Introduction
Hepatocellular carcinoma and cholangiocarcinoma are
the two most common cancer types in the hepatobiliary system
(1). Extrahepatic cholangiocarcinoma
is a malignancy that originates from the confluence of right and
left hepatic ducts to the distal end of the common bile duct
(2–4).
Clinically, the treatment options include surgery, radiotherapy and
chemotherapy; however, the prognosis remains poor following these
treatments (5). Recent studies have
indicated that autophagy is able to protect cells by degrading
misfolded proteins, which may shed new light on the treatment of
cholangiocarcinoma (6,7).
Chloroquine (CQ), which is frequently used
clinically as an antimalarial agent, is a classic inhibitor of
autophagy that blocks the binding of autophagosomes to lysosomes by
altering the acidic environment of lysosomes, resulting in the
accumulation of a large number of degraded proteins in cells
(8). The inhibition of autophagy by
CQ results in the accumulation of a large number of damaged
proteins in the cytoplasm and induces endoplasmic reticulum (ER)
stress (9). Persistent ER stress
eventually results in cell death (10,11). A
study confirmed that CQ is able to induce tumor necrosis factor
(TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis
of glioma cells (12). Our previous
experiments demonstrated that cisplatin increases the ER stress and
autophagy level in HeLa cells, whereas blocking autophagy
significantly increased the chemosensitivity of HeLa cells to
cisplatin (13); therefore, CQ may
serve an antitumor role by regulating the ER-autophagy network,
although the specific mechanism is currently unclear.
Growth arrest and DNA damage 153 [C/EBP homologous
protein (CHOP)] is considered to be one of the most important
molecules mediating ER stress-induced apoptosis (14). Activated CHOP exerts a pro-apoptotic
effect by upregulating the expression of B-cell lymphoma-2
(Bcl-2)-associated X (Bax), a pro-apoptotic member of the Bcl-2
family (15,16). Additionally, CHOP promotes the
transcription of death receptor 5 (DR5), also known as
TRAIL-receptor, and activates the caspase-8-dependent extrinsic
apoptosis pathway (17). Cells may be
protected from ER stress-induced apoptosis by inhibition of CHOP
expression. Furthermore, a previous study indicated that
CHOP-deficient mice were more tolerant to tunicamycin (an inducer
of endoplasmic reticulum stress) (18); therefore, CHOP may serve a key role in
targeted cell apoptosis induced by ER stress. Further studies have
demonstrated the involvement of an autophagy-dependent
death-inducing signaling complex known as intracellular
death-inducing signaling complex (iDISC) in the ER stress-apoptosis
signaling pathway. iDISC is associated with the autophagy substrate
p62 and initiates apoptosis through activation of caspase-8
(19,20). CHOP may be the core molecule
associated with multiple ER stress-induced apoptotic pathways. By
investigating the regulatory mechanisms of the autophagy inhibitor
CQ underlying ER stress and apoptosis, the role of autophagy-ER
stress in antitumor therapy may be further elaborated, potentially
indicating the development of novel targets.
In the present study, the autophagy inhibitor CQ was
used to regulate the ER-autophagy network, in order to examine
whether autophagy provides data on novel treatments of
cholangiocarcinoma. Additionally, by elucidating the mechanism by
which CQ exerts cytotoxicity against the cholangiocarcinoma cell
line QBC939, CQ was considered a novel and effective strategy for
the treatment of cholangiocarcinoma.
Materials and methods
Materials
CQ diphosphate salt was purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany) and was dissolved in water to
produce a 10 mM stock solution.
Hoechst 33342 and MTT were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Anti-PARP
(sc-7150), anti-Bak (sc-832), anti-Bax (sc-4239), and anti-HSPA5
(sc-13968) antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Anti-LC3 (3868), anti-CHOP (2895),
anti-Caspase-3 (9662), anti-Cleaved-Caspase-3 (9664) and
anti-β-actin (3700) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Anti-p62 (ab109012) was purchased from
Abcam (Cambridge, UK). Horseradish peroxidase-conjugated goat
anti-rabbit (H+L) immunoglobulin (Ig)G (SA00001-2) and goat
anti-mouse (H+L) IgG (SA00001-1) secondary antibodies were
purchased from Proteintech (Chicago, IL).
Cell lines and culture
The human cholangiocarcinoma cell line QBC939 was
purchased from America Tissue Culture Collection (Manassas, VA,
USA) and cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (Hyclone, Logan, UT, USA), 100 U/ml penicillin, and
100 mg/ml streptomycin (complete medium) in a humidified cell
culture incubator with an atmosphere containing 5% CO2
at 37°C.
MTT assay
Cellular viability was assessed using MTT assays.
Briefly, cells were seeded in 96-well plates at a density of
1×104 cells/well in 100 µl complete medium at 37°C in an
atmosphere containing with 5% CO2 and exposed to
different doses of CQ (0, 1.5, 3.1, 6.2, 12.5, 25, 50, 100 and 200
µM) for 24 h. At the end of the treatment 10 µl 10 mg/ml MTT
reagent (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in PBS was
added to each well and incubated at 37°C for a further 4 h. Any
formazan crystals that formed were dissolved in 150 µl dimethyl
sulfoxide and absorbance was recorded at a wavelength of 490 nm.
Each treatment was repeated in five separate wells.
Analysis of apoptosis by Hoechst 3324
staining
Apoptotic morphological alterations in nuclear
chromatin were detected by Hoechst 3324 staining. Briefly, QBC939
cells were cultured in 24-well plates and treated with 50 µM CQ for
12 and 24 h, with the untreated group used as a control. Cells were
washed with ice-cold PBS three times and fixed with 4% (w/v)
paraformaldehyde at 37°C for 15 min. The plates were then incubated
with 10 M Hoechst 33258 staining solution for 10 min at 37°C and
the cells were visualized under a fluorescence microscope
(magnification, ×10) (IX-71; Olympus Corporation, Tokyo,
Japan).
Western blot analysis
Subsequent treating with 50 µM CQ for 6, 12 and 24
h, with the untreated group used as a control, cells were collected
and lysed with radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology, Haimen, China). The lysate was agitated
with ultrasound for 20 sec, and placed on ice for 45 min. The
lysate was then centrifuged at 3,170 × g for 5 min at 4°C and the
precipitate was discarded. Briefly, following quantification of
protein in each sample with the Bio-Rad protein reagent (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), 40 µg total protein per
well was separated by 12% SDS-PAGE and transferred onto an
Immun-Blot polyvinylidene fluoride membrane (Bio-Rad Laboratories,
Inc.). The membranes were blocked in Tris buffered saline
containing 5% (w/v) non-fat dry milk at room temperature for 1 h,
and then incubated with specific primary antibodies overnight at
4°C. Following washing with PBS Tween-20 (0.1%, v/v) for three
times, membranes were incubated with
horseradish-peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at room temperature for 1 h.
Membranes were then incubated in enhanced chemiluminescent reagents
(Thermo Fisher Scientific, Inc.) and images were captured by
Syngene Bio Imaging (Synoptics, Ltd., Cambridge, UK). Densitometric
quantitation of bands was performed using Syngene Bio Imaging tools
(version 1.2.2.0; Synoptics, Ltd., Cambridge, UK).
Quantitative polymerase chain reaction
(qPCR)
Cells were harvested and total RNA was extracted
with TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). qPCR was performed using
SYBR®-Green Real-Time PCR Master Mix (Takara
Biotechnology Co., Ltd., Dalian, China) with relevant primers using
the MX300P instrument (Agilent Technologies, Inc., Santa Clara, CA,
USA), according to the manufacturer's protocol in the following
conditions: 94.0°C for 30 sec, 40 cycles of 94.0°C for 5 sec and
60.0°C for 30 sec. A melting curve was detected between 60 and 94°C
to confirm the PCR product of interest. The primer sequences used
are as follows: TNF receptor superfamily member 10c (DCR1),
5′-CCTCCTTGCTTCCCATGTAC-3′ (forward), and
5′-CTTCAACACACTGGTATCATC-3′ (reverse); TNF receptor superfamily
member 10d (DCR2), 5′-TCACGTCCTTCACGAGTTCC-3′ (forward), and
5′-CTCCGACACCCTTCAGCTTC-3′ (reverse); TNF receptor superfamily
member 25 (DR3), 5′-CCTCAATGTGCCAGGCTCTT-3′ (forward), and
5′-ATGACGGCACGCTCACACT-3′ (reverse); TNF receptor superfamily
member 10a (DR4); 5′-CTCGCTGTCCACTTTCGTCTC-3′ (forward), and
5′-GTACCAGCTCTGACCACATC-3′ (reverse); TNF receptor superfamily
member 10b (DR5), 5′-AAGACCCTTGTGCTCGTTGT-3′ (forward), and
5′-AGGCGGACACAATCCCTCTG-3′ (reverse); tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL), 5′-GAGCTGAAGCAGATGCAGGAC-3′
(forward), and 5′-TGACGGAGTTGCCACTTGACT-3′ (reverse); TNF receptor
2 (TNFR2), 5′-GCCCCACCAGATCTGCAACGTG-3′ (forward), and
5′-TGAGGCACCTTGGCTTCTCTC-3′ (reverse); and Fas cell surface death
receptor (FAS), 5′-TGTAGATTGTGTGATGAAGG-3′ (forward), and
5′-GATCCCATGTTCACATTTGG-3′ (reverse). Analysis of relative gene
expression data using qPCR and the 2−ΔΔCq method
(21).
Caspase-8 activity
Caspase-8 activity was measured using the
Caspase-Glo 8 assay kit (Promega Corporation, Madison, WI, USA),
according to the manufacturer's protocols. Each experiment was
performed in quintuplicate.
Flow cytometry
Apoptosis was determined by an Annexin-V and PI kit
(cat. no. 556547; BD Biosciences, San Jose, CA, USA). Cells were
seeded in 6-well plates overnight at a density of 5×105
cells/well. Following exposure to 50 µM CQ for 12 and 24 h, the
untreated group was used as a control, cells were trypsinized at
37°C for 5 min and incubated with PI and Annexin V-FITC for 15 min
at 37°C. The samples were analyzed using a BD Accuri C6 flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA). All
experiments were performed in triplicate.
RT2 Profiler PCR Array
System
The Human Unfolded Protein Response RT2
Profiler™ PCR Array (SABiosciences-Qiagen, Hilden, Germany)
profiles the expression of 84 key genes involved in unfolded
protein accumulation in the ER. Total cellular RNA was extracted
from cultured cells according to the manufacturer's instructions.
Single-stranded cDNA was obtained by reverse transcription of 1 µg
of total RNA using the SABiosciences RT2 First Strand
kit. qPCR was performed using Applied Biosystems 7300 Fast with
SYBR Green Fluorophore. The reactions were carried out using an
RT2 SYBR-Green Mastermix. cDNA was used as template and
cycling parameters were 95°C for 10 min, followed by 40 cycles of
95°C for 15 sec and 60°C for 1 min. Fluorescence intensities were
analyzed using the manufacturer's software, and relative
quantification was calculated using the 2−ΔΔCq method.
Change of expression of the 84 genes was shown by heat imaging.
GAPDH was used as a reference gene.
Immunoprecipitation assay
Cells were lysed in NP40 lysis buffer. Equal amounts
of lysates were immunoprecipitated with 2 µg of the p62-linkage
Specific Polyubiquitin antibody overnight at 4°C. A total of 25 µl
protein A and G agarose (Beyotime Institute of Biotechnology) were
used in each sample. Beads were washed with PBS three times with 1
ml each. The eluted proteins were examined by using western
blotting as described above.
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used for
analysis. All experiments were repeated at least three times. The
statistical significance of the difference between groups was
assessed using one-way analysis of variance with
Least-Significant-Difference post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
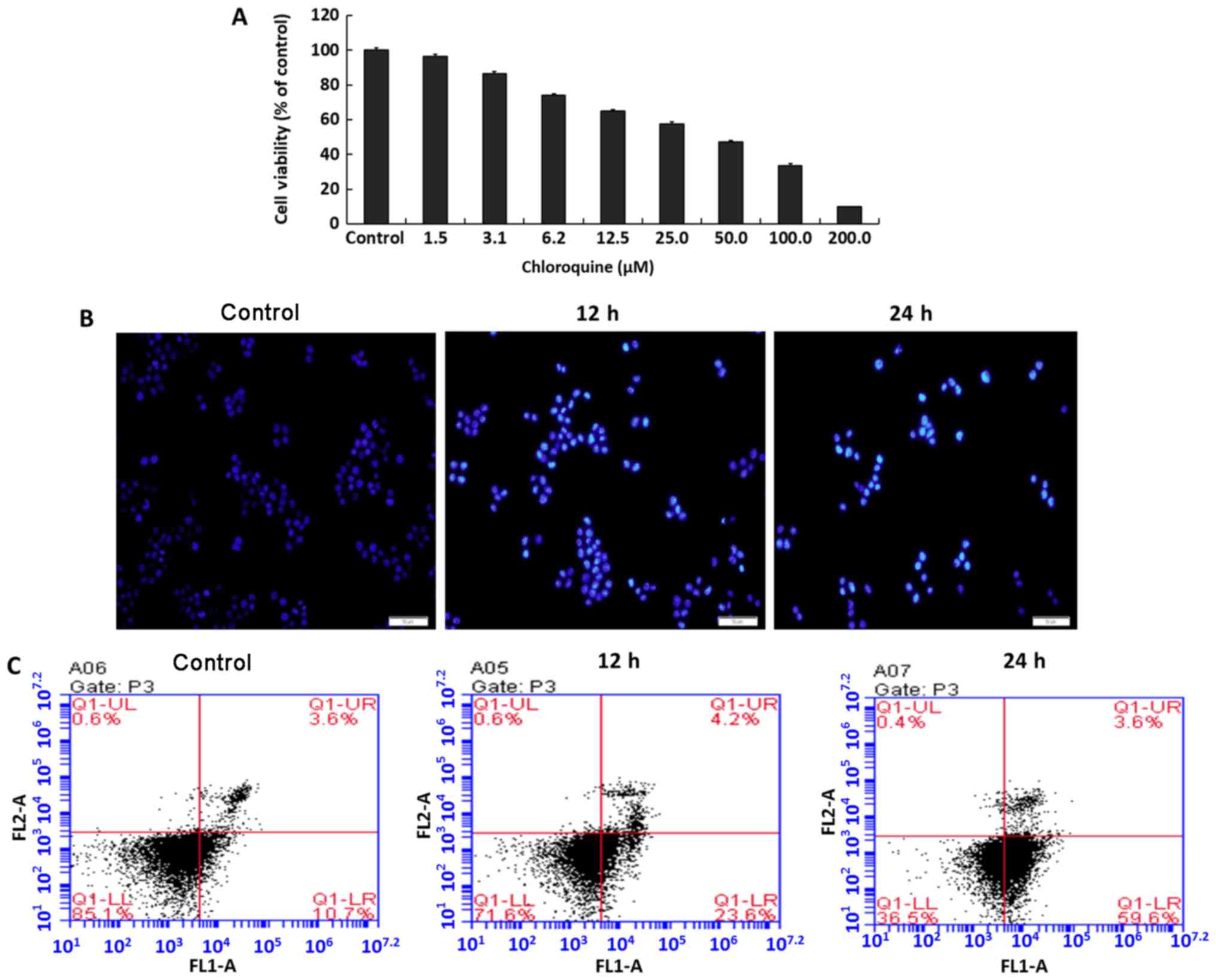
Autophagy inhibitor CQ induces
apoptosis of QBC939 cells
CQ has been frequently used as an autophagy
inhibitor. QBC939 cells died following treatment with different
doses of CQ (Fig. 1A), and the
half-maximal inhibitory concentration (IC50) was 53.01
µM (for the convenience of calculation and statistics, 50 µM was
used as the IC50 dose). However, the exact mechanism
underlying cell death caused by CQ remains unclear. Staining the
cells with Hoechst 33342 indicated typical apoptotic changes,
including nuclear fragmentation (Fig.
1B). The data produced by flow cytometry were consistent with
the fluorescence results, where the apoptosis rate of cells
gradually increased proportional to increasing CQ treatment time
(Fig. 1C).
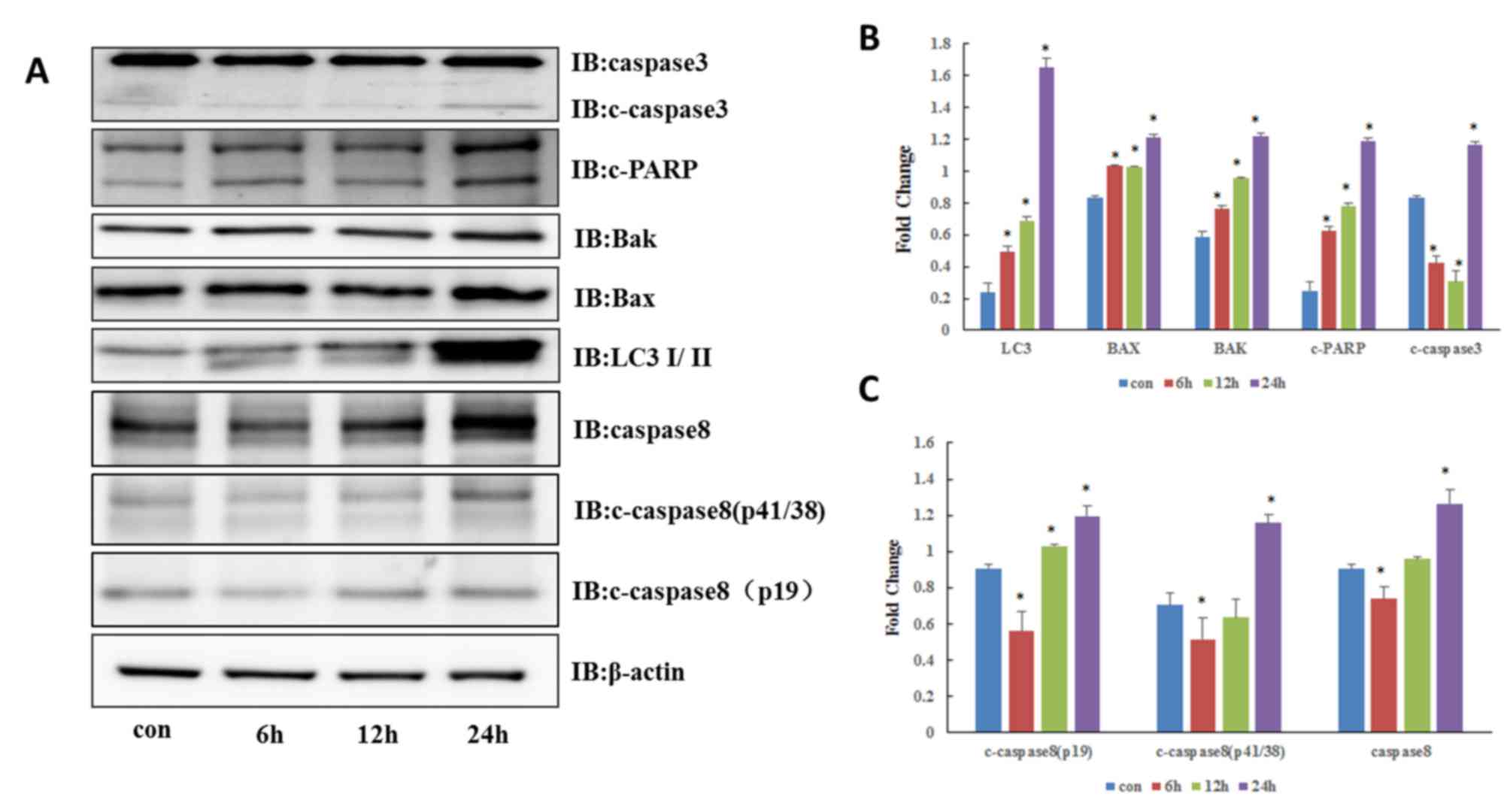
Autophagy inhibitor CQ initiates the
iDISC-associated apoptosis pathway
To verify the hypothesis that CQ induces the
apoptosis of QBC939 cells, western blotting was performed to detect
the expression of apoptosis-associated proteins. As depicted in
Fig. 2A and B, the expression of
caspase-3 increased following CQ treatment for 6, 12 and 24 h
(P<0.05). Accordingly, the level of poly(ADP-ribose) polymerase
(PARP) cleavage also increased (P<0.05). Additionally, 50 µM CQ
resulted in the notable accumulation of microtubules associated
protein light chain 3 II (P<0.05) and blocked the binding of
autophagosomes to lysosomes, resulting in the accumulation of
autophagosomes in the cytoplasm. CQ also promoted the expression of
pro-apoptotic Bcl-2 family proteins Bax and Bcl-2 antagonist/killer
(Bak) (P<0.05) and accelerated apoptosis; therefore, CQ results
in the apoptosis of QBC939 cells.
Caspase-8 is a key protease in the induction of the
extrinsic pathway of apoptosis (10).
Due to the aforementioned results indicating that CQ-induced
apoptosis may be associated with the death receptor-mediated
extrinsic apoptotic pathway, the effect of CQ on caspase-8 was
determined. As depicted in Fig. 2A and
C, activation of caspase-8 significantly increased throughout
CQ treatment 12 and 24 h (P<0.05) indicating that caspase-8 may
be involved in CQ-induced apoptosis.
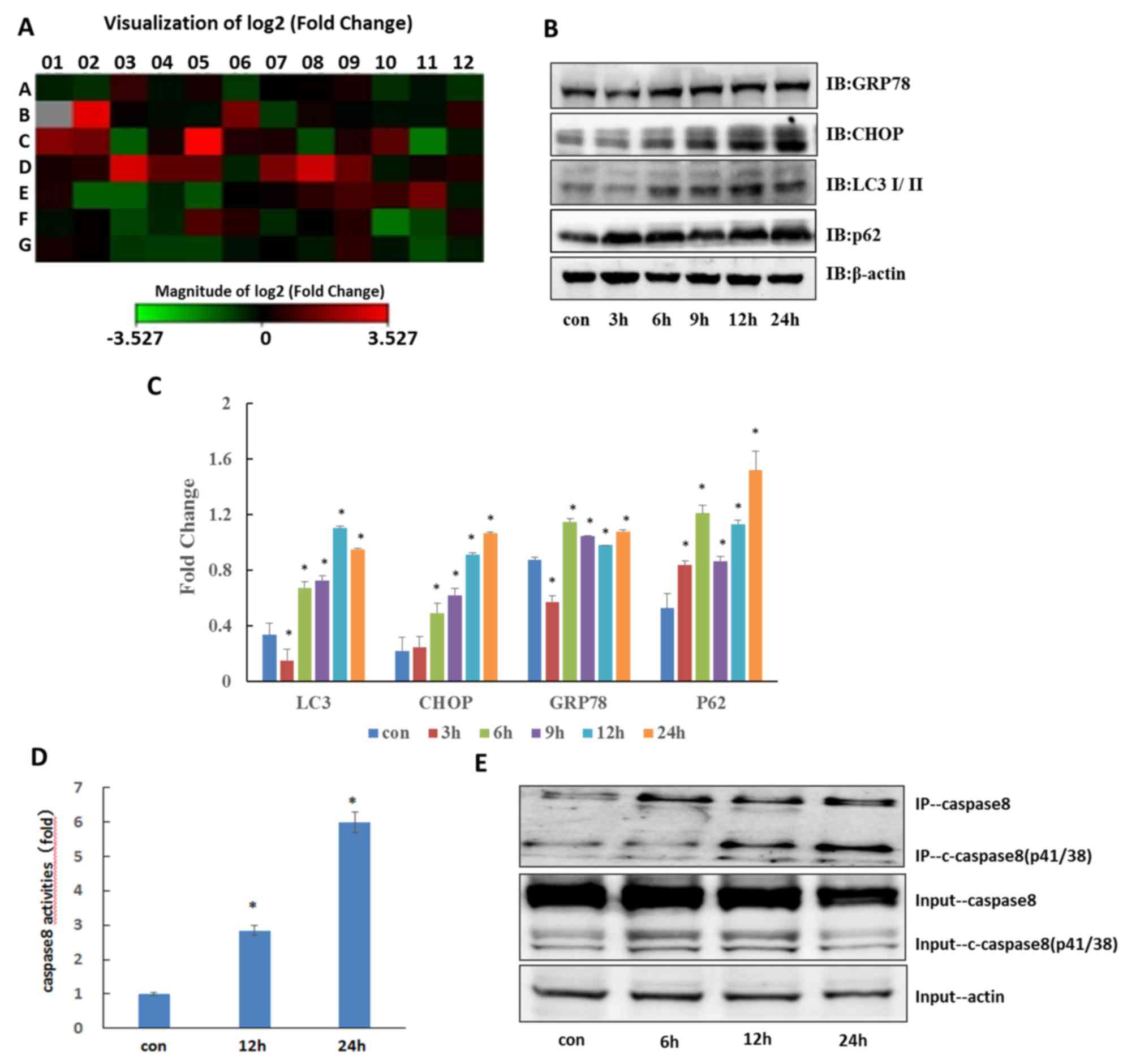
Autophagy inhibitor CQ induces ER
stress in QBC939 cells
Previous studies have demonstrated that ER stress
may be a cause of apoptosis (6,12–14); therefore, in order to examine whether
ER stress serves a key role in CQ-induced apoptosis, 84 ER
stress-associated genes were screened using a PCR Array (Table I). The results depicted in Fig. 3A demonstrated significant changes in
the expression of numerous ER stress-associated genes. Compared
with the control, the expression levels of HSPA5 (DNA damage
inducible transcript 3 and heat shock protein family A (Hsp70)
member 5) were verified to be significantly increased (Table II; P<0.05), and also demonstrated
that the protein expression of CHOP and HSPA5 were significantly
increased (Fig. 3B and C; P<0.05);
therefore, CQ induced ER stress in QBC939 cells. Particularly,
CHOP, as a downstream molecule, may serve a key role in the
mechanism of CQ-induced apoptosis.
 | Table I.84 ER stress-associated genes in PCR
array. |
Table I.
84 ER stress-associated genes in PCR
array.
|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|
| A | AMFR | ATF4 | ATF6 | ATF6B | ATXN3 | BAX | CALR | CANX | CCT4 | CCT7 | CEBPB | CREB3 |
| B | CREB3L3 | DDIT3 | DERL1 | DERL2 | DNAJB2 | DNAJB9 | DNAJC10 | DNAJC3 | DNAJC4 | EDEM1 | EDEM3 | EIF2A |
| C | EIF2AK3 | ERN1 | ERN2 | ERO1L | ERO1LB | ERP44 | FBXO6 | GANAB | GANC | HERPUD1 | HSPA1B | HSPA1L |
| D | HSPA2 | HSPA4 | HSPA4L | HSPA5 | HSPH1 | HTRA2 | HTRA4 | INSIG1 | INSIG2 | MANF | MAPK10 | MAPK8 |
| E | MAPK9 | MBTPS1 | MBTPS2 | NPLOC4 | NUCB1 | OS9 | PDIA3 | PFDN2 | PFDN5 | PPIA | PPP1R15A | PRKCSH |
| F | RNF139 | RNF5 | RPN1 | SCAP | SEC62 | SEC63 | SEL1L | SELS | SERP1 | SIL1 | SREBF1 | SREBF2 |
| G | SYVN1 | TCP1 | TOR1A | UBE2G2 | UBE2J2 | UBXN4 | UFD1L | UGGT1 | UGGT2 | USP14 | VCP | XBP1 |
| H | ACTB | B2M | GAPDH | HPRT1 | RPLP0 | HGDC | RTC | RTC | RTC | PPC | PPC | PPC |
| Table II.Upregulated genes in the polymerase
chain reaction array. |
Table II.
Upregulated genes in the polymerase
chain reaction array.
| Gene | CHOP | EIF2AK3 | ERN1 | GADD34 | HSPA5 |
|---|
| Fold-change | 9.1219 | 3.5365 | 2.8352 | 2.8954 | 2.3864 |
As an autophagy inhibitor, CQ can induce the
accumulation of the autophagy substrate p62. Johnson et al
demonstrated that ER stress can induce mTORC1-overactive cells to
produce an autophagy-dependent death-inducing signal complex iDISC
and promotes the establishment of such a death platform, a protein
complex that binds to multiple proteins during apoptosis, by
facilitating the binding between p62 and caspase-8 (10). In the present study, CQ promoted the
accumulation of p62 protein and activation of caspase-8 in QBC939
cells (Fig. 3B and C; P<0.05);
therefore, any interaction between these two factors was
investigated. Immunoprecipitation experiments demonstrated that the
binding of caspase-8 with p62 significantly increased following CQ
treatment (Fig. 3D and E; P<0.05);
therefore, it was considered that an autophagy-dependent apoptosis
mechanism was involved in CQ-induced apoptosis.
Autophagy inhibitor CQ promotes the
high expression of multiple death receptors downstream of CHOP
Previous studies demonstrated that CHOP, as a
transcription factor, has the ability to bind to a variety of death
receptor promoters, promoting their transcriptional expression
(22–24). In the present study, CQ induced ER
stress in QBC939 cells and promoted the expression of CHOP at mRNA
and protein levels (Figs. 3A and
4A; P<0.05). To determine whether
the continuous activation of CHOP resulted in the expression of
downstream death receptor genes, qPCR analysis of multiple death
receptor genes downstream of CHOP was performed. As depicted in
Fig. 4B, the gene level expression of
DCR1, DCR2, DR3, DR4, DR5 and TNFR2 significantly
increased during CQ treatment (P<0.05); however, there was no
significant change in the gene expression of TRAIL and FAS.
Therefore, CQ may initiate death receptor pathway-dependent
apoptosis by inducing CHOP, a key molecule associated with ER
stress.
 | Figure 4.Autophagy inhibitor CQ promotes high
expression of multiple DRs downstream of CHOP. (A) mRNA expression
of CHOP in QBC939 cells following treatment with 50 µM CQ
*P<0.05 vs. control. (B) Quantitative polymerase chain reaction
analysis of the mRNA expression of DR genes in QBC939 cells treated
with 50 µM CQ. *P<0.05 vs. control. DR, death receptor; CHOP,
C/EDP homologous protein; TRAIL, tumor necrosis factor-related
apoptosis-inducing ligand; FAS, Fas cell surface death receptor;
TNFR2, TNF receptor 2; CQ, chloroquine; DCR1, TNF receptor
superfamily member 10c; DCR2, TNF receptor superfamily member 10d.
DR3, TNF receptor superfamily member 25; DR4, TNF receptor
superfamily member 10a; DR5, TNF receptor superfamily member
10b. |
Discussion
CQ was initially used as an antimalarial drug, but
previous studies demonstrated that CQ has inhibitory effects on a
number of tumor types (25–27). Further investigations into the
antitumor mechanism underlying CQ demonstrated that CQ was able to
inhibit the growth of lung cancer cells by inducing the expression
of prostate apoptosis response-4 and p53 (28). In bile duct carcinoma CCKS1 and HuCCT1
cells, CQ increases cell invasion by suppressing the
autophagy-associated gene autophagy and beclin 1 regulator 1
(29). Furthermore, CQ can increase
the sensitivity of cholangiocarcinoma QBC939 cells to cisplatin by
upregulating the mitochondrial level of reactive oxygen species
(30). The present study demonstrated
that CQ reduced the viability of cholangiocarcinoma QBC939 cells.
The cells exhibited notable apoptotic changes, including nuclear
fragmentation and the increased activation of apoptotic protein
caspase-3 and its substrate PARP, and markedly increased the
apoptotic rate, indicating that the inhibitory effect of CQ on
QBC939 cholangiocarcinoma cells was achieved by inducing apoptosis.
Apoptosis is generally considered to primarily involve the
extrinsic apoptotic pathway and mitochondrial apoptosis pathway
(12,31,32). In
the present study, the expression of pro-apoptotic Bcl-2 family
proteins Bax and Bak was significantly increased following CQ
treatment of cholangiocarcinoma QBC939 cells, and the protein
expression and activity of caspase-8, which is associated with the
extrinsic apoptotic pathway, was also significantly increased.
Therefore, CQ may not trigger apoptosis through a single pathway
but rather through the co-activation of multiple apoptotic
pathways.
Inhibition of autophagy by CQ results in the
accumulation of large quantities of protein in the cytoplasm, whcih
cannot be degraded and will inevitably result in further cytotoxic
effects, including ER stress (31,33).
Whilst a low stress level assists in maintaining the normal
functions of cells, excessively high ER stress induces cell death.
CHOP is an important effector of ER stress (34). Activated CHOP, as a transcription
factor, performs numerous functions, including suppressing Bcl-2,
stimulating DR5, activating caspases, integrating mitochondrial
events and amplifying death signals (35). In models where ER stress induced the
apoptosis of colon cancer HCT116 cells and prostate cancer DU145
cells, autophagy may reduce cell vacuoles, alleviate the ER stress
and protect cells from death (36–38). In
the present study, the inhibition of autophagy in
cholangiocarcinoma QBC939 cells by CQ significantly increased the
expression of two key proteins in ER stress GRP78 and CHOP,
particularly the latter, indicating the occurrence of ER stress.
CHOP, as a transcription factor, has the ability to bind to a
variety of death receptor promoters and thus directly induce cell
death.
The expression of classic death receptor genes
including DCR1, DCR2, DCR3, DCR4, DCR5 and TNFR2 was
significantly increased in the present experimental model. Thus, CQ
may initiate the extrinsic apoptosis pathway. CQ blocks autophagy
during autolysosome formation, resulting in the accumulation of the
p62 protein. p62 and caspase-8 co-existed in QBC939 cells and their
levels increased following CQ treatment. One previous study
demonstrated that tunicamycin and thapsigargin (two classic
inducers of endoplasmic reticulum stress) are able to induce ER
stress-autophagy-dependent activation of caspase-8 in cells
deficient for caspase-9 or Bax/Bak (39). CQ treatment results in an increase in
the remaining autophagosomes in the cytoplasm. These autophagosomes
include a variety of autophagy-associated proteins, including p62,
which form iDISC, or other known stress-inducible complexes or
stressors, with caspase-8 (40,41). It is
speculated that CQ induces the establishment of a novel ER
stress-associated death platform in QBC939 cells, which results in
autophagy-dependent apoptosis.
In the present study, the cytotoxic mechanism
underlying the autophagy inhibitor CQ in cholangiocarcinoma QBC939
cells was elucidated through in vitro experiments. ER stress
and autophagy inhibition occur during the stimulation of cells by
CQ, resulting in intrinsic, extrinsic and autophagy-dependent
apoptosis. By investigating the role of CQ in the promotion of
apoptosis through regulation of the ER-autophagy network, novel
insights into the mechanism underlying CQ in antitumor therapy were
produced. Numerous studies have indicated that conventional agents
used to treat other diseases have antitumor effects, including
blocking autophagy, a process termed drug re-positioning (42–45). The
present study indicated that CQ may function synergistically with
traditional chemotherapeutics to increase apoptosis of cancer cells
by autophagy inhibition; however, there are a number of limitations
of the present study. For example, only one cholangiocarcinoma cell
line was used in the present study, and as a result it lacks
persuasiveness. Additionally, the association between the
upregulation of p53 and CHOP was not investigated. Future studies
will need to use other cell lines in order to verify the mechanism
underlying CQ and to justify the clinical application of CQ in the
chemotherapy of cholangiocarcinoma. Additionally, the role of p53
in the CQ-mediated increase of CHOP expression and apoptosis of
human cholangiocarcinoma cells will be investigated, and
experiments involving RNA interference-mediated reduction in CHOP
expression will be conducted in order to verify whether the effects
of CQ treatment are mediated by CHOP. Furthermore, the aim of
future studies will be to determine how the dose used to treat
QBC939 cells compares with the physiological dose administered to
humans. Due to there being no exact drug dose used clinically at
present, this is important for future studies to investigate.
Acknowledgments
Not applicable.
Funding
National Natural Science Foundation of China (nos.
81672948, 81772794, 81572927 and 81501982). Jilin Provincial
Research Foundation for the Development of Science and Technology
Projects (nos. 20170623021TC and 20160414005GH). Jilin Provincial
Education Department Foundation for ‘The 13th five-year’ Science
and Technology Project (nos. JJKH20170825KJ and JJKH20170834KJ).
Jilin University Bethune Plan B Projects (no. 2015222).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SLK, LYP and LYH made substantial contributions to
design. XYN, YXY, LJL, WY, GR, ZJJ and ZLC were involved in
performing the experiments, acquiring and analyzing associated
articles and data. JBX was a major contributor in designing
research program, analyzing data and writing the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Likhitrattanapisal S, Tipanee J and
Janvilisri T: Meta-analysis of gene expression profiles identifies
differential biomarkers for hepatocellular carcinoma and
cholangiocarcinoma. Tumour Biol. 37:12755–12766. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blechacz B, Komuta M, Roskams T and Gores
GJ: Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev
Gastroenterol Hepatol. 8:512–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oliveira IS, Kilcoyne A, Everett JM,
Mino-Kenudson M, Harisinghani MG and Ganesan K: Cholangiocarcinoma:
Classification, diagnosis, staging, imaging features, and
management. Abdom Radiol (NY). 42:1637–1649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matull WR, Khan SA and Pereira SP: Re:
Impact of classification of hilar cholangiocarcinomas (Klatskin
tumors) on the incidence of intra- and extrahepatic
cholangiocarcinoma in the United States. J Natl Cancer Inst.
99:407–408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roskams T: Liver stem cells and their
implication in hepatocellular and cholangiocarcinoma. Oncogene.
25:3818–3822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carew JS, Medina EC, Esquivel JA II,
Mahalingam D, Swords R, Kelly K, Zhang H, Huang P, Mita AC, Mita
MM, et al: Autophagy inhibition enhances vorinostat-induced
apoptosis via ubiquitinated protein accumulation. J Cell Mol Med.
14:2448–2459. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mushtaque M and Shahjahan: Reemergence of
chloroquine (CQ) analogs as multi-targeting antimalarial agents: A
review. Eur J Med Chem. 90:280–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fernández A, Ordóñez R, Reiter RJ,
González-Gallego J and Mauriz JL: Melatonin and endoplasmic
reticulum stress: Relation to autophagy and apoptosis. J Pineal
Res. 59:292–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson CE, Hunt DK, Wiltshire M, Herbert
TP, Sampson JR, Errington RJ, Davies DM and Tee AR: Endoplasmic
reticulum stress and cell death in mTORC1-overactive cells is
induced by nelfinavir andenhanced by chloroquine. Mol Oncol.
9:675–688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park EJ, Min KJ, Choi KS, Kubatka P,
Kruzliak P, Kim DE and Kwon TK: Chloroquine enhances TRAIL-mediated
apoptosis through up-regulation of DR5 by stabilization of mRNA and
protein in cancer cells. Sci Rep. 6:229212016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu Y, Li D, Zeng L, Wang C, Zhang L, Wang
Y, Yu Y, Liu S and Li Z: Proteasome inhibitor lactacystin enhances
cisplatin cytotoxicity by increasing endoplasmic reticulum
stress-associated apoptosis in HeLa cells. Mol Med Rep. 11:189–195.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiribau CB, Gaccioli F, Huang CC, Yuan CL
and Hatzoglou M: Molecular symbiosis of CHOP and C/EBP beta isoform
LIP contributes to endoplasmic reticulum stress-induced apoptosi.
Mol Cell Biol. 30:3722–3731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teske BF, Fusakio ME, Zhou D, Shan J,
McClintick JN, Kilberg MS and Wek RC: CHOP induces activating
transcription factor 5 (ATF5) to trigger apoptosis in response to
perturbations in protein homeostasis. Mol Biol Cell. 24:2477–2490.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moon DO, Park SY, Choi YH, Ahn JS and Kim
GY: Guggulsterone sensitizes hepatoma cells to TRAIL-induced
apoptosis through the induction of CHOP-dependent DR5: Involvement
of ROS-dependent ER-stress. Biochem Pharmacol. 82:1641–1650. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen CM, Wu CT, Chiang CK, Liao BW and Liu
SH: C/EBP homologous protein (CHOP) deficiency aggravates
hippocampal cell apoptosis and impairs memory performance. PLoS
One. 7:e408012012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Young MM, Takahashi Y, Khan O, Park S,
Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al:
Autophagosomal membrane serves as platform for intracellular
death-inducing signaling complex (iDISC)-mediated caspase-8
activation and apoptosis. J Biol Chem. 287:12455–12468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wijdeven RH, Pang B, Assaraf YG and
Neefjes J: Old drugs, novel ways out: Drug resistance toward
cytotoxic chemotherapeutics. Drug Resist Updat. 28:65–81. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He H, Ke R, Lin H, Ying Y, Liu D and Luo
Z: Metformin, an old drug, brings a new era to cancer therapy.
Cancer J. 21:70–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mudduluru G, Walther W, Kobelt D, Dahlmann
M, Treese C, Assaraf YG and Stein U: Repositioning of drugs for
intervention in tumor progression and metastasis: Old drugs for new
targets. Drug Resist Updat. 26:10–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sung B, Prasad S, Ravindran J, Yadav VR
and Aggarwal BB: Capsazepine, a TRPV1 antagonist, sensitizes
colorectal cancer cells to apoptosis by TRAIL through
ROS-JNK-CHOP-mediated upregulation of death receptors. Free Radic
Biol Med. 53:1977–1987. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He L, Jang JH, Choi HG, Lee SM, Nan MH,
Jeong SJ, Dong Z, Kwon YT, Lee KS, Lee KW, et al: Oligomycin A
enhances apoptotic effect of TRAIL through CHOP-mediated death
receptor 5 expression. Mol Carcinog. 52:85–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu M, Lawrence DA, Marsters S,
Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P
and Ashkenazi A: Opposing unfolded-protein-response signals
converge on death receptor 5 to control apoptosis. Science.
345:98–101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Burikhanov R, Hebbar N, Noothi SK, Shukla
N, Sledziona J, Araujo N, Kudrimoti M, Wang QJ, Watt DS, Welch DR,
et al: Chloroquine-inducible par-4 secretion is essential for tumor
cell apoptosis and inhibition of metastasis. Cell Rep. 18:508–519.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nitta T, Sato Y, Ren XS, Harada K, Sasaki
M, Hirano S and Nakanuma Y: Autophagy may promote carcinoma cell
invasion and correlate with poor prognosis in cholangiocarcinoma.
Int J Clin Exp Pathol. 7:4913–4921. 2014.PubMed/NCBI
|
|
30
|
Qu X, Sheng J, Shen L, Su J, Xu Y, Xie Q,
Wu Y, Zhang X and Sun L: Autophagy inhibitor chloroquine increases
sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by
mitochondrial ROS. PLoS One. 12:e01737122017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: A double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vlahopoulos S, Critselis E, Voutsas IF,
Perez SA, Moschovi M, Baxevanis CN and Chrousos GP: New use for old
drugs? Prospective targets of chloroquines in cancer therapy. Curr
Drug Targets. 15:843–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rashid HO, Yadav RK, Kim HR and Chae HJ:
ER stress: Autophagy induction, inhibition and selection.
Autophagy. 11:1956–1977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Trivedi R, Maurya R and Mishra DP:
Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells
to TRAIL-induced apoptosis through the induction of DR5 and
activation of the ROS-JNK-CHOP pathway. Cell Death Dis.
5:e14652014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mukhopadhyay S, Panda PK, Sinha N, Das DN
and Bhutia SK: Autophagy and apoptosis: Where do they meet?
Apoptosis. 19:555–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gordy C and He YW: The crosstalk between
autophagy and apoptosis: Where does this lead? Protein Cell.
3:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang S, Okamoto K, Yu C and Sinicrope FA:
p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8
aggregation/activation on the autophagosome. J Biol Chem.
288:33654–33666. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tomar D, Prajapati P, Sripada L, Singh K
and Singh R, Singh AK and Singh R: TRIM13 regulates caspase-8
ubiquitination, translocation to autophagosomes and activation
during ER stress induced cell death. Biochim Biophys Acta.
1833:3134–3144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kantari C and Walczak H: Caspase-8 and
bid: Caught in the act between death receptors and mitochondria.
Biochim Biophys Acta. 1813:558–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang H, Xie H, Pan Y, Zheng K, Xia Y and
Chen W: Plumbagin triggers ER stress-mediated apoptosis in prostate
cancer cells via induction of ROS. Cell Physiol Biochem.
45:267–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mathur A, Elmageed Abd ZY, Liu X,
Kostochka ML, Zhang H, Abdel-Mageed AB and Mondal D: Subverting
ER-stress towards apoptosis by nelfinavir and curcumin coexposure
augments docetaxel efficacy in castration resistant prostate cancer
cells. PLoS One. 9:e1031092014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Khan I, Paul S, Jakhar R, Bhardwaj M, Han
J and Kang SC: Novel quercetin derivative TEF induces ER stress and
mitochondria-mediated apoptosis in human colon cancer HCT-116
cells. Biomed Pharmacother. 84:789–799. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yoshida GJ: Therapeutic strategies of drug
repositioning targeting autophagy to induce cancer cell death: From
pathophysiology to treatment. J Hematol Oncol. 10:672017.
View Article : Google Scholar : PubMed/NCBI
|