Introduction
To maintain cellular homeostasis, cell survival is
in a state of constant equilibrium with cell death (1). When cell demise occurs in an ordered and
controlled way to cause programmed cell death, this is termed
apoptosis. As well as being part of normal tissue turnover,
apoptosis is an essential process for development, differentiation
and immune responses (2–4). It is known to be actively involved in
the removal of useless or severely damaged cells (5). In contrast, necrosis is a type of
unrequired cell death that occurs when cells are exposed to
overwhelming stresses, including radiation overdose or toxic
chemicals. Concurrently, there is an ordered type of necrosis that
is executed by signaling pathway of its associated proteins. This
type of programmed cell death, which exhibits necrotic features, is
termed programmed necrosis or necroptosis. It was initially
considered to be a specialized and regulated form of necrotic cell
death (6). At present, necroptotic
cell death is known to serve a central function in cell
development, immunity, cancer and degenerative diseases (7–10). It
exhibits typical necrotic characteristics and is under the control
of a well-defined signaling pathway. The present review begins with
a description of necroptosis-derived features, and updated
information on necroptosis regulators and their specific
inhibitors.
A list of features discriminating between apoptosis
and necroptosis are summarized in Table
I. Cells undergoing apoptosis are morphologically shrunken with
condensed cytoplasm, while necroptotic cells and nuclei are
swollen. Membrane integrity is a definite determinant parameter to
discriminate apoptosis and necroptosis (11). Cells that are dying by apoptosis or
necroptosis exhibit intact or disintegrated membranes,
respectively. At the molecular level, a cascade of signaling
pathways leading to caspase activation is required for the
mediation of apoptosis via intrinsic or extrinsic pathways, but
necroptosis is achieved by the formation of the
receptor-interacting protein kinase (RIP)1-RIP3 necrosome complex
(12). Necroptosis is different from
apoptosis as the former exhibits more marked physiological effects
compared with the latter. Specifically, necroptotic cells release
intracellular danger signaling molecules into the media to provoke
inflammation and immune responses. These endogenous molecules are
referred to as damage-associated molecular patterns (DAMPs), which
include high mobility group box 1 (HMGB1), DNA fragments and
mitochondrial contents. In particular, HMGB1 is a major DAMP
protein derived from necroptotic cells, and serves a pivotal role
in triggering inflammatory responses (11). Apoptotic cells are completely cleared
by macrophages or neighboring cells, therefore presenting no
apparent physiological responses when compared with the
consequences of necroptosis. Conclusively, the physical and
biochemical parameters that characterize apoptosis or necroptosis
contribute to different physiological outcomes in biological
systems.
 | Table I.Key features discriminating apoptosis
and necroptosis. |
Table I.
Key features discriminating apoptosis
and necroptosis.
| Feature or
characteristic | Apoptosis | Necroptosis |
|---|
| Cell &
organelles morphology | Shrinkage | Swelling |
| Membrane
integrity | Intact | Disintegrated |
| DNA ladder
fragmentation | Yes | No |
| Signaling
pathway | Intrinsic and
extrinsic routes |
RIP1/RIP3/MLKL/PGAM5 |
| Molecular
complex | Apoptosome | Necrosome |
| Biological
markers | Caspase, Poly
(ADP-ribose) polymerase | High mobility group
B1 |
| Physiological
significance | Clearance of dead
cells | Inflammation and
innate immunity |
The terminology ‘programmed’ indicates that each
processing step is developed in a well-organized way and
specifically regulated in an orchestrated manner.
Necroptosis-associated proteins have been extensively identified
through RNA interference screening to establish a series of
signaling networks (13). Notably,
certain key regulators are known to execute tumor necrosis factor α
(TNFα)-mediated necroptosis. The signaling pathway leading to
necroptosis is summarized in Fig. 1.
Upon TNFα ligation to its cognate TNFα receptor (TNFR), RIP1, TNF
receptor 1-associated death domain protein, Fas-associated death
domain and caspase-8 are assembled to form complex I (14). Subsequently, transition of the
membrane-bound complex I to the cytosolic complex II ensues,
leaving TNFR (15). Then, a tumor
suppressor cylindromatosis (CYLD) protein promotes the
deubiquitination of RIP1 in either complex I or complex II
(16,17). It is generally hypothesized that
necroptosis is mediated by formation of the RIP1-RIP3 complex when
caspase is defective (18). Since the
identification of RIP3 as a proximal protein of necroptosis, a
mitochondrial protein phosphoglycerate mutase family member 5
(PGAM5) and mixed lineage kinase domain-like (MLKL) protein have
been additionally identified as downstream proteins of TNFR
ligation. The RIP1-RIP3 complex transmits a death signal to its
downstream target, MLKL (19,20). MLKL has been suggested to be
responsible for reactive oxygen species generation and c-Jun
N-terminal kinase activation during TNFα-induced necroptosis
(20). PGAM5, an additional protein
that interacts with RIP3, has been identified in addition to RIP1
and MLKL (21). The PGAM5 gene
encodes two protein isoforms, PGAM5-long form and PGAM5-short form,
via alternative splicing (22). One
of these splice variants, PGAM5-short form, may recruit
mitochondrial fission factor dynamin-related protein 1 to cause
mitochondrial fragmentation (21).
PGAM5 functions at the convergence point of multiple necrotic death
pathways, linking extracellular stimuli derived from TNFα to the
mitochondria through ligation of the death receptor and activation
of a series of intracellular proteins (21). With the identification of
necroptosis-associated proteins, a few small molecules that may
specifically modulate necroptosis have been identified via
high-throughput screening (23).
Necrostatin-1 (Nec-1) and necrosulfonamide are specific inhibitors
of RIP1 and MLKL, respectively (23,24). These
are valuable chemical probes to confirm the presence of necroptotic
cell death and to elucidate the underlying molecular mechanisms.
Extensive identification of specific necroptotic proteins with the
development of specific small molecules may provide data to fill
the gaps in these signaling pathways.
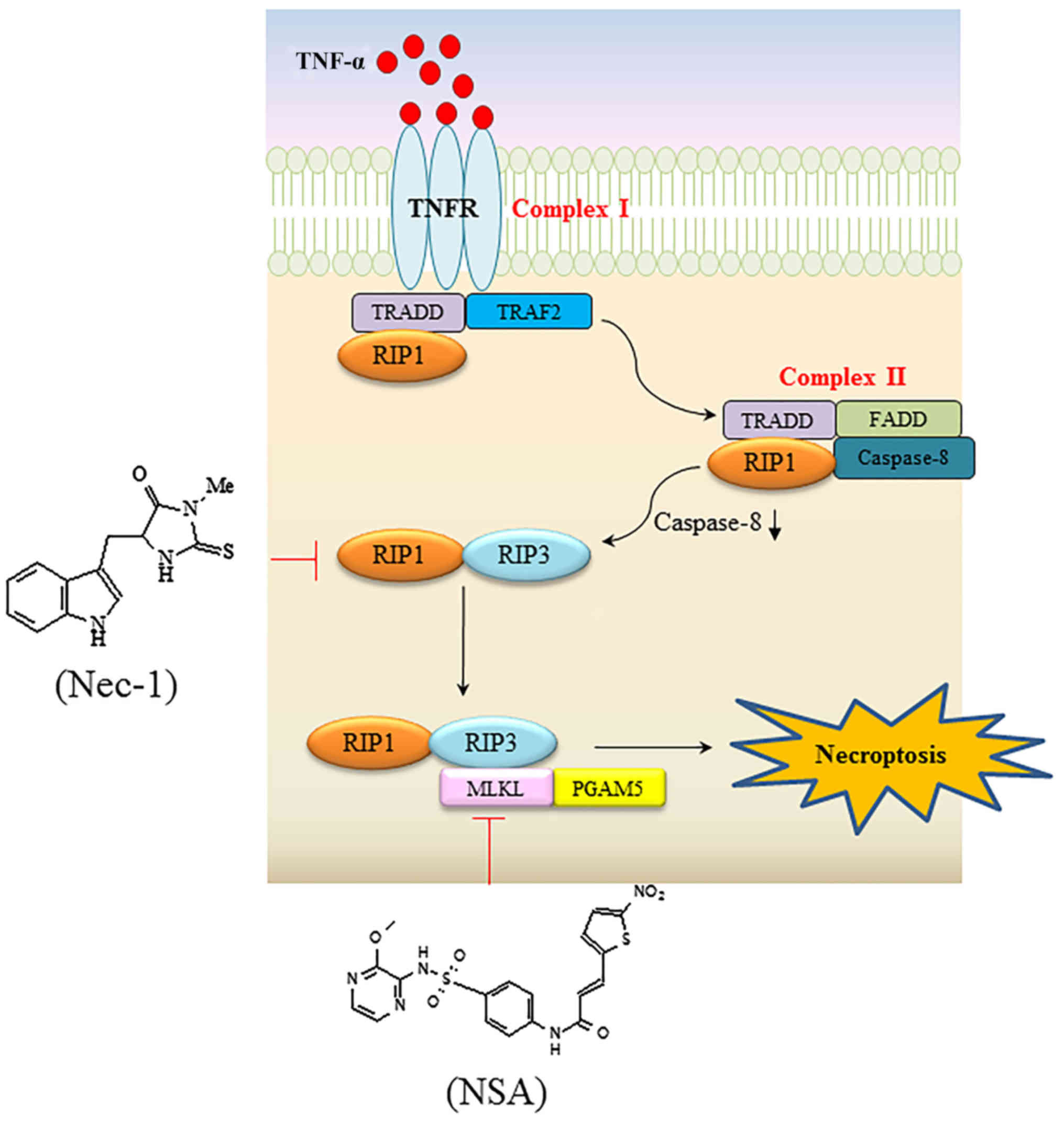 | Figure 1.Signaling pathway resulting in
TNFα-mediated necroptosis, and specific necroptotic inhibitors
targeting RIP1 and MLKL. Upon TNFα binding to its cognate receptor,
TRADD, TRAF2 and RIP1 are recruited to form complex I. In the
second step, bound proteins dissociate from the receptor when TNFR
is engulfed into the cytosol. In turn, TRADD and RIP1 are bound to
FADD and caspase 8, eventually forming the cytoplasmic complex II.
In situations where caspase is compromised, RIP1 interacts with
RIP3 to trigger consecutive downstream signaling events, including
the recruitment of MLKL and PGAM5, which transmit cytosolic death
signals to the mitochondria. Nec-1 and NSA inhibit RIP1 and MLKL,
respectively, with high specificity. TNFα, tumor necrosis factor α;
RIP, receptor-interacting protein kinase; MLKL, mixed lineage
kinase domain-like; TRADD, TNF receptor 1-associated death domain
protein; TRAF2, TNK receptor-associated factor 2; TNFR, TNFα
receptor; FADD, Fas-associated death domain; PGAM5,
phosphoglycerate mutase family member 5; Nec-1, necrostatin-1; NSA,
necrosulfonamide. |
Physiological roles and pathophysiological
conditions associated with necroptosis
Originally, necroptosis was regarded as an
alternative cell death modality to apoptosis upon TNFα stimulation.
Its activation and subsequent release of DAMPs may not only serve a
physiological function, but also cause a wide range of diseases
(Table II). Necroptotic cell death
is distinctive from necrosis in the sense that the cells actively
respond to death stresses, and is also hypothesized to be a method
of cell demise when apoptosis is compromised. Genome-wide small
interfering RNA analysis demonstrated that a set of 432 genes were
enriched in the immune and nervous systems, and that the cellular
response to necroptosis was affected by a signaling network
relevant to innate immunity (13). At
present, necroptosis is proposed to be the dominant cell death
program when apoptosis is inhibited (25). Generally, apoptosis via the intrinsic
or extrinsic pathways has been regarded as the primary mechanism
for the contraction phase of T cell immunity and the elimination of
autoreactive T cells. However, necroptosis serves a key function in
maintaining T cell homeostasis with defective B-cell
lymphoma-2-like protein 11, which is a crucial effector in the
negative selection of autoreactive thymocytes, highlighting that
caspase inactivation leads to induction of necroptosis (26). In addition, the death of host cells
through necroptosis contributes to the first defense mechanism
against infectious pathogens that may suppress or evade apoptotic
surveillance. In fact, cells infected with viruses may be removed
by apoptotic cell death or the immune response. When a virus
disarms the apoptotic machinery of host cells for the proliferation
of its progeny, necroptosis may be induced as an alternative form
of cell death to inhibit virus propagation (27). In addition, RIP3-mediated necroptosis
provides a secondary process to clear pathogens through the
induction of inflammation (12).
Intracellular pathogens, including Mycobacterium
tuberculosis and Salmonella typhimurium, transduce type
I interferon signaling to kill macrophages via the induction of
necroptosis (28,29). As demonstrated by the dynamic
functions of necroptosis during viral or bacterial infection, the
excess of intracellular molecules from cells undergoing necrosis or
necroptosis leads to a pro-inflammatory response, which may provoke
the immune system to fight against pathogens.
| Table II.Physiological and pathophysiological
significance of necroptosis. |
Table II.
Physiological and pathophysiological
significance of necroptosis.
| Physiological or
pathophysiological condition | Target or
pathway | Consequences | Comments | (Refs.) |
|---|
| Viral
infection | RIP3-dependent
pathway, RIP1/RIP3 complex | Virus
clearance | Vaccinia virus | (12,27) |
| Immune
homeostasis | Inactivation of
caspase-8 in B-cell lymphoma 2 interacting mediator of cell
death−/− | T cell
homeostasis | Suppression of
autoimmunity | (26) |
| Bacterial
infection | Type I
interferons-mediated RIP-dependent necroptosis in macrophages | Protection against
infection | Mycobacterium
tuberculosis, Salmonella enterica serovar
typhimurium | (28,29) |
| Acute
pancreatitis | RIP3 | DAMPs-provoked
inflammation | DAMPs emission | (30) |
| Inflammatory bowel
disease | RIP3 | Crohn's
disease | Caspase 8
deficiency in intestine epithelium cells | (31) |
| Retinal
degeneration | RIP3 | Photoreceptor cell
loss | Caspase 8
inhibition with retinal detachment | (32) |
| Acute kidney
injury | RIP1/RIP3 | Acute tubular
necrosis | Poly adenosine
diphosphate ribose polymerase-calpain axis or mitochondrial
permeability transition pathway involved | (33) |
| Hepatocyte
necrosis | RIP3 | APAP-induced liver
injury | RIP3: an early
mediator of APAP hepatotoxicity | (34) |
| Neurodegenerative
disease | RIP1/RIP3 insoluble
aggregates | Multiple
sclerosis | Tumor necrosis
factor.-mediated oligodendrocyte loss | (35) |
| Staphylococcus
aureus-induced necroptosis | Necroptosome
complex | Inflammatory
necrotizing pneumonia | Pore-formation and
inflammasome activation by multiple S. aureus toxins | (36) |
Conversely, excessive necroptosis in peripheral
tissues and ischemia reperfusion injury may cause an inflammatory
signal that leads to detrimental consequences, which may result
from the release of DAMPs from necroptotic cells into the
extracellular compartment. In a previous study, 33 out of 432 genes
identified were proposed to be implicated in human diseases,
including Huntington's disease, although the associations between
necroptosis-regulating genes and human diseases have remained
elusive (13). There is a growing
body of evidence suggesting that necroptosis is associated with
pathological conditions including acute pancreatitis, retinal
detachment, renal ischemic reperfusion injury, myocardial
infarction and traumatic brain injury (Table II) (12,26–36).
Necroptotic cell death was identified in cerulein-induced acute
pancreatitis, in which RIP3 overexpression was induced in the
pancreas but not in other areas (30). In addition, necroptosis of Paneth
cells in the terminal ileum was revealed to be associated with the
pathogenesis of inflammatory bowel disease (31). There is also marked RIP3 expression in
patients with inflammatory disorders (31). In addition, necroptotic cell death is
actively induced in photoreceptor cell loss and acute kidney injury
(32,33). A notable previous study suggested that
the inhibition of necroptosis was protective against
liver-associated disease conditions (34). Acetaminophen may induce RIP3
expression along with elevated levels of alanine aminotransferase
(ALT) in mice, leading to extensive necrosis. Wild type mice
subjected to morpholino antisense targeting RIP3, or RIP3-deficient
mice, are protected against acetaminophen-induced liver damage
(34). The activation of necroptosis
has previously been demonstrated to be a prerequisite for the
pathogenesis of multiple sclerosis (35). Kitur et al (36) revealed that the toxin derived from
Staphylococcus aureus caused necroptosis-associated lung
damage.
Therapeutic exploitation of necroptosis
As described above, necroptosis is a specialized
cell death mode that is does not occur in normal homeostasis. It
may be induced by external stresses in conjunction with specific
circumstances involving the absence of caspase. Necroptosis has
previously been documented in multiple diseases, including ischemic
brain injury and degenerative diseases (10). In addition, it becomes an alternative
cell death mechanism in multicellular organisms when cells are
infected with pathogens that are able to evade the apoptotic
machinery of the host. This process is associated with the innate
immune response, and may be the first line of defense against
pathogens, including viruses and bacteria. Understanding the
molecular mechanisms by which necroptosis may be activated is of
significance for the implementation of a protective strategy
against microbial infection. As well as the defensive function of
necroptosis in the host, attention has been paid to harnessing
alternative cell death pathways to fight tumor cells with acquired
resistance to cancer drugs. Along with apoptosis, necroptosis is a
promising cell death process for sensitizing tumor cells to
anticancer drugs, and its induction is expected to be a therapeutic
tool for killing tumor cells, particularly apoptosis-resistant
types of cancer. Cancer cells may evolve to multiply by evading
chemotherapy-induced apoptosis, whilst remaining inherently
susceptible to necroptosis (37).
Therefore, exploitation of the induction of necroptosis may be a
secondary therapy to counteract types of cancer resistant to
apoptosis. The potential induction of necroptosis for cancer
therapy has been paradoxically encouraged by the fact that
necroptosis is impaired during tumorigenesis (38). Chronic lymphocytic leukemia cells
exhibit defects in necroptosis regulators, including RIP3 and CYLD,
an enzyme that may regulate RIP1 ubiquitination (39). RIP3 polymorphisms in non-Hodgkin
lymphoma have been demonstrated to be correlated with tumor
progression (40). Therapeutically,
necroptosis should be induced or suppressed for anticancer therapy
or the prevention of necroptosis-associated pathological diseases,
respectively. For a more selective induction of necroptosis, the
construction of signaling pathways connecting necroptosis proteins
and the development of small molecules that may target necroptosis
regulators are required. However, this becomes difficult when
considering the interplay between apoptosis, necrosis and
autophagy-associated networks. Accordingly, the intricate molecular
crosstalk between death modalities should be fully understood prior
to the clinical application of necroptosis.
There are a number of feasible examples in which the
induction of necroptosis may be applied as a cancer treatment. For
example, a number of incurable cancers, including lung cancer, have
evolved to evade or interfere with apoptotic machinery when
challenged with repetitive treatment with anticancer drugs
(41). Accordingly, as an alternative
cell death pathway, necroptosis may be exploited to control cancer
cells with acquired anticancer drug resistance. The combined
treatment of a caspase inhibitor and an antagonist of inhibitor of
apoptosis proteins triggers TNFα-induced necroptosis in various
apoptosis-resistant cell lines and patient xenografts (42). Ovarian cancer cells undergoing
necroptosis exhibit the formation of a necrosome-like complex with
RIP1 (42). This suggests that it is
feasible to target the necroptotic signaling pathway identified in
ovarian cancer cells in a therapeutic setting. In addition,
Table III summarizes the synthetic
small molecules or natural products that may trigger necroptotic
cell death in cancer cells. Obatoclax-bearing indole bipyrrole
moiety is able to induce necroptosis via the formation of the
necrosome on the autophagosomal membrane (43). Staurosporin, a protein kinase
inhibitor, and B12536, which targets mitotic kinase pololike kinase
1, have been suggested to induce necroptosis (44,45). A
synthetic naftopidil analogue, HUHS1015, kills human gastric cancer
cells via the induction of necroptosis and caspase-independent
apoptosis (46). A amiloride
derivative, 5′-betaenzylglycinyl-amiloride, was identified to be an
inducer of caspase-independent necroptosis in glioma cells
(47). The Chinese medicine shikonin
promotes necroptotic cell death of glioma cells in a RIP1-dependent
manner (48). Shikonin has also been
suggested to exert antitumor effects on osteosarcoma by inducing
RIP1- and RIP3-dependent necroptosis (49). The use of an additional natural
compound, honokiol, in combination with chemotherapeutic agents
synergistically kills drug-resistant cell lines via apoptosis and
necroptosis (50). As well as
chemotherapy, photodynamic therapy using a photosensitizer
talaporfin sodium has been indicated to mediate necroptotic cell
death in glioblastoma T98G cells via a signaling pathway consisting
of RIP1, RIP3 and MLKL (51). In
addition, radiotherapy was performed to induce necroptosis in
anaplastic thyroid and adrenocortical cancers (52). Specifically, Nec-1 and zVAD
effectively protect cells from radiotherapy, indicating that
necroptosis is partly involved in radiation-induced cell death.
| Table III.Use of necroptosis for anticancer
therapy. |
Table III.
Use of necroptosis for anticancer
therapy.
| Treatment | Target or
pathway | Cancer type | (Refs.) |
|---|
| Combination
treatment |
|
|
|
|
zVAD+SMAC memetics | RIP3-dependent | Triggering
necroptosis in apoptosis-resistant ovarian carcinoma | (42) |
| Chemotherapy |
|
|
|
|
Obatoclax (GX15-070) | Necrosome
complex | Inducing
non-apoptotic form of cell death in rhabdomyosarcoma cells | (43) |
|
Staurosporin | Nonspecific kinase
inhibitor | Necroptotic cell
death in caspase-compromised U937 | (44) |
|
B12536 | Protein kinase
Plk1 | Plk1 leads to
necroptosis in androgen-insensitive prostate cancer cell | (45) |
|
HUHS1015 | AMID
accumulation | Human gastric
cancer cells due to AMID accumulation in the nucleus | (46) |
|
5′-betaenzylglycinyl-amiloride | AIF | AIF-mediated
malignant glioma cells | (47) |
|
Shikonin | RIP1, oxidative
stress | Glioma cells
primarily via necroptosis | (48) |
|
| Induction of RIP1
and RIP3 | Osteosarcoma | (49) |
|
Honokiol | Enhanced
apoptosis/additional necroptosis | Multidrug
resistance breast cancer cells | (50) |
| Photodynamic
therapy |
|
|
|
| Using
talaporfin sodium | Necroptosis
pathway | Glioblastoma
T98G | (51) |
| Radiation-induced
cell death |
|
|
|
|
6Gy | RIP1 dependent | Anaplastic thyroid
and adrenocortical cancer | (52) |
Perspectives of necroptosis
As aforementioned, necroptosis was initially
considered a secondary cell death pathway to TNFα-induced apoptosis
under a caspase-deficient condition. At present, it is hypothesized
that necroptosis may be triggered to evoke physiological and
pathological consequences to diverse stimuli, although its
regulatory mechanism remains unknown. From physiological and
pathological aspects, its activation may be beneficial or harmful
depending on the stimulus context, and on the cell-specific
responses to it. Therefore, the induction of necroptosis may not
only provide a secondary safety mechanism against pathogenic
infection, but may also be associated with various diseases: Apart
from innate immune surveillance, an increasing number of diseases
associated with necroptosis, including tissue inflammation and
degeneration, have been identified. Necroptosis has been
demonstrated to be involved in various neurological disorders,
including trauma, strokes, multiple sclerosis and Huntington's
disease (53). In addition, the
conversion of cholesterol to 24(S)-hydroxycholesterol and its
consequent passage through blood-brain barrier is hypothesized to
induce necroptosis in neuronal cells that are caspase-8-defective
(54). Therefore, protection against
necroptotic cell death is of primary concern to prevent the
pathogenesis of these diseases. Genetic or pharmacological
interference with necroptosis signaling results in neuroprotection
against ischemic heart or brain injury (53,55). RIP3
deficiency or administration of Nec-1 has been demonstrated to
exhibit protective effects on necroptosis-based heart or brain
damage (55,56). Apart from RIP3, additional potent
target proteins have been identified as regulators of necroptotic
cell death, including RIP1, MLKL, PGAM5 and CYLD (17,21,23,57),
which comprise a cascade of signaling pathways for necroptosis
(Fig. 1). Subsequently, a small
number of inhibitors targeting RIP1 or MLKL have been developed to
effectively protect against necroptotic cell death (23,57).
Hence, additional identification and validation of more potential
targets will be crucial for the development of drugs that may
improve pathological conditions.
Beyond necroptosis-associated pathological
consequences, necroptosis may be exploited as an alternative
therapy to overcome drug-resistant types of cancer. It is based on
the hypothesis that a failure in cancer management may be caused by
the acquired ability of cancer cells to evade cancer drug-induced
apoptosis. Therefore, inducing necroptosis may kill cancer cells
and improve immune responses to the danger molecules derived from
dying cells, although the release of intracellular contents from
dying cells may also promote neoplasia. Although not described in
the present review, autophagy complicates the processes of cell
death or survival depending on the cell types or the context of the
stress. For example, exposure to Obatoclax in rhabdomyosarcoma
results in substantial autophagy, which in turn causes RIP3
activation and then necroptotic cell death (43). Notably, caspase-8 does not inhibit
RIP3 activation in the autophagosome-driven necroptotic process,
unlike receptor-mediated necroptosis (43). In this situation, Obatoclax mediates
necroptosis by forming necrosome complexes on autophagosomes. These
data highlight that understanding the crosstalk between necroptosis
and other cell death types is a prerequisite for selecting optimal
treatments customized to specific types of cancer and
necroptosis-associated diseases. In conclusion, the comprehensive
regulation of cell death is expected to provide clinical
opportunities to use cell death programs to treat various diseases,
including different types of cancer and degenerative disorders.
References
|
1
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haanen C and Vermes I: Apoptosis:
Programmed cell death in fetal development. Eur J Obstet Gynecol
Reprod Biol. 64:129–133. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Opferman JT: Apoptosis in the development
of the immune system. Cell Death Differ. 15:234–242. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duval D, Trouillas M, Thibault C, Dembelé
D, Diemunsch F, Reinhardt B, Mertz AL, Dierich A and Boeuf H:
Apoptosis and differentiation commitment: Novel insights revealed
by gene profiling studies in mouse embryonic stem cells. Cell Death
Differ. 13:564–575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elliott MR and Ravichandran KS: Clearance
of apoptotic cells: Implications in health and disease. J Cell
Biol. 189:1059–1070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van den Berghe T, Linkermann A,
Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis:
The expanding network of non-apoptotic cell death pathways. Nat Rev
Mol Cell Biol. 15:135–147. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giampietri C, Starace D, Petrungaro S,
Filippini A and Ziparo E: Necroptosis: Molecular signalling and
translational implications. Int J Cell Biol. 2014:4902752014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu JV, Chen HC and Walsh CM: Necroptotic
signaling in adaptive and innate immunity. Semin Cell Dev Biol.
35:33–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fulda S: The mechanism of necroptosis in
normal and cancer cells. Cancer Biol Ther. 14:999–1004. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou W and Yuan J: Necroptosis in health
and diseases. Semin Cell Dev Biol. 35:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: The release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hitomi J, Christofferson DE, Ng A, Yao J,
Degterev A, Xavier RJ and Yuan J: Identification of a molecular
signaling network that regulates a cellular necrotic cell death
pathway. Cell. 135:1311–1323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Festjens N, Berghe T Vanden, Cornelis S
and Vandenabeele P: RIP1, a kinase on the crossroads of a cell's
decision to live or die. Cell Death Differ. 14:400–410. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: Backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moquin DM, McQuade T and Chan FK: CYLD
deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate
kinase activation and programmed necrosis. PLoS One. 8:e768412013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moriwaki K and Chan FK: RIP3: A molecular
switch for necrosis and inflammation. Genes Dev. 27:1640–1649.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q,
Luo J and Liu ZG: Mixed lineage kinase domain-like is a key
receptor interacting protein 3 downstream component of TNF-induced
necrosis. Proc Natl Acad Sci USA. 109:pp. 5322–5327. 2012;
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z, Jiang H, Chen S, Du F and Wang X:
The mitochondrial phosphatase PGAM5 functions at the convergence
point of multiple necrotic death pathways. Cell. 148:228–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Remijsen Q, Goossens V, Grootjans S, Van
den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman
I, Goncalves A, Bertrand MJ, et al: Depletion of RIPK3 or MLKL
blocks TNF-driven necroptosis and switches towards a delayed RIPK1
kinase-dependent apoptosis. Cell Death Dis. 5:e10042014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liao D, Sun L, Liu W, He S, Wang X and Lei
X: Necrosulfonamide inhibits necroptosis by selectively targeting
the mixed lineage kinase domain-like protein. Med Chem Comm.
5:333–337. 2014. View Article : Google Scholar
|
|
25
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bohgaki T, Mozo J, Salmena L,
Matysiak-Zablocki E, Bohgaki M, Sanchez O, Strasser A, Hakem A and
Hakem R: Caspase-8 inactivation in T cells increases necroptosis
and suppresses autoimmunity in Bim-/-mice. J Cell Biol.
195:277–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Upton JW, Kaiser WJ and Mocarski ES: Virus
inhibition of RIP3-dependent necrosis. Cell Host Microbe.
7:302–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Desvignes L, Wolf AJ and Ernst JD: Dynamic
roles of type I and type II IFNs in early infection with
Mycobacterium tuberculosis. J Immunol. 188:6205–6215. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robinson N, McComb S, Mulligan R, Dudani
R, Krishnan L and Sad S: Type I interferon induces necroptosis in
macrophages during infection with Salmonella enterica serovar
Typhimurium. Nat Immunol. 13:954–962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gunther C, Martini E, Wittkopf N, Amann K,
Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF
and Becker C: Caspase-8 regulates TNF-α-induced epithelial
necroptosis and terminal ileitis. Nature. 477:335–339. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trichonas G, Murakami Y, Thanos A,
Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW and Vavvas
DG: Receptor interacting protein kinases mediate retinal
detachment-induced photoreceptor necrosis and compensate for
inhibition of apoptosis. Proc Natl Acad Sci USA. 107:pp.
21695–21700. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Linkermann A, De Zen F, Weinberg J,
Kunzendorf U and Krautwald S: Programmed necrosis in acute kidney
injury. Nephrol Dial Transplant. 27:3412–3419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ramachandran A, McGill MR, Xie Y, Ni HM,
Ding WX and Jaeschke H: Receptor interacting protein kinase 3 is a
critical early mediator of acetaminophen-induced hepatocyte
necrosis in mice. Hepatology. 58:2099–2108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ofengeim D, Ito Y, Najafov A, Zhang Y,
Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg
H, et al: Activation of necroptosis in multiple sclerosis. Cell
Rep. 10:1836–1849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kitur K, Parker D, Nieto P, Ahn DS, Cohen
TS, Chung S, Wachtel S, Bueno S and Prince A: Toxin-induced
necroptosis is a major mechanism of Staphylococcus aureus lung
damage. PLoS Pathog. 11:e10048202015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su Z, Yang Z, Xie L, DeWitt JP and Chen Y:
Cancer therapy in the necroptosis era. Cell Death Differ.
23:748–756. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY,
Zaorsky NG, Deng L, Yuan ZY, Lu Y and Wang P: Necroptosis in
tumorigenesis, activation of anti-tumor immunity, and cancer
therapy. Oncotarget. 7:57391–57413. 2016.PubMed/NCBI
|
|
39
|
Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y,
Chen H, Dong H, Zhu Y, Miao K, et al: Dysregulation of TNFα-induced
necroptotic signaling in chronic lymphocytic leukemia: Suppression
of CYLD gene by LEF1. Leukemia. 26:1293–1300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu W, Liu P and Li J: Necroptosis: An
emerging form of programmed cell death. Crit Rev Oncol Hemato.
82:249–258. 2012. View Article : Google Scholar
|
|
41
|
Housman G, Byler S, Heerboth S, Lapinska
K, Longacre M, Snyder N and Sarkar S: Drug resistance in cancer: An
overview. Cancers (Basel). 6:1769–1792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
McCabe KE, Bacos K, Lu D, Delaney JR,
Axelrod J, Potter MD, Vamos M, Wong V, Cosford ND, Xiang R and
Stupack DG: Triggering necroptosis in cisplatin and IAP
antagonist-resistant ovarian carcinoma. Cell Death Dis.
5:e14962014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Basit F, Cristofanon S and Fulda S:
Obatoclax (GX15-070) triggers necroptosis by promoting the assembly
of the necrosome on autophagosomal membranes. Cell Death Differ.
20:1161–1173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dunai ZA, Imre G, Barna G, Korcsmaros T,
Petak I, Bauer PI and Mihalik R: Staurosporine induces necroptotic
cell death under caspase-compromised conditions in U937 cells. PLoS
One. 7:e419452012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Deeraksa A, Pan J, Sha Y, Liu XD, Eissa
NT, Lin SH and Yu-Lee LY: Plk1 is upregulated in
androgen-insensitive prostate cancer cells and its inhibition leads
to necroptosis. Oncogene. 32:2973–2983. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaku Y, Tsuchiya A, Kanno T and Nishizaki
T: HUHS1015 induces necroptosis and caspase-independent apoptosis
of MKN28 human gastric cancer cells in association with AMID
accumulation in the nucleus. Anticancer Agents Med Chem.
15:242–247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pasupuleti N, Leon L, Carraway KL III and
Gorin F: 5-Benzylglycinyl-amiloride kills proliferating and
nonproliferating malignant glioma cells through caspase-independent
necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp
Ther. 344:600–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang C, Luo Y, Zhao J, Yang F, Zhao H,
Fan W and Ge P: Shikonin kills glioma cells through necroptosis
mediated by RIP-1. PLoS One. 8:e663262013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang
Z, Zeng H, Zuo D, Hua Y and Cai Z: The anti-tumor effect of
shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent
necroptosis. BMC Cancer. 13:5802013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tian W, Deng Y, Li L, He H, Sun J and Xu
D: Honokiol synergizes chemotherapy drugs in multidrug resistant
breast cancer cells via enhanced apoptosis and additional
programmed necrotic death. Int J Oncol. 42:721–732. 2013.PubMed/NCBI
|
|
51
|
Miki Y, Akimoto J, Moritake K, Hironaka C
and Fujiwara Y: Photodynamic therapy using talaporfin sodium
induces concentration-dependent programmed necroptosis in human
glioblastoma T98G cells. Lasers Med Sci. 30:1739–1745. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nehs MA, Lin CI, Kozono DE, Whang EE, Cho
NL, Zhu K, Moalem J, Moore FD Jr and Ruan DT: Necroptosis is a
novel mechanism of radiation-induced cell death in anaplastic
thyroid and adrenocortical cancers. Surgery. 150:1032–1039. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu T, Bao YH, Wang Y and Jiang JY: The
role of necroptosis in neurosurgical diseases. Braz J Med Biol Res.
48:292–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yamanaka K, Saito Y, Yamamori T, Urano Y
and Noguchi N: 24(S)-hydroxycholesterol induces neuronal cell death
through necroptosis, a form of programmed necrosis. J Biol Chem.
286:24666–24673. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Luedde M, Lutz M, Carter N, Sosna J,
Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F,
et al: RIP3, a kinase promoting necroptotic cell death, mediates
adverse remodelling after myocardial infarction. Cardiovasc Res.
103:206–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
King MD, Whitaker-Lea WA, Campbell JM,
Alleyne CH Jr and Dhandapani KM: Necrostatin-1 reduces
neurovascular injury after intracerebral hemorrhage. Int J Cell
Biol. 2014:4958172014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang H, Sun L, Su L, Rizo J, Liu L, Wang
LF, Wang FS and Wang X: Mixed lineage kinase domain-like protein
MLKL causes necrotic membrane disruption upon phosphorylation by
RIP3. Mol Cell. 54:133–146. 2014. View Article : Google Scholar : PubMed/NCBI
|














