinson’s
disease: an autoimmune hypothesis. Cell Transplant. 17:363–372.
2008.
|
|
16
|
Hirsch EC and Hunot S: Neuroinflammation
in Parkinson’s disease: a target for neuroprotection? Lancet
Neurol. 8:382–397. 2009.
|
|
17
|
Gao HM, Zhou H, Zhang F, Wilson BC, Kam W
and Hong JS: HMGB1 acts on microglia Mac1 to mediate chronic
neuroinflammation that drives progressive neurodegeneration. J
Neurosci. 31:1081–1092. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McGeer PL, Schwab C, Parent A and Doudet
D: Presence of reactive microglia in monkey substantia nigra years
after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration.
Ann Neurol. 54:599–604. 2003.
|
|
19
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao HM, Liu B, Zhang W and Hong JS: Novel
anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol
Sci. 24:395–401. 2003.
|
|
21
|
Wu DC, Teismann P, Tieu K, et al: NADPH
oxidase mediates oxidative stress in the
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s
disease. Proc Natl Acad Sci USA. 100:6145–6150. 2003.
|
|
22
|
Zhang F, Qian L, Flood PM, Shi JS, Hong JS
and Gao HM: Inhibition of IkappaB kinase-beta protects dopamine
neurons against lipopolysaccharide-induced neurotoxicity. J
Pharmacol Exp Ther. 333:822–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang W, Yang Y, Ying C, et al: Inhibition
of glycogen synthase kinase-3beta protects dopaminergic neurons
from MPTP toxicity. Neuropharmacology. 52:1678–1684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
King TD, Bijur GN and Jope RS: Caspase-3
activation induced by inhibition of mitochondrial complex I is
facilitated by glycogen synthase kinase-3beta and attenuated by
lithium. Brain Res. 919:106–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Woodgett JR: Molecular cloning and
expression of glycogen synthase kinase-3/factor A. EMBO J.
9:2431–2438. 1990.PubMed/NCBI
|
|
26
|
Parker PJ, Embi N, Caudwell FB and Cohen
P: Glycogen synthase from rabbit skeletal muscle. State of
phosphorylation of the seven phosphoserine residues in vivo in the
presence and absence of adrenaline. Eur J Biochem. 124:47–55. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kockeritz L, Doble B, Patel S and Woodgett
JR: Glycogen synthase kinase-3 - an overview of an over-achieving
protein kinase. Curr Drug Targets. 7:1377–1388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miura T, Tanno M and Sato T: Mitochondrial
kinase signalling pathways in myocardial protection from
ischaemia/reperfusion-induced necrosis. Cardiovasc Res. 88:7–15.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Medina M, Garrido JJ and Wandosell FG:
Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies.
Front Mol Neurosci. 4:242011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
King MR, Anderson NJ, Guernsey LS and
Jolivalt CG: Glycogen synthase kinase-3 inhibition prevents
learning deficits in diabetic mice. J Neurosci Res. 91:506–514.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng H, Li W, Wang Y, et al: Glycogen
synthase kinase-3 beta regulates Snail and beta-catenin expression
during Fas-induced epithelial-mesenchymal transition in
gastrointestinal cancer. Eur J Cancer. 2013. View Article : Google Scholar
|
|
33
|
Dajani R, Fraser E, Roe SM, et al: Crystal
structure of glycogen synthase kinase 3 beta: structural basis for
phosphate-primed substrate specificity and autoinhibition. Cell.
105:721–732. 2001.PubMed/NCBI
|
|
34
|
Xavier IJ, Mercier PA, McLoughlin CM, Ali
A, Woodgett JR and Ovsenek N: Glycogen synthase kinase 3beta
negatively regulates both DNA-binding and transcriptional
activities of heat shock factor 1. J Biol Chem. 275:29147–29152.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bijur GN and Jope RS: Proapoptotic stimuli
induce nuclear accumulation of glycogen synthase kinase-3 beta. J
Biol Chem. 276:37436–37442. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bijur GN and Jope RS: Glycogen synthase
kinase-3 beta is highly activated in nuclei and mitochondria.
Neuroreport. 14:2415–2419. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoshi M, Sato M, Kondo S, et al: Different
localization of tau protein kinase I/glycogen synthase kinase-3
beta from glycogen synthase kinase-3 alpha in cerebellum
mitochondria. J Biochem. 118:683–685. 1995.PubMed/NCBI
|
|
38
|
Senatorov VV, Ren M, Kanai H, Wei H and
Chuang DM: Short-term lithium treatment promotes neuronal survival
and proliferation in rat striatum infused with quinolinic acid, an
excitotoxic model of Huntington’s disease. Mol Psychiatry.
9:371–385. 2004.PubMed/NCBI
|
|
39
|
Bijur GN, De Sarno P and Jope RS: Glycogen
synthase kinase-3beta facilitates staurosporine- and heat
shock-induced apoptosis. Protection by lithium. J Biol Chem.
275:7583–7590. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alvarez AR, Godoy JA, Mullendorff K,
Olivares GH, Bronfman M and Inestrosa NC: Wnt-3a overcomes
beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res.
297:186–196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu Y, Shang Y, Sun S, Liang H and Liu R:
Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium
ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated
signaling pathway. Apoptosis. 12:1365–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petit-Paitel A, Brau F, Cazareth J and
Chabry J: Involvment of cytosolic and mitochondrial GSK-3beta in
mitochondrial dysfunction and neuronal cell death of
MPTP/MPP-treated neurons. PLoS One. 4:e54912009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
King TD, Clodfelder-Miller B, Barksdale KA
and Bijur GN: Unregulated mitochondrial GSK3beta activity results
in NADH: ubiquinone oxidoreductase deficiency. Neurotox Res.
14:367–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang WC, Lin YS, Wang CY, et al: Glycogen
synthase kinase-3 negatively regulates anti-inflammatory
interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis
and RANTES production in microglial cells. Immunology.
128:e275–e286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yuskaitis CJ and Jope RS: Glycogen
synthase kinase-3 regulates microglial migration, inflammation, and
inflammation-induced neurotoxicity. Cell Signal. 21:264–273. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Camello-Almaraz C, Gomez-Pinilla PJ, Pozo
MJ and Camello PJ: Mitochondrial reactive oxygen species and
Ca2+signaling. Am J Physiol Cell Physiol.
291:C1082–C1088. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Grivennikova VG and Vinogradov AD:
Generation of superoxide by the mitochondrial Complex I. Biochim
Biophys Acta. 1757:553–561. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Abou-Sleiman PM, Muqit MM and Wood NW:
Expanding insights of mitochondrial dysfunction in Parkinson’s
disease. Nat Rev Neurosci. 7:207–219. 2006.
|
|
49
|
Langston JW, Ballard P, Tetrud JW and
Irwin I: Chronic Parkinsonism in humans due to a product of
meperidine-analog synthesis. Science. 219:979–980. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chiba K, Trevor A and Castagnoli N Jr:
Metabolism of the neurotoxic tertiary amine, MPTP, by brain
monoamine oxidase. Biochem Biophys Res Commun. 120:574–578. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Javitch JA, D’Amato RJ, Strittmatter SM
and Snyder SH: Parkinsonism-inducing neurotoxin,
N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the
metabolite N-methyl-4-phenylpyridine by dopamine neurons explains
selective toxicity. Proc Natl Acad Sci USA. 82:2173–2177. 1985.
View Article : Google Scholar
|
|
52
|
Bindoff LA, Birch-Machin M, Cartlidge NE,
Parker WD Jr and Turnbull DM: Mitochondrial function in Parkinson’s
disease. Lancet. 2:491989.
|
|
53
|
Kussmaul L and Hirst J: The mechanism of
superoxide production by NADH: ubiquinone oxidoreductase (complex
I) from bovine heart mitochondria. Proc Natl Acad Sci USA.
103:7607–7612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sipos I, Tretter L and Adam-Vizi V:
Quantitative relationship between inhibition of respiratory
complexes and formation of reactive oxygen species in isolated
nerve terminals. J Neurochem. 84:112–118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cassarino DS, Fall CP, Swerdlow RH, et al:
Elevated reactive oxygen species and antioxidant enzyme activities
in animal and cellular models of Parkinson’s disease. Biochim
Biophys Acta. 1362:77–86. 1997.PubMed/NCBI
|
|
57
|
Pastorino JG, Hoek JB and Shulga N:
Activation of glycogen synthase kinase 3beta disrupts the binding
of hexokinase II to mitochondria by phosphorylating
voltage-dependent anion channel and potentiates
chemotherapy-induced cytotoxicity. Cancer Res. 65:10545–10554.
2005. View Article : Google Scholar
|
|
58
|
Watcharasit P, Bijur GN, Song L, Zhu J,
Chen X and Jope RS: Glycogen synthase kinase-3beta (GSK3beta) binds
to and promotes the actions of p53. J Biol Chem. 278:48872–48879.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Valerio A, Bertolotti P, Delbarba A, et
al: Glycogen synthase kinase-3 inhibition reduces ischemic cerebral
damage, restores impaired mitochondrial biogenesis and prevents ROS
production. J Neurochem. 116:1148–1159. 2011. View Article : Google Scholar
|
|
60
|
Vila M and Przedborski S: Targeting
programmed cell death in neurodegenerative diseases. Nat Rev
Neurosci. 4:365–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Perier C, Tieu K, Guegan C, et al: Complex
I deficiency primes Bax-dependent neuronal apoptosis through
mitochondrial oxidative damage. Proc Natl Acad Sci USA.
102:19126–19131. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Roucou X and Martinou JC: Conformational
change of Bax: a question of life or death. Cell Death Differ.
8:875–877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Obame FN, Plin-Mercier C, Assaly R, et al:
Cardioprotective effect of morphine and a blocker of glycogen
synthase kinase 3 beta, SB216763
[3-(2,4-dichlorophenyl)-4(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione],
via inhibition of the mitochondrial permeability transition pore. J
Pharmacol Exp Ther. 326:252–258. 2008.PubMed/NCBI
|
|
64
|
Nishihara M, Miura T, Miki T, et al:
Modulation of the mitochondrial permeability transition pore
complex in GSK-3beta-mediated myocardial protection. J Mol Cell
Cardiol. 43:564–570. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Feng J, Lucchinetti E, Ahuja P, Pasch T,
Perriard JC and Zaugg M: Isoflurane postconditioning prevents
opening of the mitochondrial permeability transition pore through
inhibition of glycogen synthase kinase 3beta. Anesthesiology.
103:987–995. 2005. View Article : Google Scholar
|
|
66
|
Park SS, Zhao H, Mueller RA and Xu Z:
Bradykinin prevents reperfusion injury by targeting mitochondrial
permeability transition pore through glycogen synthase kinase
3beta. J Mol Cell Cardiol. 40:708–716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gomez L, Paillard M, Thibault H, Derumeaux
G and Ovize M: Inhibition of GSK3beta by postconditioning is
required to prevent opening of the mitochondrial permeability
transition pore during reperfusion. Circulation. 117:2761–2768.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhou K, Zhang L, Xi J, Tian W and Xu Z:
Ethanol prevents oxidant-induced mitochondrial permeability
transition pore opening in cardiac cells. Alcohol Alcohol.
44:20–24. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Youdim MB and Arraf Z: Prevention of MPTP
(N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic
neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and
Bax. Neuropharmacology. 46:1130–1140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Linseman DA, Butts BD, Precht TA, et al:
Glycogen synthase kinase-3beta phosphorylates Bax and promotes its
mitochondrial localization during neuronal apoptosis. J Neurosci.
24:9993–10002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Maurer U, Charvet C, Wagman AS, Dejardin E
and Green DR: Glycogen synthase kinase-3 regulates mitochondrial
outer membrane permeabilization and apoptosis by destabilization of
MCL-1. Mol Cell. 21:749–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tsujimoto Y and Shimizu S: VDAC regulation
by the Bcl-2 family of proteins. Cell Death Differ. 7:1174–1181.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Martinou JC and Green DR: Breaking the
mitochondrial barrier. Nat Rev Mol Cell Biol. 2:63–67. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Armstrong JS: Mitochondrial membrane
permeabilization: the sine qua non for cell death. Bioessays.
28:253–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gollapudi S, McCormick MJ and Gupta S:
Changes in mitochondrial membrane potential and mitochondrial mass
occur independent of the activation of caspase-8 and caspase-3
during CD95-mediated apoptosis in peripheral blood T cells. Int J
Oncol. 22:597–600. 2003.PubMed/NCBI
|
|
76
|
Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET
and Yu Q: Pharmacologic modulation of glycogen synthase
kinase-3beta promotes p53-dependent apoptosis through a direct
Bax-mediated mitochondrial pathway in colorectal cancer cells.
Cancer Res. 65:9012–9020. 2005. View Article : Google Scholar
|
|
77
|
Chen G, Zeng WZ, Yuan PX, et al: The
mood-stabilizing agents lithium and valproate robustly increase the
levels of the neuroprotective protein bcl-2 in the CNS. J
Neurochem. 72:879–882. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kaga S, Zhan L, Altaf E and Maulik N:
Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and
anti-apoptotic signaling through the induction of VEGF, Bcl-2 and
survivin expression in rat ischemic preconditioned myocardium. J
Mol Cell Cardiol. 40:138–147. 2006. View Article : Google Scholar
|
|
79
|
Chen RW and Chuang DM: Long term lithium
treatment suppresses p53 and Bax expression but increases Bcl-2
expression. A prominent role in neuroprotection against
excitotoxicity. J Biol Chem. 274:6039–6042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ohori K, Miura T, Tanno M, et al: Ser9
phosphorylation of mitochondrial GSK-3beta is a primary mechanism
of cardiomyocyte protection by erythropoietin against
oxidant-induced apoptosis. Am J Physiol Heart Circ Physiol.
295:H2079–H2086. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ge XH, Zhu GJ, Geng DQ, Zhang ZJ and Liu
CF: Erythropoietin attenuates 6-hydroxydopamine-induced apoptosis
via glycogen synthase kinase 3b-mediated mitochondrial
translocation of Bax in PC12 cells. Neurol Sci. 33:1249–1256. 2012.
View Article : Google Scholar
|
|
82
|
Ngok-Ngam P, Watcharasit P, Thiantanawat A
and Satayavivad J: Pharmacological inhibition of GSK3 attenuates
DNA damage-induced apoptosis via reduction of p53 mitochondrial
translocation and Bax oligomerization in neuroblastoma SH-SY5Y
cells. Cell Mol Biol Lett. 18:58–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Samii A, Nutt JG and Ransom BR:
Parkinson’s disease. Lancet. 363:1783–1793. 2004.
|
|
84
|
Spillantini MG, Schmidt ML, Lee VM,
Trojanowski JQ, Jakes R and Goedert M: Alpha-synuclein in Lewy
bodies. Nature. 388:839–840. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
85
|
Singleton AB, Farrer M, Johnson J, et al:
alpha-Synuclein locus triplication causes Parkinson’s disease.
Science. 302:8412003.
|
|
86
|
Liu D, Jin L, Wang H, et al: Silencing
alpha-synuclein gene expression enhances tyrosine hydroxylase
activity in MN9D cells. Neurochem Res. 33:1401–1409. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Baptista MJ, O’Farrell C, Daya S, et al:
Co-ordinate transcriptional regulation of dopamine synthesis genes
by alpha-synuclein in human neuroblastoma cell lines. J Neurochem.
85:957–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Perez RG, Waymire JC, Lin E, Liu JJ, Guo F
and Zigmond MJ: A role for alpha-synuclein in the regulation of
dopamine biosynthesis. J Neurosci. 22:3090–3099. 2002.
|
|
89
|
Yu S, Zuo X, Li Y, et al: Inhibition of
tyrosine hydroxylase expression in alpha-synuclein-transfected
dopaminergic neuronal cells. Neurosci Lett. 367:34–39. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Danzer KM, Haasen D, Karow AR, et al:
Different species of alpha-synuclein oligomers induce calcium
influx and seeding. J Neurosci. 27:9220–9232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Periquet M, Fulga T, Myllykangas L,
Schlossmacher MG and Feany MB: Aggregated alpha-synuclein mediates
dopaminergic neurotoxicity in vivo. J Neurosci. 27:3338–3346. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Desplats P, Lee HJ, Bae EJ, et al:
Inclusion formation and neuronal cell death through
neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad
Sci USA. 106:13010–13015. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Masliah E, Rockenstein E, Veinbergs I, et
al: Dopaminergic loss and inclusion body formation in
alpha-synuclein mice: implications for neurodegenerative disorders.
Science. 287:1265–1269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Parihar MS, Parihar A, Fujita M, Hashimoto
M and Ghafourifar P: Mitochondrial association of alpha-synuclein
causes oxidative stress. Cell Mol Life Sci. 65:1272–1284. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Parihar MS, Parihar A, Fujita M, Hashimoto
M and Ghafourifar P: Alpha-synuclein overexpression and aggregation
exacerbates impairment of mitochondrial functions by augmenting
oxidative stress in human neuroblastoma cells. Int J Biochem Cell
Biol. 41:2015–2024. 2009. View Article : Google Scholar
|
|
96
|
Hsu LJ, Sagara Y, Arroyo A, et al:
alpha-synuclein promotes mitochondrial deficit and oxidative
stress. Am J Pathol. 157:401–410. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Feng LR, Federoff HJ, Vicini S and
Maguire-Zeiss KA: Alpha-synuclein mediates alterations in membrane
conductance: a potential role for alpha-synuclein oligomers in cell
vulnerability. Eur J Neurosci. 32:10–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lee EJ, Woo MS, Moon PG, et al:
Alpha-synuclein activates microglia by inducing the expressions of
matrix metalloproteinases and the subsequent activation of
protease-activated receptor-1. J Immunol. 185:615–623. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Su X, Federoff HJ and Maguire-Zeiss KA:
Mutant alpha-synuclein overexpression mediates early
proinflammatory activity. Neurotox Res. 16:238–254. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Theodore S, Cao S, McLean PJ and Standaert
DG: Targeted overexpression of human alpha-synuclein triggers
microglial activation and an adaptive immune response in a mouse
model of Parkinson disease. J Neuropathol Exp Neurol. 67:1149–1158.
2008. View Article : Google Scholar
|
|
101
|
Kozikowski AP, Gaisina IN, Petukhov PA, et
al: Highly potent and specific GSK-3beta inhibitors that block tau
phosphorylation and decrease alpha-synuclein protein expression in
a cellular model of Parkinson’s disease. ChemMedChem. 1:256–266.
2006.PubMed/NCBI
|
|
102
|
Haggerty T, Credle J, Rodriguez O, et al:
Hyperphosphorylated Tau in an alpha-synuclein-overexpressing
transgenic model of Parkinson’s disease. Eur J Neurosci.
33:1598–1610. 2011.PubMed/NCBI
|
|
103
|
Wills J, Jones J, Haggerty T, Duka V,
Joyce JN and Sidhu A: Elevated tauopathy and alpha-synuclein
pathology in postmortem Parkinson’s disease brains with and without
dementia. Exp Neurol. 225:210–218. 2010.
|
|
104
|
Cho JH and Johnson GV: Glycogen synthase
kinase 3 beta induces caspase-cleaved tau aggregation in situ. J
Biol Chem. 279:54716–54723. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Peng JH, Zhang CE, Wei W, Hong XP, Pan XP
and Wang JZ: Dehydroevodiamine attenuates tau hyperphosphorylation
and spatial memory deficit induced by activation of glycogen
synthase kinase-3 in rats. Neuropharmacology. 52:1521–1527. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chun W and Johnson GV: Activation of
glycogen synthase kinase 3beta promotes the intermolecular
association of tau. The use of fluorescence resonance energy
transfer microscopy. J Biol Chem. 282:23410–23417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Greco SJ, Sarkar S, Casadesus G, et al:
Leptin inhibits glycogen synthase kinase-3beta to prevent tau
phosphorylation in neuronal cells. Neurosci Lett. 455:191–194.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Crouch PJ, Hung LW, Adlard PA, et al:
Increasing Cu bioavailability inhibits Abeta oligomers and tau
phosphorylation. Proc Natl Acad Sci USA. 106:381–386. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Engel T, Lucas JJ, Gomez-Ramos P, Moran
MA, Avila J and Hernandez F: Cooexpression of FTDP-17 tau and
GSK-3beta in transgenic mice induce tau polymerization and
neurodegeneration. Neurobiol Aging. 27:1258–1268. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Engel T, Hernandez F, Avila J and Lucas
JJ: Full reversal of Alzheimer’s disease-like phenotype in a mouse
model with conditional overexpression of glycogen synthase
kinase-3. J Neurosci. 26:5083–5090. 2006.
|
|
111
|
Perez M, Hernandez F, Lim F, Diaz-Nido J
and Avila J: Chronic lithium treatment decreases mutant tau protein
aggregation in a transgenic mouse model. J Alzheimers Dis.
5:301–308. 2003.PubMed/NCBI
|
|
112
|
Nakashima H, Ishihara T, Suguimoto P, et
al: Chronic lithium treatment decreases tau lesions by promoting
ubiquitination in a mouse model of tauopathies. Acta Neuropathol.
110:547–556. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Engel T, Goni-Oliver P, Lucas JJ, Avila J
and Hernandez F: Chronic lithium administration to FTDP-17 tau and
GSK-3beta overexpressing mice prevents tau hyperphosphorylation and
neurofibrillary tangle formation, but pre-formed neurofibrillary
tangles do not revert. J Neurochem. 99:1445–1455. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gao HM and Hong JS: Why neurodegenerative
diseases are progressive: uncontrolled inflammation drives disease
progression. Trends Immunol. 29:357–365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kim SU and de Vellis J: Microglia in
health and disease. J Neurosci Res. 81:302–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mrak RE and Griffin WS: Glia and their
cytokines in progression of neurodegeneration. Neurobiol Aging.
26:349–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
McGeer PL and McGeer EG: Glial reactions
in Parkinson’s disease. Mov Disord. 23:474–483. 2008.
|
|
118
|
Ouchi Y, Yagi S, Yokokura M and Sakamoto
M: Neuroinflammation in the living brain of Parkinson’s disease.
Parkinsonism Relat Disord. 15 Suppl 3:S200–S204. 2009.
|
|
119
|
Hunot S, Dugas N, Faucheux B, et al:
FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces,
in vitro, production of nitric oxide and tumor necrosis
factor-alpha in glial cells. J Neurosci. 19:3440–3447.
1999.PubMed/NCBI
|
|
120
|
Mogi M, Harada M, Narabayashi H, Inagaki
H, Minami M and Nagatsu T: Interleukin (IL)-1 beta, IL-2, IL-4,
IL-6 and transforming growth factor-alpha levels are elevated in
ventricular cerebrospinal fluid in juvenile parkinsonism and
Parkinson’s disease. Neurosci Lett. 211:13–16. 1996.PubMed/NCBI
|
|
121
|
Koziorowski D, Tomasiuk R, Szlufik S and
Friedman A: Inflammatory cytokines and NT-proCNP in Parkinson’s
disease patients. Cytokine. 60:762–766. 2012.
|
|
122
|
Przedborski S: Inflammation and
Parkinson’s disease pathogenesis. Mov Disord. 25 Suppl 1:S55–S57.
2010.
|
|
123
|
Frankola KA, Greig NH, Luo W and Tweedie
D: Targeting TNF-alpha to elucidate and ameliorate
neuroinflammation in neurodegenerative diseases. CNS Neurol Disord
Drug Targets. 10:391–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Qian L, Flood PM and Hong JS:
Neuroinflammation is a key player in Parkinson’s disease and a
prime target for therapy. J Neural Transm. 117:971–979. 2010.
|
|
125
|
Lofrumento DD, Nicolardi G, Cianciulli A,
et al: Neuroprotective effects of resveratrol in an MPTP mouse
model of Parkinson’s-like disease: Possible role of SOCS-1 in
reducing pro-inflammatory responses. Innate Immun. 2013.PubMed/NCBI
|
|
126
|
Martin M, Rehani K, Jope RS and Michalek
SM: Toll-like receptor-mediated cytokine production is
differentially regulated by glycogen synthase kinase 3. Nat
Immunol. 6:777–784. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
127
|
Jope RS, Yuskaitis CJ and Beurel E:
Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and
therapeutics. Neurochem Res. 32:577–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Beurel E and Jope RS:
Lipopolysaccharide-induced interleukin-6 production is controlled
by glycogen synthase kinase-3 and STAT3 in the brain. J
Neuroinflammation. 6:92009. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Cheng YL, Wang CY, Huang WC, et al:
Staphylococcus aureus induces microglial inflammation via a
glycogen synthase kinase 3beta-regulated pathway. Infect Immun.
77:4002–4008. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang MJ, Huang HY, Chen WF, Chang HF and
Kuo JS: Glycogen synthase kinase-3beta inactivation inhibits tumor
necrosis factor-alpha production in microglia by modulating nuclear
factor kappaB and MLK3/JNK signaling cascades. J Neuroinflammation.
7:992010. View Article : Google Scholar
|
|
131
|
Colasanti M, Persichini T, Di Pucchio T,
Gremo F and Lauro GM: Human ramified microglial cells produce
nitric oxide upon Escherichia coli lipopolysaccharide and tumor
necrosis factor alpha stimulation. Neurosci Lett. 200:144–146.
1995. View Article : Google Scholar
|
|
132
|
Block ML and Hong JS: Chronic microglial
activation and progressive dopaminergic neurotoxicity. Biochem Soc
Trans. 35:1127–1132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Członkowska A, Kohutnicka M,
Kurkowska-Jastrzebska I and Członkowski A: Microglial reaction in
MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced
Parkinson’s disease mice model. Neurodegeneration. 5:137–143.
1996.
|
|
134
|
Przedborski S and Vila M: The
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to
explore the pathogenesis of Parkinson’s disease. Ann NY Acad Sci.
991:189–198. 2003.PubMed/NCBI
|
|
135
|
Duka T, Duka V, Joyce JN and Sidhu A:
Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau
phosphorylation in Parkinson’s disease models. FASEB J.
23:2820–2830. 2009.PubMed/NCBI
|
|
136
|
Watcharasit P, Thiantanawat A and
Satayavivad J: GSK3 promotes arsenite-induced apoptosis via
facilitation of mitochondria disruption. J Appl Toxicol.
28:466–474. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Cookson MR: The biochemistry of
Parkinson’s disease. Annu Rev Biochem. 74:29–52. 2005.
|