Introduction
The TP53-induced glycolysis and apoptosis
regulator (TIGAR), which contains six coding exons and two p53
binding sites, is the protein product of a p53 target gene,
C12orf5, located on chromosome 12p13-3 (1). Although p53 has already been
established as a tumor suppressor protein, recent studies have
demonstrated that by promoting cellular metabolism and blocking
glycolysis via the TIGAR-mediated pentose phosphate pathway (PPP),
p53 is also able to control cellular metabolism. In normal cells,
this results in increased nicotinamide adenine dinucleotide
phosphate (NADPH) production, enhanced scavenging of intracellular
reactive oxygen species (ROS) and inhibition of oxidative
stress-induced apoptosis. Therefore, the activation of TIGAR by p53
promotes an antioxidant response that enables cells to survive
during stressful conditions (2–4).
However, recent studies have revealed that
deregulated TIGAR expression enhances the development of cancer by
promoting the survival of cancer cells. In breast cancer, TIGAR
expression was identified to protect the cells from undergoing
apoptosis (5). In multiple myeloma
cells, TIGAR was revealed to be necessary for the maintenance of
redox homeostasis, whereas the downregulation of TIGAR resulted in
myeloma cell death (6). In cases of
hepatocellular carcinoma, the suppression of TIGAR expression was
identified to induce apoptosis and autophagy (7). Furthermore, in a mouse model of
intestinal adenoma, TIGAR-deficient mice exhibited reduced adenoma
size and tumor burden compared with wild-type mice. Overall, no
significant difference was observed in the number of tumors, which
suggested that TIGAR is primarily involved in tumor progression,
rather than tumor initiation (4).
In addition, the reduced tumor burden was correlated with an
improved survival rate of the TIGAR-deficient mice (4). This evidence suggested that TIGAR
confers a protective function to cancer cells within multiple
tissue types.
Nasopharyngeal carcinoma (NPC) is a metastatic and
highly invasive Epstein-Barr virus (EBV)-associated cancer of the
nasopharynx. The disease is particularly prevalent in China, with
an annual incidence of up to 25 cases per 100,000 individuals
(8). At diagnosis, >60% of
patients present with advanced stages of the disease, which due to
distant recurrence or metastasis, are commonly unresponsive to
treatment (9). Therefore,
additional effective therapies are required for the treatment of
NPC.
Our previous study revealed that a novel nucleoside
analog inhibited cellular growth and induced apoptosis in NPC cell
lines via downregulation of TIGAR expression (10). A further study demonstrated that the
growth inhibitory effects of c-Met tyrosine kinase inhibitors were
ameliorated by the overexpression of TIGAR in NPC cell lines
(11). These results indicate a
significant role for TIGAR expression in the survival of NPC cells.
However, functional studies examining the role of TIGAR in NPC are
lacking. The present study sought to investigate the expression
pattern of TIGAR in NPC tumor tissues, and to analyze the
consequences of TIGAR overexpression and knockdown on NPC cell
growth and invasion.
Materials and methods
Antibodies
The antibodies used in the present study were rabbit
anti-human TIGAR polyclonal antibody (cat no. ab37910, dilution,
1:8,000; Abcam, Cambridge, UK), rabbit anti-human fibronectin
polyclonal antibody (cat no. sc-9068, dilution, 1:2,000; Santa Cruz
Biotechnology Inc., Dallas, TX, USA), mouse anti-pig vimentin
monoclonal antibody (cat no. V6389, dilution, 1:1,000;
Sigma-Aldrich, St. Louis, MO, USA), mouse anti-chicken actin
monoclonal antibody (cat no. MAB1501, dilution, 1:100,000; Merck
Millipore, Darmstadt, Germany), goat anti-mouse IgG polyclonal
antibody HRP conjugate (cat no. 170-6516, dilution, 1:10,000;
Bio-Rad Laboratories, Hercules, CA, USA) and goat anti-rabbit IgG
polyclonal antibody HRP conjugate (cat no. 81-6120, dilution,
1:10,000; Thermo Fisher Scientific, Waltham, MA, USA).
Immunohistochemistry (IHC) staining
In total, 36 formalin-fixed, paraffin-embedded
specimens of undifferentiated NPC, with adjacent normal epithelium,
were retrieved from the archives of the Department of Pathology,
Queen Elizabeth Hospital (Hong Kong, China).
The 4-μm thick, formalin-fixed, paraffin-embedded
serial tissue sections were cut, and antigen retrieval was
performed at 100°C for 25 min using Bond Epitope Retrieval Solution
2 on the Bond-max automated immunostainer (Leica Microsystems,
Wetzlar, Germany). The immunostaining was performed using a polymer
detection system in the immunostainer with a rabbit polyclonal
TIGAR antibody (1:500 dilution; Abcam), according to the
manufacturer’s instructions. Lymphoid cells were used as internal
positive controls for TIGAR expression, and negative controls were
constructed by replacing the antibody with Tris-buffered saline.
The stained slides were analyzed in five fields using a light
microscope (Leica DMLS; Leica Microsystems) at ×400 magnification.
The two independent observers were without knowledge of the
clinical outcomes, and in the case of a disagreement, a consensus
was reached following thorough discussion and slide examination
using a multi-headed microscope (Leica DMLS; Leica Microsystems).
In total, ~250 cells were counted in each field, and therefore at
least 1,250 cells were counted for each tissue specimen. All the
slides were scored semi-quantitatively and expressed as an IHC
score by multiplying the percentage of positive cells by the
staining intensity, as previously described (12). The staining intensity was scored as
follows: 0, negative; 1, weak; 2, moderate; 3, strong; and 4, very
strong. The IHC score ranged from 0 to 400.
Cell lines
The HONE-1 NPC cell line was derived from patients
with poorly-differentiated NPC (13,14).
The HONE-1-EBV cell line was derived from the introduction of the
EBV genome into the HONE-1 parental NPC cell line (15). The prototype latent membrane protein
1 (LMP1) was cloned from a B95.8 cell line. All the NPC cell lines
were maintained in RPMI-1640 medium supplemented with 10% fetal
bovine serum (Thermo Fisher Scientific), 100 U/ml penicillin, 100
μg/ml streptomycin and 1 mm sodium pyruvate (Thermo Fisher
Scientific). The cells were cultured at 37°C with 5% CO2
in a cell culture incubator. The HONE-1-EBV cell line was
maintained in a selection media containing 400 μg/ml G418 reagent
(Thermo Fisher Scientific). The NP460 hTert cell line (obtained
from Professor S.W. Tsao, Department of Anatomy, University of Hong
Kong, Hong Kong, China) was maintained in a 1:1 dilution of Epilife
medium and defined keratinocyte serum-free medium (KSFM; Thermo
Fisher Scientific). The Het-1A cells were obtained from the
American Type Culture Collection (Manassas, VA, USA), and
maintained in the KSFM supplemented with 25 μg/ml bovine pituitary
extract and 0.15 ng/ml epidermal growth factor (Thermo Fisher
Scientific). The HONE-LMP1 TIGAR-expressing cell line was
established by the transient co-transfection of the
TIGAR-overexpressing plasmid, or the respective control (OriGene,
Rockville, MD, USA), into the parental HONE-1-LMP1 cell line using
a pcDNA3.1(+) vector (Thermo Fisher Scientific). The cell line was
then subjected to G418 selection for 3 months for single clone
development, and maintained in a selection media containing 400
μg/ml G418 (Thermo Fisher Scientific).
Western blotting
The cell lysates were prepared as previously
described (16). For the
preparation of the NPC tumor biopsy lysates, six frozen
endoscopy-guided biopsies, obtained from treatment-naïve NPC
patients, were collected at the time of diagnosis. The patients
consented to tissue collection for research purposes at the Tumor
Bank, Department of Clinical Oncology, The Chinese University of
Hong Kong (Hong Kong, China) according to the approved Ethics
Approval of Research Protocol. The frozen tumor samples were
homogenized using a pellet-pestle, disposable, cordless, hand-held
homogenizer (Sigma-Aldrich) on ice in a western lysis buffer
containing 1.25 mm DTT, 5 mm phenylmethanesulfonylfluoride, 30
μg/ml leupeptin and 30 μg/ml aprotinin. In total, 50 μg protein was
subjected to SDS-PAGE and immunoblotting, as previously described
(17). Actin was used as the
loading control.
Plasmid, siRNA and transfection
The pCMV-XL5 plasmid was used as the control vector
for stable clone development in the transfection experiments. The
HONE-1-LMP1 cells were plated at a density of 0.8×105
cells per well in a 10-cm2 plate. After 24 h, the cells
were co-transfected using the pcDNA3.1(+) vector with 10 μg
pCMV-XL5- or TIGAR-overexpressing plasmids (OriGene). Subsequent to
48 h of transfection, the cells were subjected to 400 μg/ml G418
selection for 3 months for clone development. The HONE-1-LMP1 cells
were plated at a density of 1.2×105 cells per well in a
6-well plate. After 24 h, the cells were transfected with 20 nM
TIGAR or negative control siRNA (GE Healthcare Dharmacon, Inc.,
Lafayette, CO, USA), using Lipofectamine 2000 (Thermo Fisher
Scientific). Subsequent to 48 h of transfection, the cells were
harvested for western blotting to confirm TIGAR-knockdown, and for
the cell counting and Matrigel invasion assays.
Cell viability assay
The viable cell number of the stable clone- and
siRNA-transfected cells was determined by a trypan blue exclusion
assay. The cells were harvested and the cell number was determined
by counting with 50% trypan blue (Thermo Fisher Scientific) on a
hemocytometer. The experiments were performed at least three times,
and triplicate wells were counted in each experiment.
Intracellular NADPH determination
The cellular NADPH production was determined using
the EnzyChromTM NADP+/NADPH assay kit
(Bioassay Systems, Hayward, CA, USA), as previously described
(10). The protein concentration of
the samples was determined using protein quantification, as
previously described (17). The
NADPH concentration was normalized to the total protein, and
presented as μM/min/mg total protein. In total, at least three
independent experiments were performed.
Matrigel invasion assay
Matrigel-coated Boyden inserts, with a pore size of
8 μm, were used for the invasion assay (BD Biosciences). The cells
were seeded into the upper chamber at a density of 7×104
cells and maintained in serum-free medium. The cell-containing
chamber was immersed in a lower chamber containing complete medium.
The cells were incubated for 24 h at 37°C in a 5% CO2
incubator. The non-invaded cells, which remained in the upper
chamber, were removed with a cotton swab. The invaded cells were
then stained with 1% toluidine blue O in 1% borax (Sigma-Aldrich)
and counted under a microscope (magnification, ×200). In total, 10
random fields were counted, and each experiment was performed in
triplicate.
Statistical analysis
Statistical analyses were performed using PRISM4
software (GraphPad, San Diego, CA, USA). P-values were obtained
using an unpaired t-test with Welch’s correction. P<0.05 was
considered to indicate a statistically significant difference.
Results
TIGAR is overexpressed in NPC tumors and
cell lines
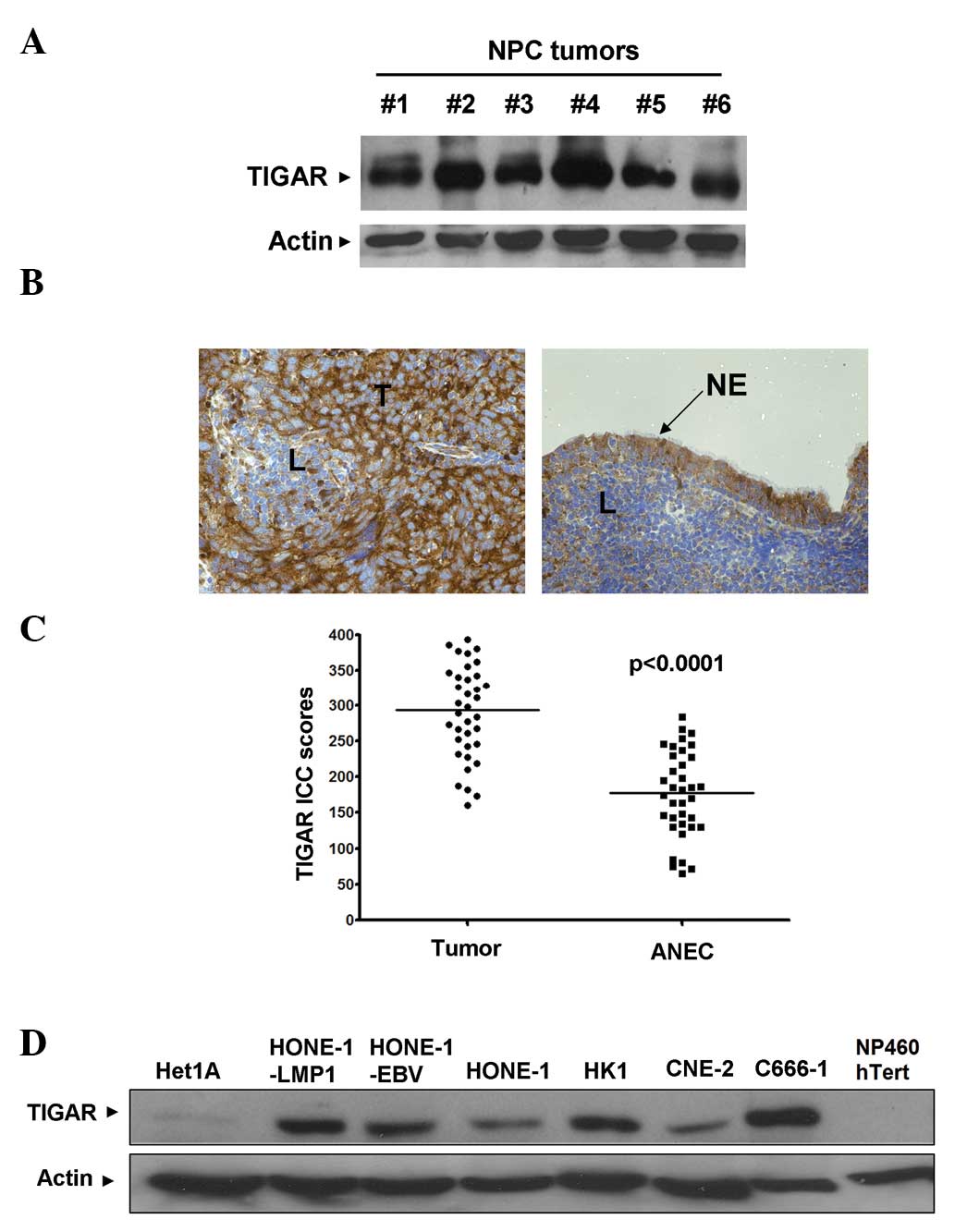
TIGAR protein expression was detected by western
blotting in all the NPC tissue samples (Fig. 1A). In order to gain an understanding
of the expression pattern of TIGAR in NPC tumors, IHC was used. Of
the 36 NPC specimens, 27 (75%) demonstrated higher TIGAR IHC scores
compared with the respective adjacent normal epithelial cells
(ANECs) (Fig. 1B and C). With
respect to the remaining nine NPC specimens, five (13.9%)
demonstrated similar TIGAR IHC scores (±5) and 4 (11.1%) exhibited
lower TIGAR IHC scores compared with the respective ANECs. In
summary, the median TIGAR IHC scores of the tumor cells and the
ANECs were 293.5 and 178, respectively. Furthermore, the
differences between the TIGAR IHC scores of the tumor and adjacent
normal cells were statistically significant (P<0.0001;
Mann-Whitney U test).
Similar to the results obtained from the IHC,
western blotting identified TIGAR overexpression across a panel of
six NPC cell lines of varying differentiation statuses (HK-1 was
from differentiated from NPC; HONE-1, HONE-1-LMP1, HONE-1-EBV and
CNE2 were from poorly-differentiated NPC; and C666-1 was from
undifferentiated NPC) compared with the normal NP460 hTert
nasopharyngeal cell line and the normal Het-1A esophageal
epithelial cell line (Fig. 1D).
TIGAR expression appeared to be increased by EBV infection or
expression of the EBV LMP1 oncoprotein, as HONE-1-EBV and
HONE-1-LMP1 expressed higher levels of TIGAR compared with the
parental HONE-1 cells.
TIGAR promotes cell proliferation in NPC
cells
The biological consequences of TIGAR upregulation in
NPCs are unclear. Therefore, to investigate the functional role of
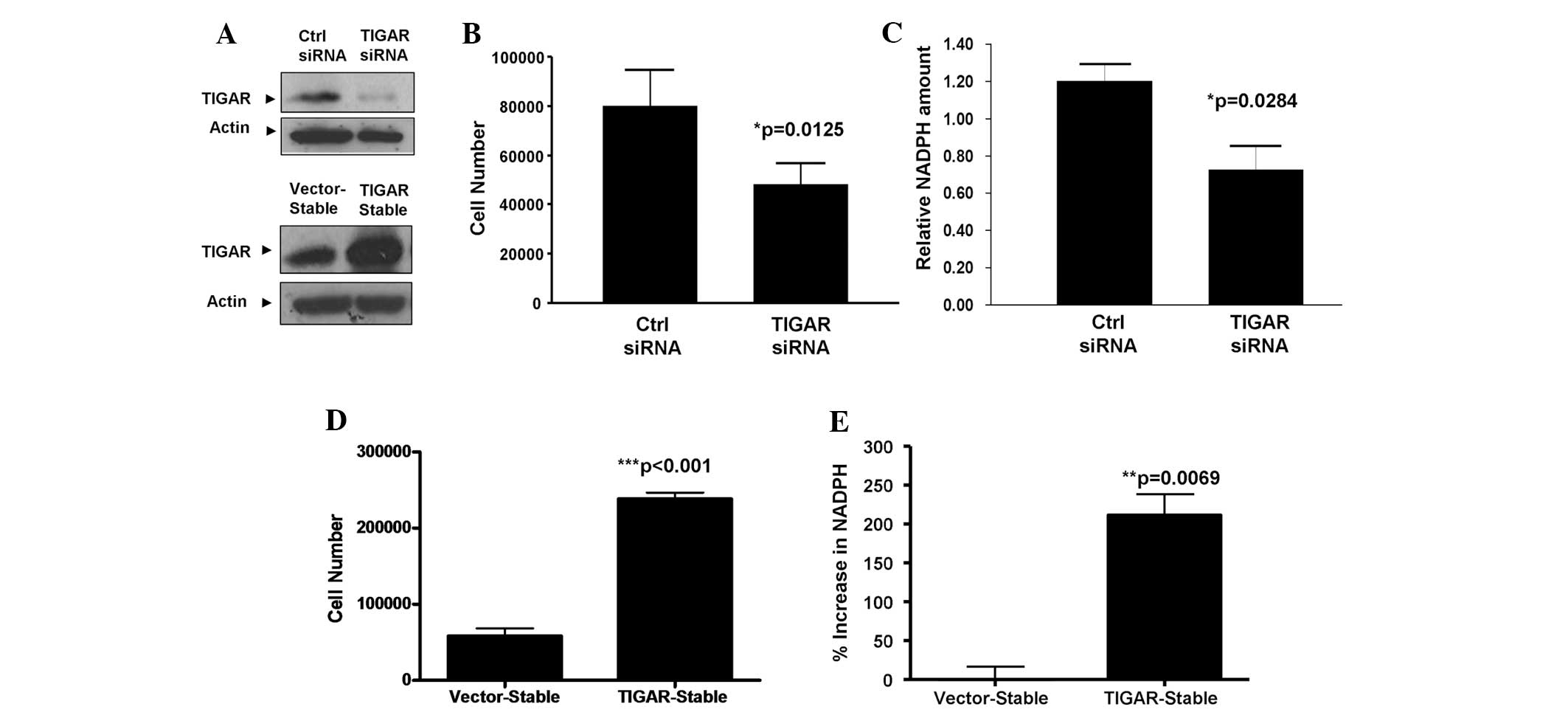
TIGAR within NPC cells, endogenous TIGAR expression was knocked
down in HONE-1-LMP1 NPC cells (Fig.
2A, upper panel). The results revealed that the knockdown of
TIGAR expression led to significant growth inhibition of the
HONE-1-LMP1 cell line (P=0.0125; unpaired t-test; Fig. 2B). To further confirm the regulatory
effect of TIGAR upon NPC cell proliferation, stable cells that
overexpressed TIGAR were created with the HONE-1-LMP1 background
(Fig. 2A, lower panel). As
demonstrated in Fig. 2C,
HONE-1-LMP1-TIGAR cells exhibited a four-fold increase in cell
growth compared with the HONE-1-LMP1-vector control cells
(P<0.001; unpaired t-test). These findings indicate that TIGAR
is capable of regulating NPC cell proliferation.
TIGAR expression promotes NADPH
production in NPC cells
TIGAR has been reported to increase the production
of cellular NADPH, an antioxidant that is required for ROS
scavenging and the inhibition of apoptosis through the PPP
(2). In order to investigate
whether TIGAR regulates NADPH production within NPC cells, TIGAR
expression was knocked down in HONE-1-LMP1 cells by siRNA
transfection. The results in Fig.
2D reveal that the silencing of TIGAR in the HONE-1-LMP1 cells
led to reduced amounts of cellular NADPH (P=0.0284; unpaired
t-test). Furthermore, the overexpression of TIGAR in the
HONE-1-LMP1-TIGAR stable cells led to a two-fold increase in NADPH
production compared with the vector-stable cells (P=0.0069;
unpaired t-test; Fig. 2E). These
results indicate that TIGAR expression is capable of promoting
NADPH production in NPC cells.
TIGAR promotes the invasiveness of NPC
cells
In order to investigate whether TIGAR promotes the
malignant properties of NPC cells, the effects of TIGAR
overexpression on the invasiveness of the HONE-1-LMP1-TIGAR NPC
cell line, compared with the vector stable cells, was examined. As
revealed in Fig. 3A, TIGAR
overexpression led to a five-fold increase in the number of NPC
cells that invaded through the Matrigel (P<0.001; unpaired
t-test). Furthermore, knockdown of TIGAR expression in HONE-1-LMP1
cells by RNAi reduced the number of cells that invaded through the
Matrigel by ten-fold (P=0.035; unpaired t-test; Fig. 3B). These findings demonstrate that
TIGAR is involved in the promotion of NPC cell invasion.
TIGAR induces the expression of
mesenchymal markers in NPC cells
During invasion and metastasis, tumor cells often
undergo a process known as epithelial-mesenchymal transition (EMT)
(18). In order to investigate
whether TIGAR-induced NPC cells also undergo this process, the
expression of several epithelial and mesenchymal markers was
examined by western blotting. Fig.
4A reveals that the expression of the mesenchymal markers,
fibronectin and vimentin, was upregulated in the HONE-1-LMP1-TIGAR
cells. This finding is consistent with the changes associated with
a mesenchymal phenotype. Conversely, Fig. 4B demonstrates that the expression of
fibronectin and vimentin was reduced upon TIGAR-knockdown.
Together, these results indicate that TIGAR promotes the expression
of mesenchymal markers in NPC cells, which may explain the
increased invasiveness of TIGAR-expressing NPC cells.
Discussion
The present study demonstrated that TIGAR expression
is upregulated in NPC tissues and cell lines. To the best of our
knowledge, this study is the first to examine the expression of
TIGAR in NPC tissues. The significant increase in TIGAR expression
in the tumor cells compared with ANECs may indicate that TIGAR is
involved in the development of NPC. The results correspond with
those from recent studies in which TIGAR was revealed to be
involved in the tumorigenesis of intestinal cancer and
glioblastomas (4,19). This evidence suggests that TIGAR is
a potential oncogene involved in various cancers. Further studies,
which will include a larger cohort of specimens, are required to
validate these findings and investigate the association between
TIGAR expression and the clinical and histopathological features of
NPC. Furthermore, functional tests, performed in a number of cell
lines, are required in order to examine whether TIGAR is a
potential therapeutic target for NPC.
In the HONE-1-LMP1 EBV-related NPC cell line, the
overexpression of TIGAR promoted cellular NADPH production,
proliferation and invasion, and resulted in a concomitant
upregulation of fibronectin and vimentin expression, which was
indicative of a mesenchymal phenotype. Conversely, the knockdown of
TIGAR by siRNA led to a reduction in cellular proliferation,
invasiveness and NADPH production, and the reduced expression of
fibronectin and vimentin. Together, these findings indicate that
TIGAR promotes NPC cellular survival and invasiveness, and induces
a mesenchymal phenotype.
TIGAR is the protein product of a p53 target gene,
it exhibits fructose-2 and 6-bisphosphatase activity, and lowers
the cellular levels of fructose-2,6-bisphosphate. This inhibits
glycolysis and promotes the PPP, which increases the production of
NADPH, and reduces the expression of glutathione (2). The synthesis of biomolecules and
protection against oxidative stressors are processes necessary for
cellular survival and that rely upon the antioxidant, NADPH
(20,21). The results from the present study
revealed that the overexpression of TIGAR upregulates cellular
NADPH production. One of the major sources of oxidative stress
within cells originates from the accumulation of ROS, which have
the potential to lead to cell cycle arrest or cell death. In order
to counteract the detrimental effects of ROS, cells increase the
production of antioxidants, such as NADPH, which is a major source
of the cellular reducing capability. In line with the pro-survival
role of NADPH, the present study identified that TIGAR
overexpression increased NPC cell proliferation, while
TIGAR-knockdown inhibited NPC cell proliferation. This demonstrated
the specificity of the TIGAR-mediating growth effect in NPC cells.
These results are consistent with those from a previous study,
which identified that TIGAR prevented cell death via modulation of
PPP activity (2). In the study,
TIGAR reduced ROS levels and protected the cells from
ROS-associated apoptosis, while TIGAR-knockdown sensitized the
cells to p53-induced cell death. Consistent with the role of TIGAR
in NPC cell survival, our previous studies revealed that TIGAR
expression reversed the anti-proliferative effects of c-met
tyrosine kinase inhibition on NPC cell growth (10,11).
In summary, these findings provide significant evidence to support
the role of TIGAR in NPC cell survival.
In the present study, TIGAR overexpression promoted
NPC cellular invasion through the Matrigel, an effect that was
ameliorated by the siRNA-mediated knockdown of TIGAR. This novel
finding is significant in that distant metastases are the
predominant cause of treatment failure in patients with NPC
(8). The present study revealed
that a downregulation of TIGAR expression inhibited NPC cell
invasiveness. This finding may provide a novel therapeutic target
for the treatment of patients with advanced stages of NPC, and one
that may improve clinical prognoses. Furthermore, the regulation of
TIGAR to prevent NPC cell invasion into surrounding tissues and
prevent progression of the disease to more advanced stages may be
investigated in future studies. At present, the precise mechanisms
that underlie TIGAR-mediated NPC cell invasion are unclear. During
tumor progression, carcinoma cells lose epithelial properties and
undergo EMT to gain invasiveness. This process promotes tumor
intravasation into lymph or blood vessels, and metastasis in
distant organs (18). In cases of
NPC, invasiveness and metastasis have been associated with EMT
(22,23). In order to gain insight into the
molecular changes associated with TIGAR-induced invasion,
alterations in the expression of EMT markers were investigated
within the present study. The results demonstrated that the
overexpression of TIGAR in the HONE-1-LMP1 NPC cell line
upregulated the expression of the mesenchymal markers, fibronectin
and vimentin, whereas the silencing of TIGAR led to downregulation.
The role of TIGAR in the regulation of EMT is supported by a recent
study, which revealed that the modulation of the p53/TIGAR pathway
induced changes in the EMT status of a cervical carcinoma cell line
(24). These findings suggested
that the expression of TIGAR promoted NPC cellular invasion by
initiating changes in the EMT phenotype.
In conclusion, the present study identified that
TIGAR is overexpressed in NPC, where the protein is involved in the
promotion of cellular proliferation, NADPH production and invasion,
and in the expression of mesenchymal markers. Given that the
involvement of TIGAR within these cellular processes may promote
tumor progression, further investigations that examine how TIGAR
supports NPC tumor growth, and the associated molecular pathways,
are warranted.
Acknowledgements
This study was funded by the Research Grants
Council, Hong Kong (no. 2140717).
References
|
1
|
Lee P, Vousden KH and Cheung EC: TIGAR,
TIGAR, burning bright. Cancer Metab. 2:12014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bensaad K, Tsuruta A, Selak MA, et al:
TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell.
126:107–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bensaad K, Cheung EC and Vousden KH:
Modulation of intracellular ROS levels by TIGAR controls autophagy.
EMBO J. 28:3015–3026. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheung EC, Athineos D, Lee P, et al: TIGAR
is required for efficient intestinal regeneration and
tumorigenesis. Dev Cell. 25:463–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martinez-Outschoorn UE, Goldberg A, Lin Z,
et al: Anti-estrogen resistance in breast cancer is induced by the
tumor microenvironment and can be overcome by inhibiting
mitochondrial function in epithelial cancer cells. Cancer Biol
Ther. 12:924–938. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yin L, Kosugi M and Kufe D: Inhibition of
the MUC1-C oncoprotein induces multiple myeloma cell death by
down-regulating TIGAR expression and depleting NADPH. Blood.
119:810–816. 2012. View Article : Google Scholar :
|
|
7
|
Ye L, Zhao X, Lu J, et al: Knockdown of
TIGAR by RNA interference induces apoptosis and autophagy in HepG2
hepatocellular carcinoma cells. Biochem Biophys Res Commun.
437:300–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chan AT: Nasopharyngeal carcinoma. Ann
Oncol. 21(suppl 7): vii308–vii312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu MC and Yuan JM: Epidemiology of
nasopharyngeal carcinoma. Semin Cancer Biol. 12:421–429. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lui VW, Lau CP, Cheung CS, Ho K, Ng MH, et
al: An RNA-directed nucleoside anti-metabolite,
1-(3-C-ethynyl-beta-d-ribo-pentofuranosyl)cytosine (ECyd), elicits
antitumor effect via TP53-induced glycolysis and apoptosis
regulator (TIGAR) downregulation. Biochem Pharmacol. 79:1772–1780.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lui VW, Wong EY, Ho K, et al: Inhibition
of c-Met downregulates TIGAR expression and reduces NADPH
production leading to cell death. Oncogene. 30:1127–1134. 2011.
View Article : Google Scholar
|
|
12
|
Wong SC, He CW, Chan CM, et al: Clinical
significance of frizzled homolog 3 protein in colorectal cancer
patients. PLoS One. 8:e794812013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glaser R, Zhang HY, Yao KT, et al: Two
epithelial tumor cell lines (HNE-1 and HONE-1) latently infected
with Epstein-Barr virus that were derived from nasopharyngeal
carcinomas. Proc Natl Acad Sci USA. 86:9524–9528. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sizhong Z, Xiukung G and Yi Z: Cytogenetic
studies on an epithelial cell line derived from poorly
differentiated nasopharyngeal carcinoma. Int J Cancer. 31:587–590.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lo AK, Lo KW, Tsao SW, et al: Epstein-Barr
virus infection alters cellular signal cascades in human
nasopharyngeal epithelial cells. Neoplasia. 8:173–180. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lui VW, He Y and Huang L: Specific
down-regulation of HER-2/neu mediated by a chimeric U6 hammerhead
ribozyme results in growth inhibition of human ovarian carcinoma.
Mol Ther. 3:169–177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lui VW, Boehm AL, Koppikar P, et al:
Antiproliferative mechanisms of a transcription factor decoy
targeting signal transducer and activator of transcription (STAT)
3: the role of STAT1. Mol Pharmacol. 71:1435–1443. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wanka C, Steinbach JP and Rieger J:
Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects
glioma cells from starvation-induced cell death by up-regulating
respiration and improving cellular redox homeostasis. J Biol Chem.
287:33436–33446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fico A, Paglialunga F, Cigliano L, et al:
Glucose-6-phosphate dehydrogenase plays a crucial role in
protection from redox-stress-induced apoptosis. Cell Death Differ.
11:823–831. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tian WN, Braunstein LD, Apse K, et al:
Importance of glucose-6-phosphate dehydrogenase activity in cell
death. Am J Physiol. 276:C1121–C1131. 1999.PubMed/NCBI
|
|
22
|
Li XJ, Peng LX, Shao JY, et al: As an
independent unfavorable prognostic factor, IL-8 promotes metastasis
of nasopharyngeal carcinoma through induction of
epithelial-mesenchymal transition and activation of AKT signaling.
Carcinogenesis. 33:1302–1309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo W, Fang W, Li S and Yao K: Aberrant
expression of nuclear vimentin and related epithelial-mesenchymal
transition markers in nasopharyngeal carcinoma. Int J Cancer.
131:1863–1873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin CC, Cheng TL, Tsai WH, et al: Loss of
the respiratory enzyme citrate synthase directly links the Warburg
effect to tumor malignancy. Sci Rep. 2:7852012. View Article : Google Scholar : PubMed/NCBI
|