Gastric cancer is a leading cause of cancer-related
mortality worldwide, with nearly 1 million new cases and
approximately 750,000 mortalities annually (1). Gastric cancer is a multifactorial
disease and the risk factors for this include environmental factors
and factors that influence host-pathogen interaction and complex
interplay between these factors. Gastric cancer occurrence is more
predominant in developing countries in Eastern Europe, South
America, and Asia, accounting for approximately two thirds of all
cases globally, with China representing approximately 42% of all
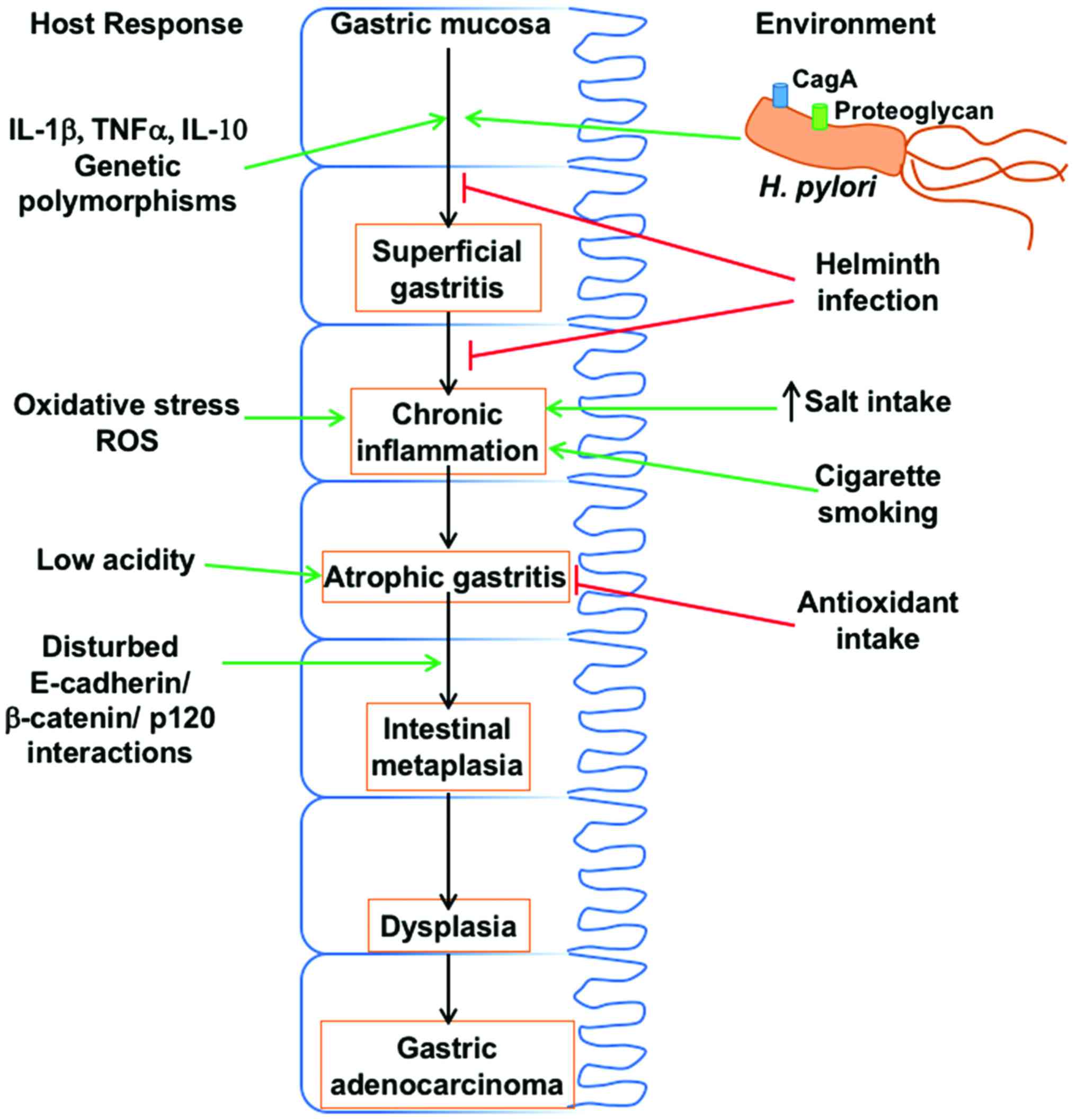
new cases (2). Development of gastric
cancer likely originates with the onset of chronic active gastritis
and follows with atrophic gastritis, intestinal metaplasia, and
dysplasia, eventually leading to gastric cancer (3). Besides environmental, diet and genetic
factors, gastric cancer is closely associated with Helicobacter
pylori (H. pylori) infection (4) and related host gene polymorphisms
(5). Gastric adenocarcinomas
constitute 90–95% of gastric cancers and are of two types,
intestinal and diffuse type. Although there is no known precursor
lesion for the development of diffuse type of gastric cancers,
H. pylori infection has been suspected of being causally
linked to the initiation of chronic active gastritis, which leads
to adenocarcinoma (6). Infection of
H. pylori is one of the thoroughly studied risk factors of
gastric cancer.
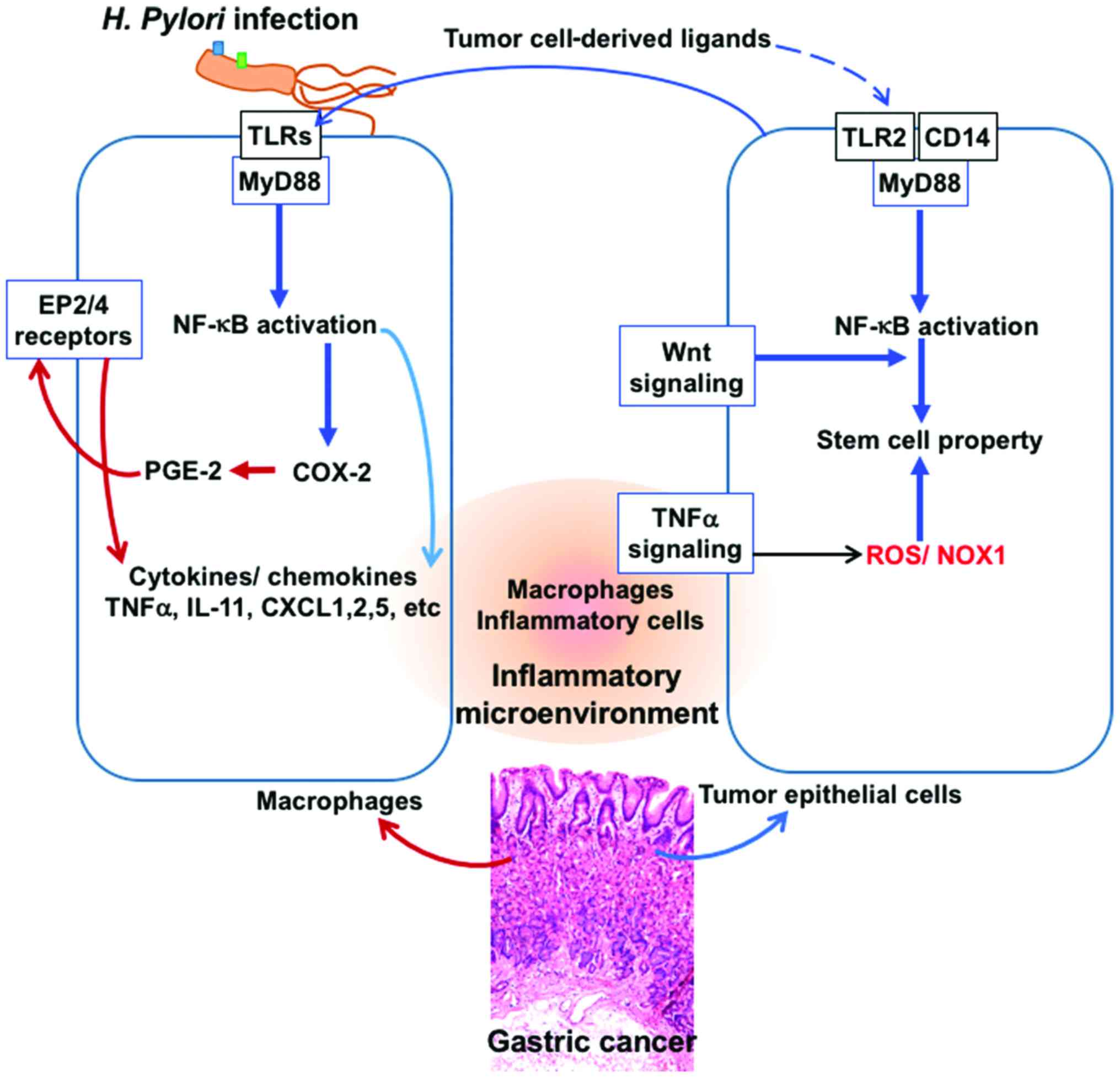
Inflammation of gastric epithelium is known to be
associated with the development of gastric cancer (29). There are several mechanisms by which
inflammation may promote cancer development and the induction of
the cyclooxygenase-2/prostaglandin E2 (COX-2⁄PGE2) pathway and
activation of NF-κB and Stat3 appear to be major pathways (Fig. 2) (30).
Besides these, innate immune responses through the TLR/MyD88
adapter signaling also play a role in tumorigenesis (31,32). In
fact, it has been shown that almost all the gastric tumors show an
induction of COX-2 expression (33)
and H. pylori infection is known to lead to COX-2 induction
(34). Inflammation in combination
with oncogenic activation, promotes tumorigenesis and also Wnt
signaling activation (Fig. 2) with
the accumulation of β-catenin, which facilitate tumor growth and
this altered signaling has been observed in over 50% of gastric
cancers (35). PGE2 signaling,
through the EP4 receptor, is known to induce the expansion of
CD133+ CD44+ cancer stem cells in intestinal
tumors through the activation of PI3K and MAPK signaling (36), which potentially aggravates tumor
growth.
Another important cytokine, IL-1β is known to play a
role in a variety of cellular activities such as inflammatory
response and acid secretion by gastric epithelium (37). Disturbances in the regulation of IL-1β
are observed in several cancer types and in particular, in IL-1β
gene polymorphisms including IL-1β −31 (T>C) and IL-1β −511
(C>T) which are closely related to gastric cancer (Fig. 1) (38,39). Of
note, it has been shown that IL-1β-511T polymorphism is present in
all the Mozambican subjects with intestinal metaplasia (40). This polymorphism is also associated
with the prevalence of dysplasia (41), indicating that the IL-1β T alleles are
related to premalignant gastric lesions. Apparently, the same
polymorphism of IL-1β is involved in the intestinal type of gastric
cancers, which are triggered by H. pylori infection and not
diffusive type (42). IL-1β gene
polymorphisms also increase the production of IL-1β, which
suppresses gastric acid secretion, and is related to the grade of
gastric atrophy in patients with H. pylori infection
(43). Additionally, H. pylori
infection leads to elevated secretion of IL-1β and reduction in
acid secretion (44). It has been
suggested that a combination of IL-1β-511T/T polymorphism and H.
pylori infection aggravates the development of gastric tumor
more than either of these agents alone (45). Thus, infection of H. pylori
promotes the expression of IL-1β, which leads to gastric
carcinogenesis through its actions on both inflammatory and
epithelial cells (46). Even though
the precise molecular basis of these actions is not clear, it seems
that hypochlorhydria and atrophic gastritis induced by IL-1β
polymorphisms, which depends on H. pylori infection are
critical in gastric cancer development (47).
A primary factor that is important in the events
that lead to the progression of the inflammation-to-carcinoma is
oxidative DNA damage induced by H. pylori infection
(48), which is probably due to
infiltrating neutrophils, and also direct effects of H.
pylori (49). Production of
reactive oxygen species in the H. pylori-infected gastric
epithelium is linked to the presence of cagPAI and contribute to
the oxidative stress response in gastric epithelial cells (50). It is well known that H. pylori
infection causes elevated level of polyamines, in particular
spermine and this is associated with an induction of spermine
oxidase (51). Action of spermine
oxidase on spermine leads to the production of elevated levels of
hydrogen peroxide, which is a powerful oxidizing agent and also
contributes to the production of free radicals such as hydroxyl
radical (52). Besides, H.
pylori also activates macrophages which show a significant
upregulation of spermine oxidase, contributing to oxidative stress
and damage to the gastric epithelial cells (53). Besides, altered polyamine metabolism
and overexpression of arginase enzyme in the infected gastric
epithelium leads to lowered NO production and increased production
of spermine and hydrogen peroxide.
E-cadherin, which is an adhesion molecule in
epithelial tissues that is important in maintaining proper cellular
architecture, is regulated by the binding of p120 to the cadherin
juxtamembrane domain (54).
Furthermore, the cytoplasmic domain of E-cadherin interacts with
β-catenin and p120, which, in turn, interact with the cytoskeletal
component actin. It has been documented that there is a loss of
E-cadherin function in gastric cancer, and in fact promoter
methylation of E-cadherin gene is induced by H. pylori
infection, leading to reduction in E-cadherin expression (55). Following H. pylori infection,
the translocated CagA in the gastric epithelial cells binds with
E-cadherin, resulting in the dissociation of the
E-cadherin-β-catenin complex and accumulation of β-catenin in
cytoplasm and nucleus, where it transactivates β-catenin-dependent
genes involved in carcinogenesis (23,56). Along
with the downregulation of E-cadherin, a decreased expression or
aberrant subcellular localization of p120, from membrane to the
cytosol or nucleus, is commonly seen in gastric cancer (57). In the cytoplasm, p120 interacts with
Rho GTPases and promotes motility and metastasis (58). Aberrant localization of p120 to the
nucleus in gastric epithelia infected with H. pylori has
been reported and p120 in nucleus can relieve transcriptional
repression of the mmp-7 gene, which is involved in gastric
tumorigenesis, leading to its enhanced expression (59).
Gastric adenocarcinoma is strongly influenced by
dietary salt intake, with high salt intake aggravating
tumorigenesis (60). Epidemiological
studies indicated that high salt intake increases the prevalence of
H. pylori infection (61) and
the incidence of gastric adenocarcinoma in infected patients
(62). Experimental studies indicated
that a high-salt diet and H. pylori infection exert
synergistic effects on the development of premalignant lesions or
gastric cancer (63), probably by
elevating the production of inflammatory cytokines IL-1, IL-6 and
TNF-α (64). However, the precise
molecular events that underlie this synergistic effect on cancer
development are not known. It has been suggested that high salt
increases the expression of CagA, the potential carcinogen in H.
pylori (65), which may be the
reason for the observed synergy between H. pylori and salt
for gastric cancer induction (Fig.
1).
|
1
|
Ferro A, Peleteiro B, Malvezzi M, Bosetti
C, Bertuccio P, Levi F, Negri E, La Vecchia C and Lunet N:
Worldwide trends in gastric cancer mortality (1980–2011), with
predictions to 2015, and incidence by subtype. Eur J Cancer.
50:1330–1344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Correa P: Human gastric carcinogenesis: a
multistep and multifactorial process - first American cancer
society award lecture on cancer epidemiology and prevention. Cancer
Res. 52:6735–6740. 1992.PubMed/NCBI
|
|
4
|
Herrera V and Parsonnet J: Helicobacter
pylori and gastric adenocarcinoma. Clin Microbiol Infect.
15:971–976. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He C, Tu H, Sun L, Xu Q, Li P, Gong Y,
Dong N and Yuan Y: Helicobacter pylori-related host gene
polymorphisms associated with susceptibility of gastric
carcinogenesis: a two-stage case-control study in Chinese.
Carcinogenesis. 34:1450–1457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Souza RF and Spechler SJ: Concepts in the
prevention of adenocarcinoma of the distal esophagus and proximal
stomach. CA Cancer J Clin. 55:334–351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marshall BJ and Warren JR: Unidentified
curved bacilli in the stomach of patients with gastritis and peptic
ulceration. Lancet. 1:1311–1315. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weeks DL, Eskandari S, Scott DR and Sachs
G: A H+-gated urea channel: the link between
Helicobacter pylori urease and gastric colonization. Science.
287:482–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peek RM Jr and Blaser MJ: Helicobacter
pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer.
2:28–37. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peek RM Jr and Crabtree JE: Helicobacter
infection and gastric neoplasia. J Pathol. 208:233–248. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sipponen P and Marshall BJ: Gastritis and
gastric cancer. Western countries. Gastroenterol Clin North Am.
29:579–592. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Correa P and Houghton J: Carcinogenesis of
Helicobacter pylori. Gastroenterology. 133:659–672. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Atherton JC: The pathogenesis of
Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev
Pathol. 1:63–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimoyama T, Fukuda S, Tanaka M, Mikami T,
Munakata A and Crabtree JE: CagA seropositivity associated with
development of gastric cancer in a Japanese population. J Clin
Pathol. 51:225–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Torres J, Pérez-Pérez GI, Leal-Herrera Y
and Muñoz O: Infection with CagA+ Helicobacter pylori
strains as a possible predictor of risk in the development of
gastric adenocarcinoma in Mexico. Int J Cancer. 78:298–300. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stein M, Bagnoli F, Halenbeck R, Rappuoli
R, Fantl WJ and Covacci A: c-Src/Lyn kinases activate Helicobacter
pylori CagA through tyrosine phosphorylation of the EPIYA motifs.
Mol Microbiol. 43:971–980. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Segal ED, Cha J, Lo J, Falkow S and
Tompkins LS: Altered states: involvement of phosphorylated CagA in
the induction of host cellular growth changes by Helicobacter
pylori. Proc Natl Acad Sci USA. 96:14559–14564. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Basso D, Zambon CF, Letley DP, Stranges A,
Marchet A, Rhead JL, Schiavon S, Guariso G, Ceroti M, Nitti D, et
al: Clinical relevance of Helicobacter pylori cagA and vacA gene
polymorphisms. Gastroenterology. 135:91–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Higashi H, Tsutsumi R, Muto S, Sugiyama T,
Azuma T, Asaka M and Hatakeyama M: SHP-2 tyrosine phosphatase as an
intracellular target of Helicobacter pylori CagA protein. Science.
295:683–686. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wroblewski LE, Peek RM Jr and Wilson KT:
Helicobacter pylori and gastric cancer: factors that modulate
disease risk. Clin Microbiol Rev. 23:713–739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bagnoli F, Buti L, Tompkins L, Covacci A
and Amieva MR: Helicobacter pylori CagA induces a transition from
polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci
USA. 102:16339–16344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murata-Kamiya N, Kurashima Y, Teishikata
Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM
Jr, Azuma T, et al: Helicobacter pylori CagA interacts with
E-cadherin and deregulates the beta-catenin signal that promotes
intestinal transdifferentiation in gastric epithelial cells.
Oncogene. 26:4617–4626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohnishi N, Yuasa H, Tanaka S, Sawa H,
Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, et
al: Transgenic expression of Helicobacter pylori CagA induces
gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl
Acad Sci USA. 105:1003–1008. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Viala J, Chaput C, Boneca IG, Cardona A,
Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et
al: Nod1 responds to peptidoglycan delivered by the Helicobacter
pylori cag pathogenicity island. Nat Immunol. 5:1166–1174. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nagy TA, Frey MR, Yan F, Israel DA, Polk
DB and Peek RM Jr: Helicobacter pylori regulates cellular migration
and apoptosis by activation of phosphatidylinositol 3-kinase
signaling. J Infect Dis. 199:641–651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miehlke S, Kirsch C, AghaAmiri K, Günther
T, Lehn N, Malfertheiner P, Stolte M, Ehninger G and Bayerdörffer
E: The Helicobacter pylori vacA s1, m1 genotype and cagA is
associated with gastric carcinoma in Germany. Int J Cancer.
87:322–327. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dossumbekova A, Prinz C, Gerhard M,
Brenner L, Backert S, Kusters JG, Schmid RM and Rad R: Helicobacter
pylori outer membrane proteins and gastric inflammation. Gut.
55:1360–1361. 2006.PubMed/NCBI
|
|
29
|
Echizen K, Hirose O, Maeda Y and Oshima M:
Inflammation in gastric cancer: interplay of the
COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways.
Cancer Sci. 107:391–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pradere JP, Dapito DH and Schwabe RF: The
yin and yang of toll-like receptors in cancer. Oncogene.
33:3485–3495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maeda Y, Echizen K, Oshima H, Yu L,
Sakulsak N, Hirose O, Yamada Y, Taniguchi T, Jenkins BJ, Saya H, et
al: Myeloid differentiation factor 88 signaling in bone
marrow-derived cells promotes gastric tumorigenesis by generation
of inflammatory microenvironment. Cancer Prev Res (Phila).
9:253–263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saukkonen K, Rintahaka J, Sivula A,
Buskens CJ, Van Rees BP, Rio MC, Haglund C, Van Lanschot JJ,
Offerhaus GJ and Ristimaki A: Cyclooxygenase-2 and gastric
carcinogenesis. APMIS. 111:915–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sung JJ, Leung WK, Go MY, To KF, Cheng AS,
Ng EK and Chan FK: Cyclooxygenase-2 expression in Helicobacter
pylori-associated premalignant and malignant gastric lesions. Am J
Pathol. 157:729–735. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oshima H, Matsunaga A, Fujimura T,
Tsukamoto T, Taketo MM and Oshima M: Carcinogenesis in mouse
stomach by simultaneous activation of the Wnt signaling and
prostaglandin E2 pathway. Gastroenterology. 131:1086–1095. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang D, Fu L, Sun H, Guo L and DuBois RN:
Prostaglandin E2 promotes colorectal cancer stem cell expansion and
metastasis in mice. Gastroenterology. 149:1884–1895. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jayaraman P, SadaOvalle I, Nishimura T,
Anderson AC, Kuchroo VK, Remold HG and Behar SM: IL-1β promotes
antimicrobial immunity in macrophages by regulating TNFR signaling
and caspase-3 activation. J Immunol. 190:4196–4204. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumar S, Kumar A and Dixit VK: Evidences
showing association of interleukin-1B polymorphisms with increased
risk of gastric cancer in an Indian population. Biochem Biophys Res
Commun. 387:456–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Garza-González E, BosquesPadilla FJ,
ElOmar E, Hold G, TijerinaMenchaca R, MaldonadoGarza HJ and
Pérez-Pérez GI: Role of the polymorphic IL-1B, IL-1RN and TNF-A
genes in distal gastric cancer in Mexico. Int J Cancer.
114:237–241. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Peleteiro B, Lunet N, Carrilho C, Durães
C, Machado JC, La Vecchia C and Barros H: Association between
cytokine gene polymorphisms and gastric precancerous lesions:
systematic review and meta-analysis. Cancer Epidemiol Biomarkers
Prev. 19:762–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marcos-Pinto R, DinisRibeiro M, Carneiro
F, Wen X, Lopes C, Figueiredo C, Machado JC, Ferreira RM, Reis CA,
Canedo P, et al: First-degree relatives of early-onset gastric
cancer patients show a high risk for gastric cancer: phenotype and
genotype profile. Virchows Arch. 463:391–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kamangar F, Cheng C, Abnet CC and Rabkin
CS: Interleukin-1B polymorphisms and gastric cancer risk - a
meta-analysis. Cancer Epidemiol Biomarkers Prev. 15:1920–1928.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Waghray M, Zavros Y, SaquiSalces M,
ElZaatari M, Alamelumangapuram CB, Todisco A, Eaton KA and Merchant
JL: Interleukin-1beta promotes gastric atrophy through suppression
of Sonic Hedgehog. Gastroenterology. 138:562–572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang M, Furuta T, Takashima M, Futami H,
Shirai N, Hanai H and Kaneko E: Relation between interleukin-1beta
messenger RNA in gastric fundic mucosa and gastric juice pH in
patients infected with Helicobacter pylori. J Gastroenterol.
34:(Suppl 11). 10–17. 1999.PubMed/NCBI
|
|
45
|
Zeng ZR, Hu PJ, Hu S, Pang RP, Chen MH, Ng
M and Sung JJ: Association of interleukin 1B gene polymorphism and
gastric cancers in high and low prevalence regions in China. Gut.
52:1684–1689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shigematsu Y, Niwa T, Rehnberg E, Toyoda
T, Yoshida S, Mori A, Wakabayashi M, Iwakura Y, Ichinose M, Kim YJ,
et al: Interleukin-1β induced by Helicobacter pylori infection
enhances mouse gastric carcinogenesis. Cancer Lett. 340:141–147.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Furuta T, ElOmar EM, Xiao F, Shirai N,
Takashima M and Sugimura H: Interleukin 1beta polymorphisms
increase risk of hypochlorhydria and atrophic gastritis and reduce
risk of duodenal ulcer recurrence in Japan. Gastroenterology.
123:92–105. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Farinati F, Cardin R, Degan P, Rugge M,
Mario FD, Bonvicini P and Naccarato R: Oxidative DNA damage
accumulation in gastric carcinogenesis. Gut. 42:351–356. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Obst B, Wagner S, Sewing KF and Beil W:
Helicobacter pylori causes DNA damage in gastric epithelial cells.
Carcinogenesis. 21:1111–1115. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes
VE, Patel J, DirdenKramer B, Boldogh I, Ernst PB and Crowe SE:
Helicobacter pylori infection induces oxidative stress and
programmed cell death in human gastric epithelial cells. Infect
Immun. 75:4030–4039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cheng Y, Chaturvedi R, Asim M, Bussière
FI, Scholz A, Xu H, Casero RA Jr and Wilson KT: Helicobacter
pylori-induced macrophage apoptosis requires activation of
ornithine decarboxylase by c-Myc. J Biol Chem. 280:22492–22496.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu H, Chaturvedi R, Cheng Y, Bussiere FI,
Asim M, Yao MD, Potosky D, Meltzer SJ, Rhee JG, Kim SS, et al:
Spermine oxidation induced by Helicobacter pylori results in
apoptosis and DNA damage: implications for gastric carcinogenesis.
Cancer Res. 64:8521–8525. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chaturvedi R, Cheng Y, Asim M, Bussière
FI, Xu H, Gobert AP, Hacker A, Casero RA Jr and Wilson KT:
Induction of polyamine oxidase 1 by Helicobacter pylori causes
macrophage apoptosis by hydrogen peroxide release and mitochondrial
membrane depolarization. J Biol Chem. 279:40161–40173. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jo TY, Jeon TY, Chae KH, Kim DH, Sim MS,
Park DY and Suh KS: RImmunohistochemical evaluation of
E-cadherin/catenin (alpha-, beta-, gamma-catenin and p120CTN)
complex expression in early gastric cancer. Cancer Res Treat.
35:16–24. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Perri F, Cotugno R, Piepoli A, Merla A,
Quitadamo M, Gentile A, Pilotto A, Annese V and Andriulli A:
Aberrant DNA methylation in non-neoplastic gastric mucosa of H.
pylori infected patients and effect of eradication. Am J
Gastroenterol. 102:1361–1371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Oliveira MJ, Costa AM, Costa AC, Ferreira
RM, Sampaio P, Machado JC, Seruca R, Mareel M and Figueiredo C:
CagA associates with c-Met, E-cadherin, and p120-catenin in a
multiproteic complex that suppresses Helicobacter pylori-induced
cell-invasive phenotype. J Infect Dis. 200:745–755. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jawhari AU, Noda M, Pignatelli M and
Farthing M: Up-regulated cytoplasmic expression, with reduced
membranous distribution, of the src substrate p120(ctn) in gastric
carcinoma. J Pathol. 189:180–185. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Noren NK, Liu BP, Burridge K and Kreft B:
p120 catenin regulates the actin cytoskeleton via Rho family
GTPases. J Cell Biol. 150:567–580. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ogden SR, Wroblewski LE, Weydig C,
RomeroGallo J, O'Brien DP, Israel DA, Krishna US, Fingleton B,
Reynolds AB, Wessler S, et al: p120 and Kaiso regulate Helicobacter
pylori-induced expression of matrix metalloproteinase-7. Mol Biol
Cell. 19:4110–4121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tsugane S: Salt, salted food intake, and
risk of gastric cancer: epidemiologic evidence. Cancer Sci. 96:1–6.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Beevers DG, Lip GY and Blann AD: Salt
intake and Helicobacter pylori infection. J Hypertens.
22:1475–1477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shikata K, Kiyohara Y, Kubo M, Yonemoto K,
Ninomiya T, Shirota T, Tanizaki Y, Doi Y, Tanaka K, Oishi Y, et al:
A prospective study of dietary salt intake and gastric cancer
incidence in a defined Japanese population: the Hisayama study. Int
J Cancer. 119:196–201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Gamboa-Dominguez A, Ubbelohde T,
SaquiSalces M, RomanoMazzoti L, Cervantes M, Domínguez-Fonseca C,
de la Luz Estreber M and Ruíz-Palacios GM: Salt and stress
synergize H. pylori-induced gastric lesions, cell proliferation,
and p21 expression in Mongolian gerbils. Dig Dis Sci. 52:1517–1526.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun J, Aoki K, Zheng JX, Su BZ, Ouyang XH
and Misumi J: Effect of NaCl and Helicobacter pylori vacuolating
cytotoxin on cytokine expression and viability. World J
Gastroenterol. 12:2174–2180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gancz H, Jones KR and Merrell DS: Sodium
chloride affects Helicobacter pylori growth and gene expression. J
Bacteriol. 190:4100–4105. 2008. View Article : Google Scholar : PubMed/NCBI
|