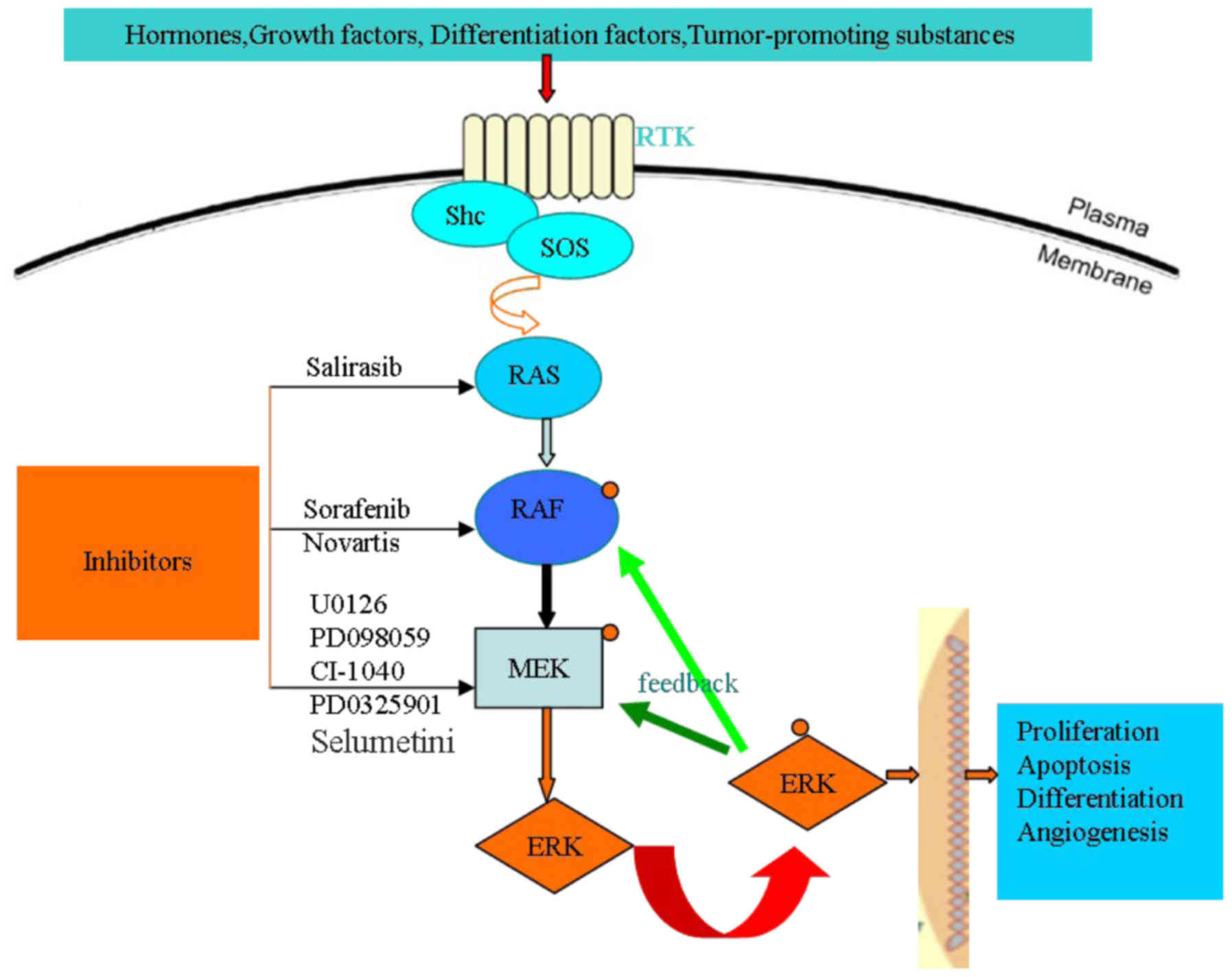
Hepatocellular carcinoma (HCC) is one of the most
common cancers, with an incidence rate ranking sixth highest and a
mortality rate ranking third highest, accounting for 7% of all
cancers in the world (1). In
particular, there is a high incidence in China, with ~4/10,000
cases per year. Current treatment options for HCC, including
surgical approaches, locoregional ablative techniques and
interventional ablation treatments, could increase the
5-year-survival rate to 75% (vs. 30% prior to these treatments),
however, <20% of HCC patients qualify for these treatments
(2). Although the promising
5-year-survival rate of HCC cases has been increased due to
advances in surgical techniques, nutritional support and
perioperative management, long-term survival after surgical
resection remains low due to the high rate of recurrence and
metastasis (3,4). Novel evidence-based therapies for HCC
are urgently required. Recently, biological studies have pointed to
aberrant rapidly accelerated fibrosarcoma (Raf)/mitogen-activated
protein kinase kinase (MEK)/extracellular signal-regulated kinase
(ERK) signaling pathway activation as being central for cancer
growth, survival and motility, as well as for targeted therapy
resistance mechanisms (5–7). For example, sorafenib is a Raf-1 kinase
inhibitor and is the only approved drug therapy for HCC. In
patients with advanced or metastatic HCC and compensated cirrhosis,
sorafenib offers disease control in ~40% of treated patients, with
a time to progression of 5.5 months and a median survival time of
10.7 months, ~3 months longer than that of placebo-treated patients
(8). Hence, there is an eagerness to
dissect the molecular mechanisms of invasion and metastasis for
novel insights and interventions against the recurrence of HCC.
Undoubtedly, the rat sarcoma virus (Ras)/Raf/MEK/ERK
signaling pathway contributes a core effect in regulating cell
proliferation, differentiation and survival in the signaling
networks (9). On this account, it has
been studied and discussed to determine the pathogenesis of several
types of human cancers, including HCC (10). Not merely the Ras/Raf/MEK/ERK
signaling pathway, but also the phosphoinositide 3-kinase
(PI3K)/Akt signaling pathway has been studied to determine whether
it is connected with the pathogenesis of HCC (11). It is notable that the interaction
between the Ras/Raf/MEK/ERK and PI3K/Akt signaling pathways may
lead to the regulation of cell growth and development, more than
either alone.
In the present review, the function of the
Ras/Raf/MEK/ERK signaling pathway in HCC is elaborated on and its
therapeutic potential as a target for the intervention and
treatment of HCC is expounded.
The mitogen-activated protein kinase (MAPK) cascade
consists of serine/threonine kinases, which convert extracellular
molecules such as growth factors, hormones, tumor-promoting
substances and differentiation factors, into intracellular signals
for regulating cell proliferation, differentiation and survival
(12,13). There are four core protein kinases,
Ras, Raf, MEK and ERK, in the Ras/Raf/MEK/ERK signaling pathway.
Ras, Raf and MEK are members of multi-gene families; of those, Ras
has three members (Ki-Ras, N-Ras and Ha-Ras), Raf has three members
(A-Raf, B-Raf and Raf-1) and MEK has five gene family members
(MEK1, MEK2, MEK3, MEK4 and MEK5). At the cell surface, the
activation of the Ras/Raf/MEK/ERK signaling pathway is initiated by
ligand binding to receptor tyrosine kinases (RTK), then, in the
nucleus, the phosphorylation of four core protein kinases, Ras,
Raf, MEK and ERK, in turn regulating gene transcription (14). The specific activation pathways are as
follows.
There are a number of extracellular signals,
including growth factors, hormones, tumor-promoting substances and
differentiation factors, which activate the Ras/Raf/MEK/ERK
signaling pathway. When extracellular signals bond with an
appropriate RTK (an Src homology 2 domain-containing protein), the
C-terminus of the growth factor receptor [for example, FGFR, Flt-3,
platelet-derived growth factor receptor (PDGFR), insulin-like
growth factor receptor-1 (IGFR-1) and macrophage colony stimulating
receptor among others] that has been activated is linked with the
RTK. The tyrosine kinase domain of the excessive phosphorylation of
RTK acting as a carrier protein, such as sex muscle abnormal
protein-5 or organization control-1, recruits guanine nucleotide
exchange factors [for example, mammalian son-of-sevenless (SOS)] to
the cytomembrane where they stimulate Ras-GDP conversion to
Ras-GTP, resulting in Ras protein activation (14,15). Ras
phosphorylation then recruits Raf to the membrane where it becomes
activated, likely via an Src-family tyrosine (Y) kinase. Raf is
responsible for the serine/threonine phosphorylation of MEK1. The
MEK family has five genes, namely MEK1, MEK2, MEK3, MEK4 and MEK5.
The five genes are all double specificity kinases, which means that
they can phosphorylate serine/threonine residues along with
tyrosine residues. Of those, Ras and Raf activate downstream target
proteins of MEK1 and MEK2 through phosphorylating the activation
domain of two serine residues. MEK1 phosphorylates ERK1/2 at
specific T and Y residues. The ERK family has four members, namely
JNK1/2/3, ERK1/2, ERK5 and p38 MAPK. ERK1/2 is the only downstream
protein target of MEK1/2 phosphorylation. When activating ERK1/2
serine/threonine kinases, they will generate a series of effects
(for example, the phosphorylation and activation p90 ribosomal six
kinase-1) (16,17). There are 460 ERK1/2 targets, including
downstream and upstream substrates (18,19).
Therefore, the regulation of the Ras/Raf/MEK/ERK signaling pathway
plays an important role in cell proliferation, differentiation and
survival by suppressing MEK and ERK activities.
There is a feedback pathway regulating the activity
of B-Raf, Raf-1 and MEK1 through the activation of ERK. With regard
to Raf-1, ERK phosphorylation can improve or lower the its
activity, which depends on the site phosphorylated. With regard to
B-Raf and MEK1, ERK phosphorylation can lower their activity. There
is also a negative feedback pathway preventing the activation of
Ras through the phosphorylation of SOS by ERK. Target protein
phosphorylation, such as that of Ras, Raf, MEK and ERK, can enhance
or inhibit the associated signaling pathway, even phosphorylating
different sites of a target protein playing a different role in
regulating the pathway (20,21). Therefore, regulating the
Ras/Raf/MEK/ERK signaling pathway is a complex process, which plays
an important role in cell proliferation, differentiation and
survival (Fig. 1).
A large amount of preclinical and clinical evidence
has shown that the abnormal activation of the Ras/Raf/MEK/ERK
signaling pathway frequently results in HCC. Ito et al
showed that MAPK/ERK is activated and its associated gene
expression is upregulated in 58% of HCC cases (22). Hoffmann et al demonstrated that
the mRNA of Ras, MEK, ERK and MAPK was overexpressed in 33, 40, 50
and 50% of HCC patients, respectively (23). Similarly, H-ras has been found to be
activated in ~93.8% of HCC cases (24). A study further showed that the
expression Raf and its downstream genes, MEK and ERK, were
upregulated in samples of hepatocirrhosis and HCC (25). Western blot analysis demonstrated the
overexpression of Raf-1 in 91.2% of hepatocirrhosis and 100% of HCC
patients. Furthermore, the Raf-1 expression level in HCC patients
was significantly high compared with that of hepatocirrhosis
patients (26). All research results
showed that the activation of the Ras/Raf/MEK/ERK pathway may lead
to HCC development functionally.
The cellular mechanisms behind the activation of the
Ras/Raf/MEK/ERK signaling pathway are not yet completely clear in
HCC. However, activation by RTKs is hypothesized to be the main
mechanism. It has been verified that EGFR, IGFR, vascular
endothelial growth factor receptor (VEGFR) and c-Met are
overexpressed. Of those, EGFR accounts for 47.1% of cases in HCC,
and the overexpression of EGFR is responsible for the invasiveness
and recurrence of HCC (27). Wiedmann
and Mössner showed that the EGFR inhibitors erlotinib and lapatinib
inhibit not only RTKs, but also serine/threonine kinases along the
Ras/Raf/MEK/ERK pathway, in two phase III placebo-controlled trials
(28). Similarly, the overexpression
of VEGFR has been found in HCC cell lines and in the serum and
tissues of HCC patients (29,30). Furthermore, the overexpression of
c-MET accounts for 20–48% of cases in HCC (31). Activation of c-MET plays a role in
bringing the growth factor receptor-bound protein 2/SOS complex to
the plasma membrane. As a result, GTP along with Ras activates a
protein cascade that contributes to the downstream protein kinase
phosphorylation of ERK by Raf and MEK (32).
Hepatitis virus infections also play an important
role in the activation of the Ras/Raf/MEK/ERK pathway in HCC.
Hepatitis B virus X protein has also been demonstrated to have an
essential effect on the progression of HCC through activating the
Ras/Raf/MEK/ERK cascade (33) and
then contributing to the loss of function of the tumor suppressor
p53 (34). Hepatitis C virus (HCV)
core protein activates the kinase Raf-1 and MAPK/ERK pathway
through interacting with 14–3-3 protein (35). Schmitz et al confirmed that the
mechanism of HCC carcinogenesis may be via the activation of the
Ras/Raf/MEK/ERK pathway by HCV infection (36). There are at least two functions of the
HCV core protein, including the activation of the Ras/Raf/MEK/ERK
pathway and an anti-apoptotic effect. HCV core protein can activate
ERK phosphorylation alone without hepatocyte mitogen-mediated
signaling. However, HCV core protein along with tissue plasminogen
activator may contribute to the effect on MEK1 or further upstream
of the protein kinase (37).
In recent years, genomic sequencing research has
revealed the associated gene changes of the Ras/Raf/MEK/ERK
signaling pathway in HCC. The B-Raf gene, one of the human isoforms
of Raf, has been found to be mutated or deleted in HCC, accounting
for ~23% of cases. As a result, this may lead to activating
oncogenic Ras in HCC (38). In
addition to the mutation or deletion of B-Raf, codon 12 of the
K-Ras and N-Ras genes is also mutated or deleted in HCC, accounting
for 4.69 and 41.37% of cases (24,38,39).
Challen et al found that mutations of the K-ras and N-ras
genes in codon 61 occurs in 5.3 and 15.8% of HCC cases (40). All the reported mutations of K-Ras,
N-Ras and H-ras are somatic missense mutations (for example,
changes to the amino acids of codons 12, 13 and 61) in HCC.
Mutations in Ras family genes can phosphorylate the Ras/Raf/MEK/ERK
signaling pathway, then deregulate signal transduction (41). However, Taketomi et al examined
61 patients through a dot-blot and elaborated that the mutations of
Ras proto-oncogenes in codons 12, 13 and 61 had little effect in
HCC (42). Therefore, further study
is required to clarify the controversy.
As the abnormal activation of the Ras/Raf/MEK/ERK
signaling pathway plays a major role in HCC cell proliferation,
differentiation, survival and apoptosis, a number of studies have
been focused on the inhibitors of the core protein kinases Ras,
Raf, MEK and ERK in the Ras/Raf/MEK/ERK signaling pathway (Fig. 1); a number of these studies are
preclinical, while others are clinical studies (Table I).
Activating Ras mutations have been observed in ~30%
of all cancers. However, according to studies of the pathogenesis
of cancer at present, the specific function of Ras is not yet
settled. Salirasib [also known as S-trans,
trans-farnesylthiosalycilic acid (FTS)] is a synthetic low-weight
molecule of a S-farnesyl cysteine analog that expresses a potent
inhibitory effect on Ras. Its mechanism of action is likely to be
associated primarily with the dislodgment of the mature protein
from membrane domains that interact with Ras and with the
subsequent accelerated degradation of the dislodged Ras proteins
(43,44). These effects of FTS are manifested by
a decrease in the amount of cellular Ras accompanied by
interruption of the Ras-dependent Raf-1/ERK signaling cascade
(45). FTS plays an antitumoral role
in several non-hepatic cancer cell lines (46), and a phase I clinical trial of
gemcitabine and FTS has demonstrated that FTS is well tolerated in
patients with solid non-liver tumors (47). As its expression is well tolerated,
salirasib may become a drug of choice for the treatment of HCC by
targeting Ras and mammalian target of rapamycin (mTOR) protein
kinase (48). A study by da Silva
Morais et al indicated that a high concentration of
salirasib can inhibit liver cancer cell proliferation in
vivo in rats following a partial liver resection (49). Its mechanism of inhibitory effects is
mediated, at least in part, by the inhibition of ERK
phosphorylation. Furthermore, the study by Nicolas et al
demonstrated that salirasib injection can prevent the development
of liver tumors in a subcutaneous xenograft model (50).
Certain special distinct classes of compounds have
been developed as potential Raf kinase inhibitors. However, thus
far, sorafenib (Nexavar) is the most successful anti-Raf inhibitor.
Sorafenib, an orally available anti-Raf compound, is the only small
molecular target kinase to receive Food and Drug Administration
approval for the treatment of advanced HCC (51). Sorafenib suppresses the
serine/threonine kinase subtypes of Raf, which are well known to
regulate the Raf/MEK/ERK signaling pathway and inhibit tyrosine
kinase receptors, including VEGFR2, PDGFR and IGFR. For this
reason, sorafenib inhibits angiogenesis and tumor growth (52). Sorafenib has demonstrable preclinical
and clinical activity against certain types of cancer (for example,
HCC, and ovarian, breast and pancreatic cancer). However, in
pharmacokinetics, pharmacodynamics and adverse events, sorafenib
has exhibited individual differences in clinical research (53,54). An
updated meta-analysis evaluated sorafenib administration and found
that it could significantly increase OS time and TTP in patients
with advanced HCC. Additional large-scale, well-designed randomized
controlled trials are planned to further evaluate the efficacy of
treating advanced HCC with sorafenib (55).
NVP-AAL881 (Novartis, Basel, Switzerland) is an oral
Raf and VEGFR2 small molecule inhibitor that has been shown to
inhibit cell proliferation and tumor growth in a subcutaneous
xenograft model of HCC (56).
NVP-AAL881 can abnormally activate ERK and STAT3, and inhibits the
migration of HCC cells. NVP-AAL881 inhibits the growth of HCC cells
in a dose-dependent manner, as observed through
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assays. In HCC cells under low serum culture conditions, the
inhibition effect of NVP-AAL881 is enhanced. Furthermore, with
increasing concentrations of NVP-AAL881, the inhibition of ERK, MEK
and STAT3 phosphorylation is enhanced in HCC cell lines (57). More recently, it has been shown that
NVP-AAL881 administration markedly inhibits the growth of HCC
xenograft tumors compared with controls (58).
As Ras inhibitors are challenging to identify and
almost no biological functions of B-Raf inhibitors are known, more
attention is being focused on the study of MEK and ERK inhibitors.
Numerous MEK inhibitors have been developed; PD98059 was the first
MEK1/2 inhibitor to be found, which combines with the inactive
forms of MEK1/2 to prevent its phosphorylation, then inhibits the
phosphorylation of ERK1/2 and blocks cell signal transduction
(59). U0126 is the second MEK1/2
inhibitor to be identified, and its inhibitive effect is greater
than that of PD98059. U0126 is widely used in in vitro
experiments (60). In HepG2 cell
cultures in vitro, the phosphorylation of p38 and ERK1/2 was
inhibited by PD98059, and this effect was also demonstrated in
vivo (61). The two types of
MEK1/2 kinase inhibitors are non-ATP-competitive, acting by
inhibiting the MEK excitation instead of directly inhibiting the
activity of MEK. Due to the low solubility and bioavailability of
PD98059 and U0126, they have not entered into clinical trials and
are only applied in experiments in vitro (62).
The oral, non-ATP competitive MEK1/2 inhibitor
selumetinib (previously known as ARRY-142886 and AZD6244), is a
benzimidazole derivative (69). The
drug is the second MEK inhibitor to enter into clinical trials
(70). A large quantity of effective
results have been shown in preclinical studies only, using cell
cultures and animal models (71).
Selumetinib has been demonstrated to play a role in contributing to
the inhibition of ERK1/2 phosphorylation in a number of cancer cell
lines, with a half maximal inhibitory concentration as low as 8
nmol/l and the sustained inhibition of ERK activity achieved with a
concentration of 10 mg/kg/day in a subcutaneous xenograft model of
HCC (72). In a multi-center,
single-arm phase II study of selumetinib in advanced or metastatic
HCC patients, the study was stopped at the interim analysis due to
a lack of radiographic reaction. The drug has not yet been shown to
significantly decrease the time to progression. Selumetinib is well
tolerated, but the treatment effect is not ideal in advanced HCC
(73). Another phase II study showed
that 14 out of 17 evaluable HCC patients succumbed; of the
remaining 3 patients, 2 experienced progression and 1 remained
alive without progression (74). The
median progression-free survival time was 1.4 months. The median
time to progression was the same. The median survival time was 4.2
months (75).
Studying the Ras/Raf/MEK/ERK signaling pathway has
provided novel insights and novel target drugs for HCC treatment.
On the one hand, although such drugs exhibit improved therapeutic
effects compared with conventional chemotherapeutic drugs, they
present potential problems and challenges for HCC therapy, such as
adverse events and resistance. On the other hand, although the
Ras/Raf/MEK/ERK signaling pathway plays an important role in the
regulation of HCC cell proliferation, differentiation, survival and
apoptosis, its exact functional relevance in the settings of this
complex signaling network and HCC tumorigenesis are far from being
fully understood. Furthermore, a key challenge for Ras/Raf/MEK/ERK
pathway inhibition will likely be the level of cross-talk and
negative feedback along parallel pathways (such as the
PI3K/AKT/mTOR pathway). Preclinical data suggest that certain
problems and challenges may be overcome by combining
Ras/Raf/MEK/ERK pathway inhibitors with other pathway inhibitors,
but this must be confirmed in clinical studies.
|
1
|
He W, Zeng Q, Zheng Y, Chen M, Shen J, Qiu
J, Chen M, Zou R, Liao Y, Li Q, et al: The role of clinically
significant portal hypertension in hepatic resection for
hepatocellular carcinoma patients: A propensity score matching
analysis. BMC Cancer. 15:2632015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang B, Zan RY, Wang SY, Li XL, Wei ML,
Guo WH, You X, Li J and Liao ZY: Radiofrequency ablation versus
percutaneous ethanol injection for hepatocellular carcinoma: A
meta-analysis of randomized controlled trials. World J Surg Oncol.
13:962015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baek YH, Kim KT, Lee SW, Jeong JS, Park
BH, Nam KJ, Cho JH, Kim YH, Roh YH, Lee HS, et al: Efficacy of
hepatic arterial infusion chemotherapy in advanced hepatocellular
carcinoma. World J Gastroenterol. 18:3426–3434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim JY, Chung SM, Choi BO and Kay CS:
Hepatocellular carcinoma with portal vein tumor thrombosis:
Improved treatment outcomes with external beam radiation therapy.
Hepatol Res. 41:813–824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Buscà R, Pouysségur J and Lenormand P:
ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy?
Front Cell Dev Biol. 4:532016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Nie H, Zhao X, Qin Y and Gong X:
Bicyclol induces cell cycle arrest and autophagy in HepG2 human
hepatocellular carcinoma cells through the PI3K/AKT and
Ras/Raf/MEK/ERK pathways. BMC Cancer. 16:7422016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asati V, Mahapatra DK and Bharti SK:
PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as
anticancer agents: Structural and pharmacological perspectives. Eur
J Med Chem. 109:314–341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee SH, Song IH, Noh R, Kang HY, Kim SB,
Ko SY, Lee ES, Kim SH, Lee BS, Kim AN, et al: Clinical outcomes of
patients with advanced hepatocellular carcinoma treated with
sorafenib: A retrospective study of routine clinical practice in
multi-institutions. BMC Cancer. 15:2362015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu P, Ye L, Wang H, Du G, Zhang J, Zhang J
and Tian J: NSK-01105 inhibits proliferation and induces apoptosis
of prostate cancer cells by blocking the Raf/MEK/ERK and
PI3K/Akt/mTOR signal pathways. Tumour Biol. 36:2143–2153. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huynh H, Ngo VC, Koong HN, Poon D, Choo
SP, Toh HC, Thng CH, Chow P, Ong HS, Chung A, et al: AZD6244
enhances the anti-tumor activity of sorafenib in ectopic and
orthotopic models of human hepatocellular carcinoma (HCC). J
Hepatol. 52:79–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Q, Lui VW and Yeo W: Targeting the
PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol.
7:1149–1167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang M, Wen F, Cao J, Li P, She J and Chu
Z: Genome-wide exploration of the molecular evolution and
regulatory network of mitogen-activated protein kinase cascades
upon multiple stresses in Brachypodium distachyon. BMC Genomics.
16:2282015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ward AF, Braun BS and Shannon KM:
Targeting oncogenic Ras signaling in hematologic malignancies.
Blood. 120:3397–3406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Knight T and Irving JA: Ras/Raf/MEK/ERK
pathway activation in childhood acute lymphoblastic leukemia and
its therapeutic targeting. Front Oncol. 4:1602014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang YS, Liu JC, Fu HQ, Yu BT, Zou SB, Wu
QC and Wan L: Roles of targeting Ras/Raf/MEK/ERK signaling pathways
in the treatment of esophageal carcinoma. Yao Xue Xue Bao.
48:635–641. 2013.(In Chinese). PubMed/NCBI
|
|
16
|
Zebisch A and Troppmair J: Back to the
roots: The remarkable RAF oncogene story. Cell Mol Life Sci.
63:1314–1330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moodie SA and Wolfman A: The 3Rs of life:
Ras, Raf and growth regulation. Trends Genet. 10:44–48. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signaling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16:(Suppl 2).
S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steelman LS, Franklin RA, Abrams SL,
Chappell W, Kempf CR, Bäsecke J, Stivala F, Donia M, Fagone P,
Nicoletti F, et al: Roles of the Ras/Raf/MEK/ERK pathway in
leukemia therapy. Leukemia. 25:1080–1094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Trujillo JI: MEK inhibitors: A patent
review 2008–2010. Expert Opin Ther Pat. 21:1045–1069. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chung E and Kondo M: Role of
Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and
leukemia development. Immunol Res. 49:248–268. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito Y, Sasaki Y, Horimoto M, Wada S,
Tanaka Y, Kasahara A, Ueki T, Hirano T, Yamamoto H, Fujimoto J, et
al: Activation of mitogen-activated protein kinases/extracellular
signal-regulated kinases in human hepatocellular carcinoma.
Hepatology. 27:951–958. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hoffmann K, Shibo L, Xiao Z, Longerich T,
Büchler MW and Schemmer P: Correlation of gene expression of
ATP-binding cassette protein and tyrosine kinase signaling pathway
in patients with hepatocellular carcinoma. Anticancer Res.
31:3883–3890. 2011.PubMed/NCBI
|
|
24
|
Zuo Q, Huang H, Shi M, Zhang F, Sun J, Bin
J, Liao Y and Liao W: Multivariate analysis of several molecular
markers and clinicopathological features in postoperative prognosis
of hepatocellular carcinoma. Anat Rec (Hoboken). 295:423–431. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gollob JA, Wilhelm S, Carter C and Kelley
SL: Role of Raf kinase in cancer: Therapeutic potential of
targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol.
33:392–406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang Z, Qin L, Wang X, Zhou G, Liao Y,
Weng Y, Jiang X, Lin Z, Liu K and Ye S: Alterations of oncogenes,
tumor suppressor genes and growth factors in hepatocellular
carcinoma: With relation to tumor size and invasiveness. Chin Med J
(Engl). 111:313–318. 1998.PubMed/NCBI
|
|
28
|
Wiedmann MW and Mössner J: Molecular
targeted therapy of hepatocellular carcinoma-results of the first
clinical studies. Curr Cancer Drug Targets. 11:714–733. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poon RT, Ho JW, Tong CS, Lau C, Ng IO and
Fan ST: Prognostic significance of serum vascular endothelial
growth factor and endostatin in patients with hepatocellular
carcinoma. Br J Surg. 91:1354–1360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dhar DK, Naora H, Yamanoi A, Ono T, Kohno
H, Otani H and Nagasue N: Requisite role of VEGF receptors in
angiogenesis of hepatocellular carcinoma: A comparison with
angiopoietin/Tie pathway. Anticancer Res. 22:379–386.
2002.PubMed/NCBI
|
|
31
|
Tavian D, De Petro G, Benetti A, Portolani
N, Giulini SM and Barlati S: u-PA and c-MET mRNA expression is
co-ordinately enhanced while hepatocyte growth factor mRNA is
down-regulated in human hepatocellular carcinoma. Int J Cancer.
87:644–649. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giambartolomei S, Covone F, Levrero M and
Balsano C: Sustained activation of the Raf/MEK/Erk pathway in
response to EGF in stable cell lines expressing the Hepatitis C
Virus (HCV) core protein. Oncogene. 20:2606–2610. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang XW: Microinjection technique used to
study functional interaction between p53 and hepatitis B virus X
gene in apoptosis. Mol Biotechnol. 18:169–177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nakamura H, Aoki H, Hino O and Moriyama M:
HCV core protein promotes heparin binding EGF-like growth factor
expression and activates Akt. Hepatol Res. 41:455–462. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schmitz KJ, Wohlschlaeger J, Lang H,
Sotiropoulos GC, Malago M, Steveling K, Reis H, Cicinnati VR,
Schmid KW and Baba HA: Activation of the ERK and AKT signaling
pathway predicts poor prognosis in hepatocellular carcinoma and ERK
activation in cancer tissue is associated with hepatitis C virus
infection. J Hepatol. 48:83–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hayashi J, Aoki H, Kajino K, Moriyama M,
Arakawa Y and Hino O: Hepatitis C virus core protein activates the
MAPK/ERK cascade synergistically with tumor promoter TPA, but not
with epidermal growth factor or transforming growth factor alpha.
Hepatology. 32:958–961. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Colombino M, Sperlongano P, Izzo F,
Tatangelo F, Botti G, Lombardi A, Accardo M, Tarantino L, Sordelli
I, Agresti M, et al: BRAF and PIK3CA genes are somatically mutated
in hepatocellular carcinoma among patients from South Italy. Cell
Death Dis. 3:e2592012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kalinina O, Marchio A, Urbanskii AI,
Tarkova AB, Rebbani K, Granov DA, Dejean A, Generalov MI and Pineau
P: Somatic changes in primary liver cancer in Russia: A pilot
study. Mutat Res. 755:90–99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Challen C, Guo K, Collier JD, Cavanagh D
and Bassendine MF: Infrequent point mutations in codons 12 and 61
of ras oncogenes in human hepatocellular carcinomas. J Hepatol.
14:342–346. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saini KS, Loi S, de Azambuja E,
Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE and
Piccart-Gebhart MJ: Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK
pathways in the treatment of breast cancer. Cancer Treat Rev.
39:935–946. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Taketomi A, Shirabe K, Muto J, Yoshiya S,
Motomura T, Mano Y, Ikegami T, Yoshizumi T, Sugio K and Maehara Y:
A rare point mutation in the Ras oncogene in hepatocellular
carcinoma. Surg Today. 43:289–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marom M, Haklai R, Ben-Baruch G, Marciano
D, Egozi Y and Kloog Y: Selective inhibition of Ras-dependent cell
growth by farnesylthiosalisylic acid. J Biol Chem. 270:22263–22270.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haklai R, Weisz MG, Elad G, Paz A,
Marciano D, Egozi Y, Ben-Baruch G and Kloog Y: Dislodgment and
accelerated degradation of Ras. Biochemistry. 37:1306–1314. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McMahon LP, Yue W, Santen RJ and Lawrence
JC Jr: Farnesylthiosalicylic acid inhibits mammalian target of
rapamycin (mTOR) activity both in cells and in vitro by promoting
dissociation of the mTOR-raptor complex. Mol Endocrinol.
19:175–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blum R, Elkon R, Yaari S, Zundelevich A,
Jacob-Hirsch J, Rechavi G, Shamir R and Kloog Y: Gene expression
signature of human cancer cell lines treated with the ras inhibitor
salirasib (S-farnesylthiosalicylic acid). Cancer Res. 67:3320–3328.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tsimberidou AM, Rudek MA, Hong D, Ng CS,
Blair J, Goldsweig H and Kurzrock R: Phase 1 first-in-human
clinical study of S-trans, trans-farnesylthiosalicylic acid
(salirasib) in patients with solid tumors. Cancer Chemother
Pharmacol. 65:235–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Charette N, De Saeger C, Lannoy V,
Horsmans Y, Leclercq I and Stärkel P: Salirasib inhibits the growth
of hepatocarcinoma cell lines in vitro and tumor growth in vivo
through ras and mTOR inhibition. Mol Cancer. 9:2562010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
da Silva Morais A, Saliez A, Leclercq I,
Horsmans Y and Stärkel P: Inhibition of the Ras oncoprotein reduces
proliferation of hepatocytes in vitro and in vivo in rats. Clin Sci
(Lond). 114:73–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schneider-Merck T, Borbath I, Charette N,
De Saeger C, Abarca J, Leclercq I, Horsmans Y and Stärkel P: The
Ras inhibitor farnesylthiosalicyclic acid (FTS) prevents nodule
formation and development of preneoplastic foci of altered
hepatocytes in rats. Eur J Cancer. 45:2050–2060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wecksler AT, Hwang SH, Liu JY, Wettersten
HI, Morisseau C, Wu J, Weiss RH and Hammock BD: Biological
evaluation of a novel sorafenib analogue, t-CUPM. Cancer Chemother
Pharmacol. 75:161–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fucile C, Marenco S, Bazzica M, Zuccoli
ML, Lantieri F, Robbiano L, Marini V, Di Gion P, Pieri G, Stura P,
et al: Measurement of sorafenib plasma concentration by
high-performance liquid chromatography in patients with advanced
hepatocellular carcinoma: Is it useful the application in clinical
practice? A pilot study. Med Oncol. 32:3352015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Guan YS and He Q: Sorafenib: Activity and
clinical application in patients with hepatocellular carcinoma.
Expert Opin Pharmacother. 12:303–313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Arrondeau J, Mir O, Boudou-Rouquette P,
Coriat R, Ropert S, Dumas G, Rodrigues MJ, Rousseau B, Blanchet B
and Goldwasser F: Sorafenib exposure decreases over time in
patients with hepatocellular carcinoma. Invest New Drugs.
30:2046–2049. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Peng S, Zhao Y, Xu F, Jia C, Xu Y and Dai
C: An updated meta-analysis of randomized controlled trials
assessing the effect of sorafenib in advanced hepatocellular
carcinoma. PLoS One. 9:e1125302014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sathornsumetee S, Hjelmeland AB, Keir ST,
McLendon RE, Batt D, Ramsey T, Yusuff N, Rasheed BK, Kieran MW,
Laforme A, et al: AAL881, a novel small molecule inhibitor of RAF
and vascular endothelial growth factor receptor activities, blocks
the growth of malignant glioma. Cancer Res. 66:8722–8730. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lang SA, Schachtschneider P, Moser C, Mori
A, Hackl C, Gaumann A, Batt D, Schlitt HJ, Geissler EK and
Stoeltzing O: Dual targeting of Raf and VEGF receptor 2 reduces
growth and metastasis of pancreatic cancer through direct effects
on tumor cells, endothelial cells, and pericytes. Mol Cancer Ther.
7:3509–3518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lang SA, Brecht I, Moser C, Obed A, Batt
D, Schlitt HJ, Geissler EK and Stoeltzing O: Dual inhibition of Raf
and VEGFR2 reduces growth and vascularization of hepatocellular
carcinoma in an experimental model. Langenbecks Arch Surg.
393:333–341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cotrim CZ, Amado FL and Helguero LA:
Estrogenic effect of the MEK1 inhibitor PD98059 on endogenous
estrogen receptor alpha and beta. J Steroid Biochem Mol Biol.
124:25–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Han Y, Xu G, Zhang J, Yan M, Li X, Ma B,
Jun L, Wang SJ and Tan J: Leptin induces osteocalcin expression in
ATDC5 cells through activation of the MAPK-ERK1/2 signaling
pathway. Oncotarget. Aug 24–2016.(Epub ahead of print).
|
|
61
|
Davis JE, Xie X, Guo J, Huang W, Chu WM,
Huang S, Teng Y and Wu G: ARF1 promotes prostate tumorigenesis via
targeting oncogenic MAPK signaling. Oncotarget. 7:39834–39845.
2016.PubMed/NCBI
|
|
62
|
Montagut C and Settleman J: Targeting the
RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283:125–134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Barrett SD, Bridges AJ, Dudley DT, Saltiel
AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold
WR, et al: The discovery of the benzhydroxamate MEK inhibitors
CI-1040 and PD 0325901. Bioorg Med Chem Lett. 18:6501–6504. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lorusso PM, Adjei AA, Varterasian M,
Gadgeel S, Reid J, Mitchell DY, Hanson L, DeLuca P, Bruzek L, Piens
J, et al: Phase I and pharmacodynamic study of the oral MEK
inhibitor CI-1040 in patients with advanced malignancies. J Clin
Oncol. 23:5281–5293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rinehart J, Adjei AA, Lorusso PM,
Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury
P, Kaldjian EP, et al: Multicenter phase II study of the oral MEK
inhibitor, CI-1040, in patients with advanced non-small-cell lung,
breast, colon, and pancreatic cancer. J Clin Oncol. 22:4456–4462.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ciuffreda L, Del Bufalo D, Desideri M, Di
Sanza C, Stoppacciaro A, Ricciardi MR, Chiaretti S, Tavolaro S,
Benassi B, Bellacosa A, et al: Growth-inhibitory and antiangiogenic
activity of the MEK inhibitor PD0325901 in malignant melanoma with
or without BRAF mutations. Neoplasia. 11:720–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lorusso PM, Krishnamurthi SS, Rinehart JJ,
Nabell LM, Malburg L, Chapman PB, DePrimo SE, Bentivegna S, Wilner
KD, Tan W and Ricart AD: Phase I pharmacokinetic and
pharmacodynamic study of the oral MAPK/ERK kinase inhibitor
PD-0325901 in patients with advanced cancers. Clin Cancer Res.
16:1924–1937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Haura EB, Ricart AD, Larson TG, Stella PJ,
Bazhenova L, Miller VA, Cohen RB, Eisenberg PD, Selaru P, Wilner KD
and Gadgeel SM: A phase II study of PD-0325901, an oral MEK
inhibitor, in previously treated patients with advanced non-small
cell lung cancer. Clin Cancer Res. 16:2450–2457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Davies BR, Logie A, McKay JS, Martin P,
Steele S, Jenkins R, Cockerill M, Cartlidge S and Smith PD: AZD6244
(ARRY-142886), a potent inhibitor of mitogen-activated protein
kinase/extracellular signal-regulated kinase kinase 1/2 kinases:
Mechanism of action in vivo, pharmacokinetic/pharmacodynamic
relationship, and potential for combination in preclinical models.
Mol Cancer Ther. 6:2209–2219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Do K, Speranza G, Bishop R, Khin S,
Rubinstein L, Kinders RJ, Datiles M, Eugeni M, Lam MH, Doyle LA, et
al: Biomarker-driven phase 2 study of MK-2206 and selumetinib
(AZD6244, ARRY-142886) in patients with colorectal cancer. Invest
New Drugs. 33:720–728. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Grasso S, Pereira GJ, Palmeira-Dos-Santos
C, Calgarotto AK, Martínez-Lacaci I, Ferragut JA, Smaili SS and
Bincoletto C: Autophagy regulates Selumetinib (AZD6244)
induced-apoptosis in colorectal cancer cells. Eur J Med Chem.
122:611–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang QJ, Huo Y, Han YL, Wan LL, Li J,
Huang JL, Lu J, Chen PG, Gan R and Guo C. Cheng: Selumetinib
attenuate skeletal muscle wasting in murine cachexia model through
ERK inhibition and AKT activation. Mol Cancer Ther. Sep
6–2016.(Epub ahead of print).
|
|
73
|
O'Neil BH, Goff LW, Kauh JS, Strosberg JR,
Bekaii-Saab TS, Lee RM, Kazi A, Moore DT, Learoyd M, Lush RM, et
al: Phase II study of the mitogen-activated protein kinase 1/2
inhibitor selumetinib in patients with advanced hepatocellular
carcinoma. J Clin Oncol. 29:2350–2356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Holkova B, Zingone A, Kmieciak M, Bose P,
Badros AZ, Voorhees PM, Baz R, Korde N, Lin HY, Chen JQ, et al:
Phase II trial of AZD6244 (Selumetinib, ARRY-142886), an oral
MEK1/2 inhibitor, in relapsed/refractory multiple myeloma. Clin
Cancer Res. 22:1067–1075. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Coleman RL, Sill MW, Thaker PH, Bender DP,
Street D, McGuire WP, Johnston CM and Rotmensch J: A phase II
evaluation of selumetinib (AZD6244, ARRY-142886), a selective
MEK-1/2 inhibitor in the treatment of recurrent or persistent
endometrial cancer: An NRG Oncology/Gynecologic Oncology Group
study. Gynecol Oncol. 138:30–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Barrett SD, Bridges AJ, Dudley DT, Saltiel
AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold
WR, et al: The discovery of the benzhydroxamate MEK inhibitors
CI-1040 and PD 0325901. Bioorg Med Chem Lett. 18:6501–6504. 2008.
View Article : Google Scholar : PubMed/NCBI
|