Introduction
Gliomas, which originate from the predominant glial
tissue in the central nervous system, are the most common types of
malignant brain tumors in adults. Patients suffering from malignant
gliomas have a life-span between 9 and 12 months after the
diagnosis of grade IV and 2 years after the diagnosis of grade III
gliomas (1–4). More than 90% of cancers show activated
telomerase, including gliomas. Telomerase activity permits cancer
cell immortalization and promotes tumorigenesis. The expression of
telomerase and its genetic variation has been correlated with
malignant glioma progression (5)
and is therefore an important enzyme to target for improving the
prognosis and treatment of gliomas (6). Three components of human telomerase
have been identified: the RNA component (hTER) (7), the telomerase-associated protein
(TEP1) (8,9), and the telomerase reverse
transcriptase (hTERT) (6,10,11).
Although both hTER and hTERT are necessary for telomerase activity,
hTERT is the major determinant of telomerase activity. hTERT, the
catalytic subunit of telomerase, is the rate-limiting step in the
activation of telomerase and is correlated with the pathological
grade and type of human glioma.
Apoptosis plays a key role in the pathogenesis of
cancers, and the genes relating to this process are of interest in
studies on cancer onset and progression. Glioblastomas pose a
challenge in neuro-oncology because of their resistance to
apoptosis and conventional therapies (12,13).
Bax and Bcl-2 are transcriptional targets of the tumor suppression
protein p53, which is responsible for the induction of cell cycle
arrest and/or apoptosis in response to DNA damage. The progression
of cancer mainly depends on the balance between pro-apoptotic
proteins, such as Bax, and anti-apoptotic proteins, such as Bcl-2
(14). The inhibition of hTERT
rapidly induces apoptosis in gastric cancer, but this effect has
rarely been reported in gliomas.
In this study, we used siRNA to downregulate hTERT
in T98G cells and investigated the effect of hTERT on T98G cell
proliferation, apoptosis and cell cycle progression. We also
explored its possible molecular mechanism.
Materials and methods
Cell culture
The human glioblastoma cell line T98G was procured
from the Huaxi Medical Center (Sichuan University, Chengdu,
Sichuan, China). T98G was derived from a human glioblastoma
multiforme tumor. We propagated T98G cells in DMEM (Gibco-BRL,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA) and antibiotics in a humidified
incubator containing 5% CO2 at 37°C.
Small-interfering RNA design and
transfection
The cDNA sequence of the human telomerase reverse
transcriptase gene (hTERT) (GenBank accession number NM_198253) was
used to design small-interfering RNA (siRNA). The specificity of
the siRNA sequence was confirmed through BLAST searches, and the
sequence did not show any homology to other known human genes. A
random coding sequence of siRNA was used as a negative control. The
specific small-interfering RNAs (siRNAs) were synthetized and
sequenced by Shanghai GenePharma Co., Ltd. (Shanghai, China). For
hTERT, the siRNA sense sequence was 5′-CGGUGUACGCCGAGACCA ATT-3′,
and the anti-sense sequence was 5′-UUGGUCUCGG CGUACACCGGG-3′. The
negative-control siRNA sense sequence was
5′-UUCUCCGAACGUGUCACGUTT-3′, and the anti-sense sequence was
5′-ACGUGACACGUUCGGA GAATT-3′. The most effective construct was
selected based on the percentage knockdown of hTERT at both the
mRNA and protein levels. For transfection, 2×105 cells
were seeded into each well of a 6-well tissue culture plate
(Costar). The next day (when the cells were 70–80% confluent), the
culture medium was aspirated, and the cell monolayer was washed
with pre-warmed sterile phosphate-buffered saline (PBS). The cells
were transfected with Lipofectamine 2000 reagent (Invitrogen) in
accordance with the manufacturer's protocol. The cells were
continuously cultured until they were harvested for analysis. The
transfection efficiency was monitored with the expression of
carboxyfluorescein (FAM) under a phase-contrast fluorescent
microscope (Olympus IX71, Japan).
Reverse transcription-polymerase chain
reaction (RT-PCR) and western blotting to examine the hTERT mRNA
and protein levels
RT-PCR and western blotting were performed to
examine the downregulation of hTERT mRNA and protein levels,
respectively, after the knockdown of hTERT. The following primer
sequences were used for the PCR amplification of hTERT: sense
strand, 5′-ATGGCTGCGTGGTGAAC TTG-3′, and antisense strand,
5′-AGGTGAGACTGGCTCT GATGG-3′. The primers were used to amplify
1,000 ng of total RNA with a single-step RT-PCR kit (Invitrogen)
with a PCR cycler (Eppendorf, Westbury, NY, USA) at an annealing
temperature of 56°C. Western blotting was performed with an hTERT
antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) to
determine hTERT protein levels as described below. Both RT-PCR and
western blotting images were quantified using Gel-Pro Analyzer
software (Media Cybernetics, Silver Spring, MD, USA).
Flow cytometry cell cycle analysis
T98G cells (1×107) were cultured in each
well of a 6-well plate to 70–80% confluence with normal culture
medium. The cell were treated with hTERT siRNA (100 nM) for 2 or 3
days, trypsinized, and stained with propidium iodide with the
Cellular DNA Flow Cytometric Analysis Reagent Set (Boehringer
Mannheim, Indianapolis, IN, USA). The cells were harvested and
fixed with 3 ml of ice-cold 70% ethanol overnight. Then, the cells
were incubated with RNase A (1 mg/ml; Sigma, St. Louis, MO, USA)
for 10 min at room temperature. The DNA was stained with propidium
iodide (50 μg/ml) for at least 1 h at 4°C, and the DNA content was
determined with flow cytometry (Beckman Coulter, San Diego, CA,
USA). The data were analyzed with CellQuest software
(Becton-Dickinson, San Jose, CA, USA).
Telomerase activity (TRAP) assay
Telomerase activity was determined with a PCR-based
telomeric repeat amplification protocol (TRAP) enzyme-linked
immunosorbent assay (ELISA) kit (Roche, Mannheim, Germany)
according to the manufacturer's protocol. In brief, T98G cells were
collected 48 h after siRNA transfection. The cells were washed
three times with cold PBS, homogenized in 200 μl cell lysis buffer,
and incubated on ice for 30 min. For the TRAP reaction, 2 μl of
cell extract was added to 25 μl of reaction mixture, and sterile
water was added to a final volume of 50 μl. PCR was then performed
as follows: primer elongation (20 min, 25°C), telomerase
inactivation (5 min, 94°C), product amplification for 30 cycles
(94°C for 30 sec, 50°C for 30 sec, and 72°C for 90 sec) and then
balance (10 min at 72°C). A total of 5 μl of PCR products was added
to a streptavidin-coated 96-well plate and hybridized to a
digoxigenin (DIG)-labeled telomeric repeat-specific detection
probe. The immobilized PCR products were detected with
peroxidise-conjugated anti-DIG antibody. After addition of the stop
reagent, the plate was assessed with a plate reader at a wavelength
of 450 nm within 30 min.
MTT assay
T98G cells were incubated in 96-well plates, with
each well containing 200 μl of medium. The cells were divided into
the three following groups: i) blank group, ii) control siRNA
group, and ii) hTERT siRNA group. The transfection of siRNAs was
performed the following day, as previously described (15). The rate of cellular proliferation
was measured every 24 h for 120 h. At the end of each time point,
20 μl of 5 mg/ml MTT (Sigma) was added to each well. Four hours
later, 200 μl of DMSO was added to the MTT-treated wells, and the
absorption at 492 nm was determined with a spectrometer. Each
experimental condition was performed in triplicate.
Apoptosis assay
Flow cytometry assays and TUNEL assays were
performed to detect cell apoptosis. A total of 1×106
cells were transfected with siRNA. At 30 h post-transfection, the
cells were harvested, washed twice with PBS, and resuspended in 200
μl Annexin V binding buffer (10 mM HEPES, 140 mM NaCl, 2 mM
MgCl2, 5 mM KCl, and 2.5 mM CaCl2, pH 7.4). A
total of 10 μl FITC-conjugated Annexin V (Beijing Biosea
Biotechnology Co., Beijing, China) was added according to the
manufacturer's protocol. After incubation for 20 min at room
temperature in the dark, another 400 μl of binding buffer was
added, and the samples were immediately analyzed using FACSCalibur.
In total, 1×104 cells were collected and analyzed with
CellQuest software. Apoptotic cells are expressed as a percentage
of total cells.
The TUNEL assay was performed to detect apoptosis as
a marker of cell death. Briefly, T98G cells were fixed with 4%
paraformaldehyde for 10 min and then incubated for 60 min with TdT.
Streptavidin-HRP was added to the samples, which were then
incubated in the dark for 30 min and incubated for 10 min with DAB.
Positively stained cells were visualized and photographed using a
microscope.
Western blotting for molecules involved
in cell apoptosis regulation in vitro
Cells were lysed in M-PER Mammalian Protein
Extraction Reagent (Pierce Biotechnology, Inc., Rockford, IL, USA)
supplemented with a protease inhibitor cocktail (Roche,
Indianapolis, IN, USA) followed by centrifugation at 12,000 rpm for
10 min. After centrifugation, the cell lysates were collected, and
the protein concentrations of the cell lysates were measured.
Proteins (10–20 μg) were resolved through SDS-PAGE and then
transferred to PVDF membranes (Bio-Rad Laboratories, Hercules, CA,
USA). The blots were incubated with primary antibodies in 3%
BSA/TBST at 4°C overnight followed by incubation with secondary
antibodies at room temperature for 1 h. The protein signals were
detected with the ECL method (16).
Statistical analysis
The mean and standard deviation (SD) were calculated
for all of the quantitative data. The results were statistically
evaluated using a one-way analysis of variance (ANOVA). The least
significant difference method was used to compare the mean values
of control or negative control siRNA-treated groups with hTERT
siRNA-treated groups. A value of P<0.05 was considered
statistically significant.
Results
Downregulation of hTERT mRNA and protein
levels in T98G cells
We initially tested three sets of siRNAs for hTERT
knockdown. The preliminary results showed one set being particular
effective (data not shown); thus, all of the experiments described
in this report were performed with this siRNA (for the sequence,
see the methods section).
The hTERT siRNA was transiently transfected into the
T98G glioma cell line. After 48 h, the hTERT mRNA and protein
levels were quantified with real-time RT-PCR and western blotting,
respectively. As shown in Fig. 1A,
hTERT siRNA transfection significantly reduced the amount of hTERT
mRNA. The level of hTERT mRNA in the hTERT siRNA-treated group was
~40% of the blank group. The control siRNA had no effect on the
hTERT mRNA level. The hTERT siRNA was also successful in knocking
down hTERT protein expression. As shown in Fig. 1B, although the β-actin internal
control showed equal loading among the three groups, the level of
hTERT protein was noticeably lower in the hTERT siRNA-treated group
compared to both the blank and the negative siRNA-treated groups,
suggesting that the hTERT siRNA treatment could effectively reduce
the hTERT protein level.
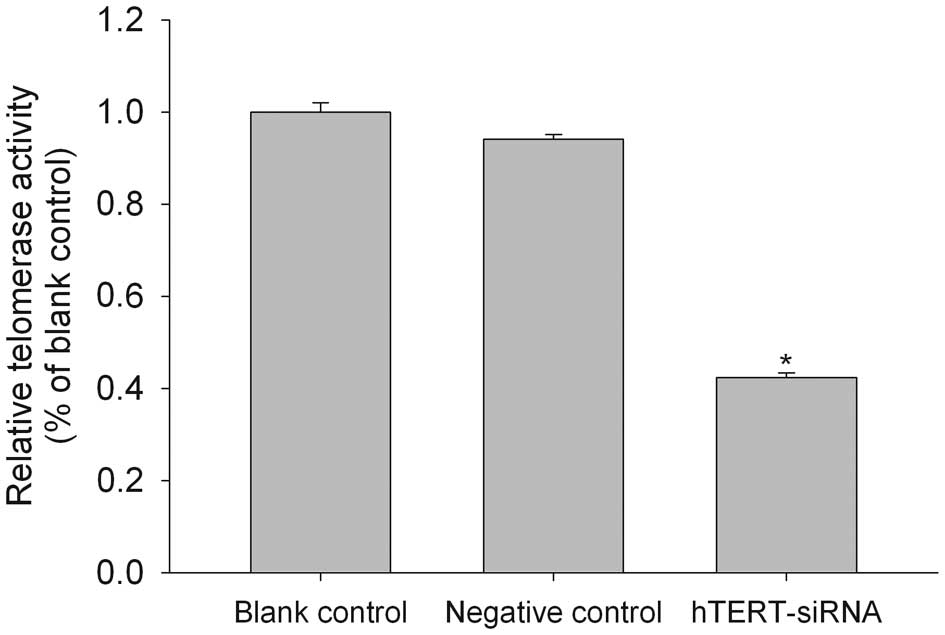
Downregulation of telomerase activity in
brain glioma cells after hTERT siRNA transfection
The level of telomerase activity in human gliomas
has been shown to correlate significantly with hTERT mRNA
expression (17). Consistent with
this result, we found that the hTERT siRNA-transfected cells showed
a 42% reduction in telomerase activity, as determined with a
PCR-based telomeric repeat amplification protocol (TRAP) ELISA
(Fig. 2).
hTERT siRNA inhibited cell viability in
vitro
Decreased telomerase activity is associated with
arrested cell growth; therefore, we determined whether the hTERT
siRNA-induced reduction in telomerase activity affected the cell
viability of the T98G cell line. The cells were transfected with
hTERT siRNA, and the number of viable cells was determined with the
MTT assay every 24 h for 4 days. As shown in Fig. 3, hTERT siRNA significantly decreased
the percentage of viable cells in the T98G cell line. The decrease
was rapid: only ~57.7% of cells were viable after 24 h, and only
47% of cells survived after 48 h. However, the inhibitory effect of
the siRNA was temporary, and the T98G cells began proliferating 3
days after the exposure to siRNA.
The effect of hTERT siRNA on the cell
cycle
To determine whether hTERT siRNA affected the cell
cycle of malignant glioma cells, flow cytometry was performed. As
shown in Fig. 4, the flow cytometry
assay showed the after transfection with hTERT siRNA, the number of
cells in G1 phase was increased, but the number of cells in S phase
was decreased. There was no alteration in the cell population in
subG1, S, or G2/M phase in the negative-control group.
The decrease in cell viability caused by
hTERT siRNA is due to an increase in apoptosis
To determine whether the decrease in cell viability
caused by the hTERT siRNA was due to an increase in apoptosis, we
determined the number of early apoptotic cells in the
untransfected, negative control- and hTERT siRNA-transfected cells
with Annexin V-FITC and propidium iodide (PI) labeling followed by
fluorescence-activated cell sorting (FACS). As shown in Fig. 5, 48 h after siRNA transfection, the
number of early apoptotic cells was increased significantly. Cell
apoptosis was then examined using a TUNEL assay. hTERT siRNA
dramatically increased the number of cells that stained positive
for TUNEL within the nucleus (Fig.
6), indicating a high incidence of apoptosis.
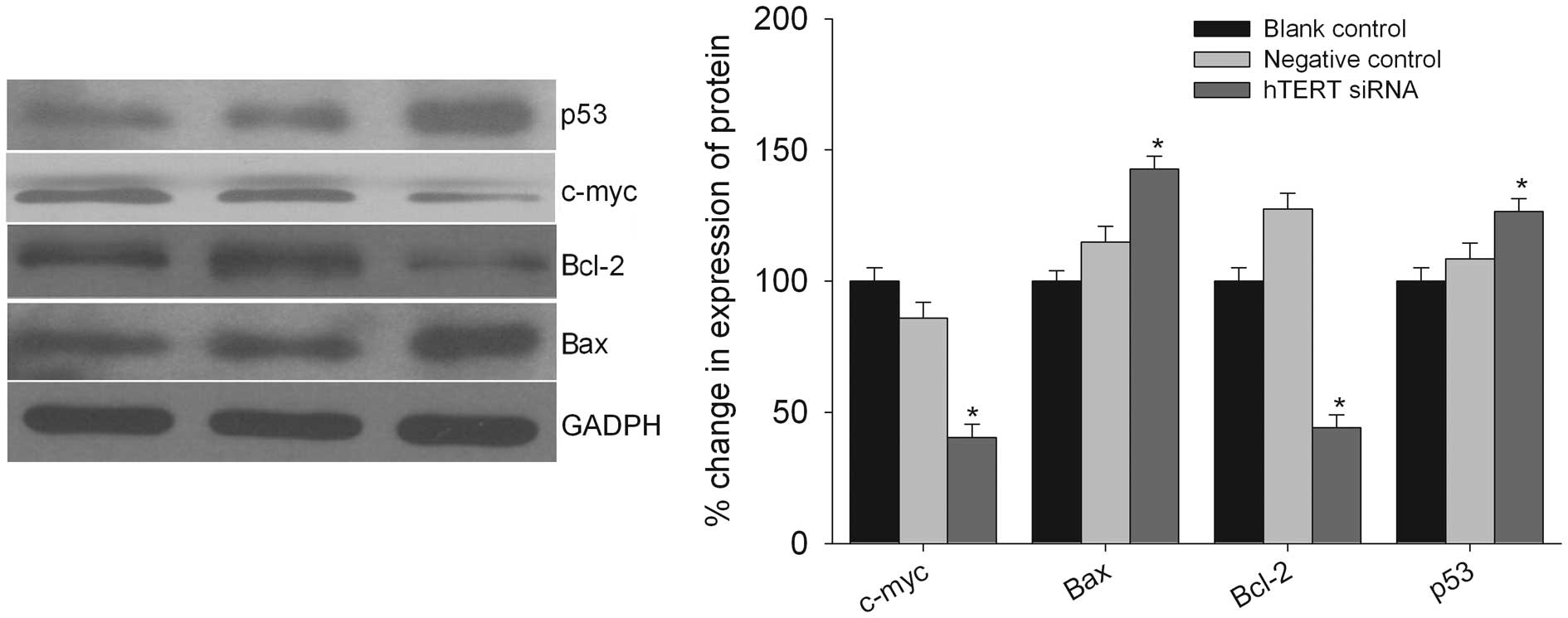
hTERT siRNA downregulates the molecules
involved in apoptosis
To confirm the molecular mechanism of the inhibition
of cell apoptosis after the downregulation of hTERT, we determined
the protein levels of the important molecules involved in this
process (Fig. 7). A western
blotting assay showed that the levels of several proteins involved
in the apoptotic pathway were different. The expression level of
Bcl-2 and c-Myc was decreased whereas the expression level of Bax
and p53 was increased after the treatment of T98G cells with hTERT
siRNA (Fig. 7).
Discussion
This study shows that in T98G glioma cells, siRNAs
targeting the hTERT gene can be efficiently delivered and results
in the rapid inhibition of telomerase activity and cell growth. The
inhibition of cell growth is associated with cell cycle arrest and
the promotion of cell apoptosis through transcriptional and/or
translational upregulation and/or downregulation of the molecules
involved in this process.
RNA interference has emerged as an effective method
for the specific inhibition of gene expression both in vitro
and in vivo. Telomerase plays a key role in cellular
immortality and tumorigenesis. Telomerase is a distinctive
candidate for the targeted gene therapy of malignant gliomas
because the vast majority of malignant gliomas express telomerase
activity, whereas normal brain tissues do not (18–20).
Telomerase and its major catalytic subunit hTERT are upregulated in
most cancers, including glioblastomas (17,21).
Moreover, hTERT expression has been correlated with poor survival
in glioblastoma patients (22).
Previous studies have demonstrated that the
downregulation of hTERT in glioblastoma cells is correlated with a
decrease in cell viability, proliferation, tumor cell migration,
and invasion through the downregulation of the molecules involved
in these processes and cell cycle inhibition (17,21).
In the present study, siRNA directed against hTERT resulted in
>70% suppression of hTERT at the mRNA and protein levels.
Furthermore, siRNA targeting hTERT significantly inhibited cell
proliferation and increased apoptosis by downregulating hTERT
expression and decreasing telomerase activity in T98G human glioma
cells.
In cancer cells, the stabilization of telomeres
through the reactivation of telomerase has been suggested to be a
crucial step during cellular immortalization and tumorigenesis.
Moreover, telomerase inhibition is associated with the induction of
apoptosis and senescence. Earlier studies have shown that the
selective silencing of hTERT using hTERT siRNA and oligonucleotides
targeting the RNA component of telomerase induces both apoptosis
and senescence in Barrett's adenocarcinoma cells (5,18). In
our present study, silencing hTERT using hTERT siRNA induced
apoptosis in T98G glioma cells.
c-Myc contributes to apoptosis via its interaction
with a number of apoptotic pathways. Pathways involving p53 and Bax
(Bcl-2-associated × protein) have been shown to be activated by
c-Myc (6). In addition, Bcl-2
suppresses c-Myc-induced apoptosis without affecting the ability of
c-Myc to regulate the progression of the cell cycle from G1 phase
to S phase. c-Myc-induced tumorigenesis is the result of the
suppression of apoptosis by cooperating oncogenes and the
activation of S phase by c-Myc, leading to cell proliferation
(23,24). siRNA-mediated c-Myc downregulation
resulted in an inhibition of cellular proliferation and clonogenic
growth, the inhibition of G1-S phase cell cycle progression, and a
decrease in human telomerase reverse transcriptase (hTERT)
expression and telomerase activity in human medulloblastoma cells
(25).
Anti-apoptotic Bcl-2 family members are highly
overexpressed in malignant gliomas. The induction of apoptosis by
downregulating hTERT expression and decreasing telomerase activity
was shown in changes in the expression levels of proteins
responsible for the regulation of apoptosis. Bax and Bcl-2 are the
two principal genes involved in the regulation of apoptosis.
Previous studies have demonstrated that during apoptosis induction,
bax protein levels are upregulated, which has a well-known
pro-apoptotic effect, Bcl-2, which protects cells from apoptosis,
is downregulated. According to our results, the anticancer cell
growth inhibition is due to the deregulation of apoptosis
induction.
The p53 tumor suppressor is another cell cycle
regulator that is frequently altered in brain tumors. During cell
DNA damage or cytotoxic stress, there is an increase in p53 protein
levels that induces cell growth arrest, DNA repair mechanisms, and
apoptosis (26–28).
In conclusion, our study demonstrated that the
knockdown of hTERT effectively inhibited the cell viability of
human glioblastoma cells by increasing the positive index of
apoptotic cells via decreasing the expression of Bcl-2 and c-Myc
and cell cycle arrest at G0/G1 phase. Therefore, hTERT siRNA offers
a potential therapeutic regimen for effectively controlling the
growth of human glioblastoma cells.
Acknowledgements
This study was supported in part by the Shaanxi
Provincial scientific and technological research projects (no.
2011K12-56).
References
|
1
|
Lino MM and Merlo A: PI3Kinase signaling
in glioblastoma. J Neurooncol. 103:417–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao P, Wang C, Fu Z, et al: Lentiviral
vector mediated siRNA knock-down of hTERT results in diminished
capacity in invasiveness and in vivo growth of human glioma
cells in a telomere length-independent manner. Int J Oncol.
31:361–368. 2007.PubMed/NCBI
|
|
3
|
Li C, Zhou C, Wang S, et al: Sensitization
of glioma cells to tamoxifen-induced apoptosis by Pl3-kinase
inhibitor through the GSK-3beta/beta-catenin signaling pathway.
PLoS One. 6:e270532011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prados MD and Levin V: Biology and
treatment of malignant glioma. Semin Oncol. 27:1–10. 2000.
|
|
5
|
Ponnala S, Chetty C, Veeravalli KK, Dinh
DH, Klopfenstein JD and Rao JS: MMP-9 silencing regulates hTERT
expression via beta1 integrin-mediated FAK signaling and induces
senescence in glioma xenograft cells. Cell Signal. 23:2065–2075.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shervington A, Cruickshanks N, Wright H,
et al: Glioma: what is the role of c-Myc, hsp90 and telomerase? Mol
Cell Biochem. 283:1–9. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng J, Funk WD, Wang SS, et al: The RNA
component of human telomerase. Science. 269:1236–1241. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Harrington L, McPhail T, Mar V, et al: A
mammalian telomerase-associated protein. Science. 275:973–977.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakayama JI, Saito M, Nakamura H, Matsuura
A and Ishikawa F: TLP1: a gene encoding a protein component of
mammalian telomerase is a novel member of WD repeats family. Cell.
88:875–884. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meyerson M, Counter CM, Eaton EN, et al:
hEST2, the putative human telomerase catalytic subunit gene, is
up-regulated in tumor cells and during immortalization. Cell.
90:785–795. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kondo S, Kanzawa T, Germano IM, Kondo Y,
Ito H and Kyo S: Inhibition of telomerase activity in malignant
glioma cells correlates with their sensitivity to temozolomide. Br
J Cancer. 89:922–929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferguson SD: Malignant gliomas: diagnosis
and treatment. Dis Mon. 57:558–569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Furnari FB, Fenton T, Bachoo RM, et al:
Malignant astrocytic glioma: genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xue Y, Li L, Zhang D, et al: Telomerase
suppression initiates PML-dependent p53 activation to inhibit
bladder cancer cell growth. Oncol Rep. 24:1551–1559.
2010.PubMed/NCBI
|
|
16
|
Zhang Y, Cheng Y, Zhang L, et al:
Inhibition of eEF-2 kinase sensitizes human glioma cells to TRAIL
and down-regulates Bcl-xL expression. Biochem Biophys Res Commun.
414:129–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
George J, Banik NL and Ray SK: Knockdown
of hTERT and concurrent treatment with interferon-gamma inhibited
proliferation and invasion of human glioblastoma cell lines. Int J
Biochem Cell Biol. 42:1164–1173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shammas MA, Koley H, Batchu RB, et al:
Telomerase inhibition by siRNA causes senescence and apoptosis in
Barrett's adenocarcinoma cells: mechanism and therapeutic
potential. Mol Cancer. 4:242005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen CH and Chen RJ: Prevalence of
telomerase activity in human cancer. J Formos Med Assoc.
110:275–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shay JW and Wright WE: Role of telomeres
and telomerase in cancer. Semin Cancer Biol. 21:349–353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
George J, Banik NL and Ray SK: Combination
of hTERT knockdown and IFN-gamma treatment inhibited angiogenesis
and tumor progression in glioblastoma. Clin Cancer Res.
15:7186–7195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang YF, Wang DL, Shi GS and Huang H:
Expressions of hTERT, HIF-1alpha and CD105 in gliomas and their
clinical significance. Zhonghua Bing Li Xue Za Zhi. 35:681–682.
2006.(In Chinese).
|
|
23
|
Packham G and Cleveland JL: c-Myc and
apoptosis. Biochim Biophys Acta. 1242:11–28. 1995.PubMed/NCBI
|
|
24
|
Lutz W, Leon J and Eilers M: Contributions
of Myc to tumorigenesis. Biochim Biophys Acta. 1602:61–71.
2002.PubMed/NCBI
|
|
25
|
von Bueren AO, Shalaby T, Oehler-Janne C,
et al: RNA interference-mediated c-MYC inhibition prevents cell
growth and decreases sensitivity to radio- and chemotherapy in
childhood medulloblastoma cells. BMC Cancer. 9:102009.PubMed/NCBI
|
|
26
|
Louis DN: The p53 gene and protein in
human brain tumors. J Neuropathol Exp Neurol. 53:11–21. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levine AJ, Hu W and Feng Z: The P53
pathway: what questions remain to be explored? Cell Death Differ.
13:1027–1036. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krakstad C and Chekenya M: Survival
signalling and apoptosis resistance in glioblastomas: opportunities
for targeted therapeutics. Mol Cancer. 9:1352010. View Article : Google Scholar : PubMed/NCBI
|