Introduction
Pheochromocytoma (PCC), a rare
catecholamine-producing tumor with an estimated incidence of
0.005–0.1% in the worldwide population (1), may result in classical symptoms,
including severe hypertension accompanied by headache and
palpitation requiring proactive preoperative medical management to
decrease morbidity and mortality. However, there are certain
exceptions that have been described traditionally as the ‘10%
rule’, as they occur at an incidence of ~10% within patients with
PCC (1,2). ‘Silent’ PCC is one of the exceptions
that does not exhibit classic PCC symptoms (3); therefore, ‘silent’ PCC often remains
undiagnosed until surgical excision occurs and the anesthesia teams
face a greater challenge. The authors report the case of a silent
giant cystic pheochromocytoma (GPCC), which was preoperatively
diagnosed as a malignant renal mass; GPCC was confirmed as a result
of the classical hypertension crisis following surgical exploration
and histopathological evaluation.
Case report
Written informed consent was obtained from the
patient and the institutional ethics review board was consulted for
approval (not deemed necessary by the Institutional Ethics Review
Board of Qilu Hospital, Jinan, China) for publishing this case.
A 36-year-old woman presented to Qilu Hospital of
Shandong University on May 9, 2013, with the primary complaint of
abdominal discomfort following eating and lumbodorsal distending
pain for 3 months, and reported weight loss of 8 kg during this
time. The patient's medical history included a caesarean section
and an ovarian cysts surgery, but no history of hypertension or
headache. The patient's vital signs included an arterial blood
pressure of 120/80 mmHg, heart rate of 80 bpm and temperature
36.8°C. The only significant finding during physical examination
was for left renal region percussion pain. Laboratory analysis
identified a slightly elevated blood glucose level of 7.73 mmol/l
(normal range, 3.90–6.10 mmol/l). Ultrasonography examination
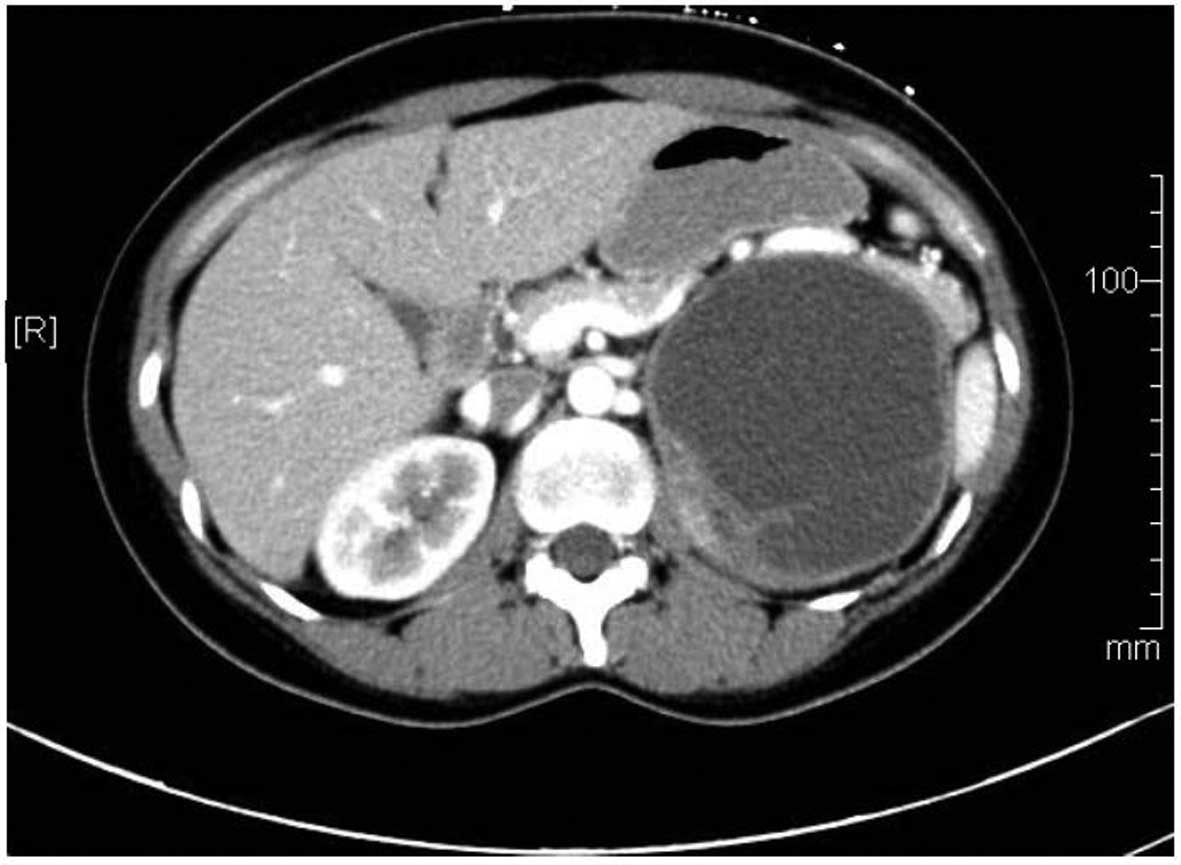
revealed a cystic space-occupying lesion (10.3×9.3 cm) in the left
upper abdomen, which was considered to be a left renal cystic mass.
Abdominal computed tomography (CT) and a contrast-enhanced CT scan
demonstrated a giant cystic-solid mass on the left kidney, which
occupied a large part of the superior abdominal cavity (Fig. 1). Based on the patient's age, gender,
history, physical examination, and preoperative imaging, a clinical
diagnosis of a malignant adrenal mass was suspected. No special
treatment was given prior to surgery because the diagnosis of a
pheochromocytoma had not been considered.
Standard monitors were applied and the blood
pressure, heart rate and peripheral capillary oxygen saturation
(SpO2) measurements were 108/70 mmHg, 80 bpm and 97% on
arrival in the operating room. General anesthesia was induced with:
Midazolam, 3 mg; etomdate, 12 mg; and fentanyl, 0.2 mg. Rocuronium
bromide, 50 mg, was given to facilitate the intubation of the
larangeal mask airway (LMA), and anesthesia was maintained with
1.5–2.5% sevoflurane. When the mass was investigated, the
anesthesiologist noted the blood pressure and heart rate rapidly
increased to >210/120 mmHg and 120 bpm, respectively, and
ventricular premature contractions became frequent. Investigation
of the mass was immediately ceased because it became apparent that
the mass may was potentially a pheochromocytoma. Intravenous (i.v.)
phentolamine (1 mg injection), nitroprusside (4 mg drip), esmolol
(30 mg injection) were administrated following deepening
anesthesia, and a radical arterial puncture catheter was inserted
to aid in the monitoring of arterial blood pressure. The patient
experienced multiple episodes of hypertension alternating with
hypotension, and this resulted in the SpO2 reducing to
89% and the PaO2 reducing to 61 mmHg 40 min later.
Meanwhile, a moist rale became obvious in the bilateral lungs
following auscultation of the chest. An endotracheal tube was
intubated to replace the LMA. PEEP (5–10 cm H2O),
dihydroxypropyl theophylline (10 g i.v. drip), dexamethasone (10 g
i.v. drip), fursemide (10 g i.v. injection) were administered to
inhibit the development of pulmonary edema. 5% NaHCO3,
10% potassium chloride were administrated according the results of
blood gas analysis. When the blood circulation was under control,
the mass was quickly excised and the blood pressure rapidly reduced
to 60/40 mmHg. Following administration of intravenous
norepinephrine and a larger volume of liquids infusion thought a
central venous catheter, the blood pressure was maintained at
>100/60 mmHg. During the dissection, the mass was identified and
appeared to originate from the left adrenal gland. The patient was
transferred to the ICU with the support of a portable breathing
machine for further monitoring and treatment of the hypotension at
the end of surgery. Inverted T waves were observed in the V1-V5
chest leads of the electrocardiogram from when the patient was
transferred to the intensive care unit for 7 days post-surgery.
Increases in serum N-terminal pro-brain natriuretic peptide
(6,316.0 pg/ml, normal value <450 pg/ml) and troponin I (0.48
ng/ml, normal value <0.06 ng/ml) were also observed, which
indicated the occurrence of myocardial damage. Following
comprehensive treatment with a continuous pump of norepinephrine (2
mg i.v. drip), the patient was transferred back to the surgical
ward a week later. Two days later, norepinephrine treatment was
stopped and the patient was discharged home on postoperative day 14
without any recurrence. During a follow-up examination performed 7
days after discharge from hospital, the patient exhibited favorable
results without any discomfort. In addition, all laboratory
examination and hemodynamic index results were within the normal
ranges.
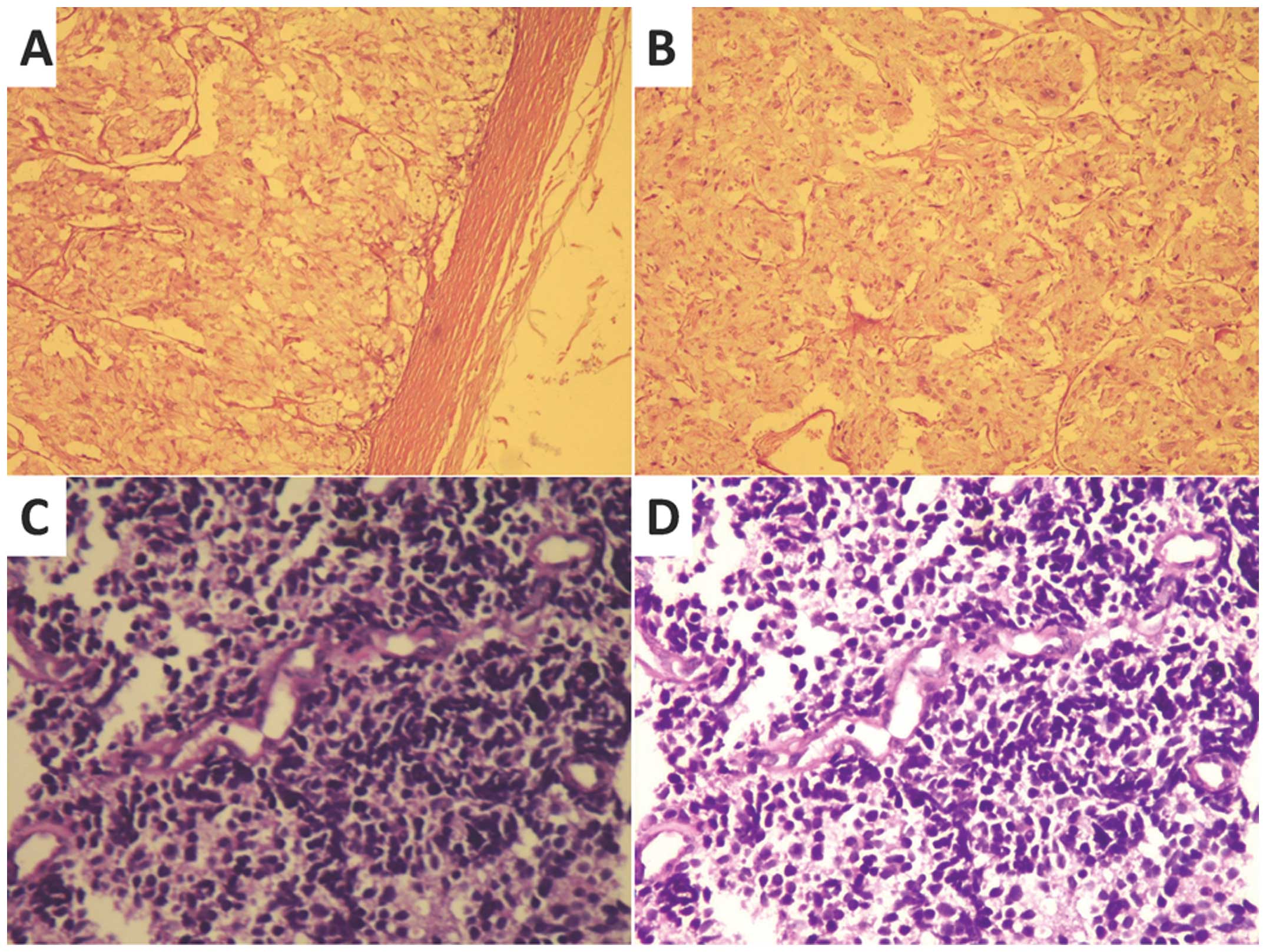
Gross histopathological evaluation revealed a 10×9
cm, cyst-solid mass in the adrenal gland. Formalin-fixed tissue was
embedded into paraffin wax and cut into 5-µm thick sections. The
sections were stained with hematoxylin and eosin for
histopathological examination or incubated with specific antibodies
against chromogranin A, synaptophysin, CD5 and Melan A and stained
with DAB for immunohistochemical evaluation. The sections were
examined and images were captured with an Olympus CX21 light
microscope (Olympus Corporation, Greenhills, Philippines) at a
magnification of x100. Representative histopathological images are
presented in Fig. 2, which confirmed
the histopathological diagnosis of PCC.
Discussion
PCCs have an estimated incidence of <0.1% in the
hypertensive population (4). They are
catecholamine-secreting tumors derived from chromaffin cells
originating in the neural crest and from cells of similar embryonic
derivation within the sympathetic ganglia (1). PCCs are highly vascular tumors, which
are commonly unilateral and solitary. However, there are certain
exceptions which have been described traditionally as the ‘10%
rule’ with a ~10% incidence of extra-adrenal location, familial
occurrence, childhood onset, malignant phenotype, recurrence
following resection, multiple or bilateral tumor formation and the
absence of symptoms. The ‘10% rule’ has been challenged by
developments in clinical and molecular research. A previous study
has indicated that as many as 25% of PCCs may be familial (5), and asymptomatic and malignant PCC occur
in >10% of PCC patients (1).
Typical PCCs are 3–5 cm in diameter, weighing ~50–90
g. GPCCs weighing >1 kg are rarely observed. Pan et al
(6) summarized 15 cases of GPCCs from
the English literature in the 30 years between 1976 and 2006: The
authors observed that GPCCs occurred more frequently in males (male
to female ratio, 2:1), the mean age was 45 years (ranging between
12 and 70 years), the classical symptoms are attacks of severe
hypertension accompanied by headache and palpitation, but the
majority of GPCCs were asymptomatic (9/15). The factors which
contributed to the GPCCs presenting as asymptomatic included the
number of cells producing catecholamine, which significantly
reduced with the presence of an extensive necrotic cystic region at
the centre of the mass; when interstitial tissue composes the
majority of the neoplasm and the cells are thus not bioactive; and
that the catecholamines and metabolic products stored in the
capsular mass are not released into the blood circulation until the
PCC mass is isolated (3). Thus,
patients with GPCC may not manifest the typical clinical
manifestations or elevated levels of urinary catecholamine
metabolites. Abdominal CT scan also may not accurately delineate
the organ site of mass in the case of GPCCs. Clinically, GPCCs may
mimic cystic neoplasms of the pancreas (7), liver (8),
pelvis (9) or kidney as in the
present case report. Large adrenal tumors may also cause atrophy of
the gland, making the residual gland unrecognizable. Therefore,
they are often not accurately diagnosed until the time of
resection, when hemodynamic instability occurs, or even following
evaluation of pathological specimens (7,9–13).
Once PCC is diagnosed, surgical removal of the tumor
is the preferred treatment. Hypertensive crises and arrhythmias,
which are directly associated with the increase of preoperative
complications or even mortality, may occur even if patients are
preoperatively normotensive and asymptomatic. Therefore, blocking
the effects of released catecholamines are recommended for all
patients with biochemically positive PCCs (14). However, in the case of silent PCCs
which are not diagnosed until the tumor is further investigated,
surgeons may prefer to complete the resection immediately following
achieving control over the hypertension during surgery (3,7,10,15,16)
compared with postponing the surgery until later (17), despite the evidence that this may
result in a hypertensive crisis (3,7),
ventricular ectopic rhythms and cardiac arrest (10), or acute myocardial damage as in the
present report. When unexpected hypertension occurs during
resection of an abdominal tumor, the anesthesiologist and surgeon
must consider the diagnosis of pheochromocytoma and clearly realize
that it is an anesthetic challenge. Good coordination between the
surgical and anesthesia teams is essential for the patient's safety
during surgery (7). A surgeon may be
able to carefully manipulate the tumor with early isolation of
tumors' venous drainage, halting intermittently to allow the
anesthesia team to make the hemodynamics stable with appropriate
vasoactive drugs, this coordincation may minimize the risk of an
intraoperative hypertensive crisis.
GPCCs are rare adrenal tumors and the majority of
them are asymptomatic. This leads to a high proportion of these
tumors being undiagnosed until surgery and surgical teams face a
great challenge in perioperative management as a result. The
present study reports a case of GPCC; the tumor was successfully
resected despite the occurrence of perioperative cardiovascular
events, and the patientwas discharged home without any
recurrence.
Acknowledgements
The present study was partly supported by Shandong
Provincial Natural Science Foundation, P.R. China (Y2007C115
ZR2011HM028, to Dr Huan-Liang Wang and 2009ZRB14031 to Dr Wei-Fu
Lei).
References
|
1
|
Dahia PL: Evolving concepts in
pheochromocytoma and paraganglioma. Curr Opin Oncol. 18:1–8. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elder EE, Elder G and Larsson C:
Pheochromocytoma and functional paraganglioma syndrome: No longer
the 10% tumor. J Surg Oncol. 89:193–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li C, Chen Y, Wang W and Teng L: A case of
clinically silent giant right pheochromocytoma and review of
literature. Can Urol Assoc J. 6:E267–E269. 2012.PubMed/NCBI
|
|
4
|
Lee TH, Slywotsky CM, Lavelle MT and
Garcia RA: Cystic pheochromocytoma. Radiographics. 22:935–940.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neumann HP, Berger DP, Sigmund G, Blum U,
Schmidt D, Parmer RJ, Volk B and Kirste G: Pheochromocytomas,
multiple endocrine neoplasia type 2 and von Hippel-Lindau disease.
N Engl J Med. 329:1531–1538. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan Z, Repertinger S, Deng C and Sharma P:
A giant cystic pheochromocytoma of the adrenal gland. Endocr
Pathol. 19:133–138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antedomenico E and Wascher RA: A case of
mistaken identity: Giant cystic pheochromocytoma. Curr Surg.
62:193–198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu JS, Ahya SN, Reploeg MD, Singer GG,
Brennan DC, Howard TK and Lowell JA: Pheochromocytoma presenting as
a giant cystic tumor of the liver. Surgery. 128:482–484. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Terk MR, de Verdier H and Colletti PM:
Giant extra-adrenal pheochromocytoma: Magnetic resonance imaging
with gadolinium-DTPA enhancement. Magn Reson Imaging. 11:47–50.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Melegh Z, Rényi-Vámos F, Tanyay Z, Köves I
and Orosz Z: Giant cystic pheochromocytoma located in the renal
hilus. Pathol Res Pract. 198:103–106. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan FK, Choi KL, Tiu SC, Shek CC and Au
Yong TK: A case of giant malignant phaeochromocytoma. Hong Kong Med
J. 6:325–328. 2000.PubMed/NCBI
|
|
12
|
Grissom JR, Yamase HT and Prosser PR:
Giant pheochromocytoma with sarcoidosis. South Med J. 72:1605–1607.
1979. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Awada SH, Grisham A and Woods SE: Large
dopamine-secreting pheochromocytoma: Case report. South Med J.
96:914–917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pacak K, Eisenhofer G, Ahlman H, Bornstein
SR, Gimenez-Roqueplo AP, Grossman AB, Kimura N, Mannelli M, McNicol
AM and Tischler AS: International Symposium on Pheochromocytoma:
Pheochromocytoma: Recommendations for clinical practice from the
First international Symposium. Nat Clin Pract Endocrinol Metab.
3:92–102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Basiri A and Radfar MH: Giant cystic
pheochromocytoma. Urol J. 7:162010.PubMed/NCBI
|
|
16
|
Costa SR, Cabral NM, Abhrão AT, Costa RB,
Silva LM and Lupinacci RA: Giant cystic malignant pheochromocytoma
invading right hepatic lobe: Report on two cases. Sao Paulo Med J.
126:229–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tarant NS, Dacanay RG, Mecklenburg BW,
Birmingham SD, Lujan E and Green R: Acute appendicitis in a patient
with undiagnosed pheochromocytoma. Anesth Analg. 102:642–643. 2006.
View Article : Google Scholar : PubMed/NCBI
|