Introduction
Lung cancer is the leading cause of cancer-related
deaths for both males and famales in the US and worldwide. An
estimated 160,340 deaths, accounting for ~28% of all cancer deaths,
were estimated to occur in the US in 2012 (American Cancer Society.
Cancer Facts & Figures 2012. Atlanta: American Cancer Society;
2012). Based on histological characteristics, lung cancer is
classified clinically as small-cell (SCLC) (15%) or non-small cell
lung cancer (NSCLC) (85%) (1).
Adenocarcinoma and squamous cell carcinoma (SQC) are the most
common histological subtypes accounting for ~50 and 30% of NSCLC
cases, respectively (2). During the
past decade, several oncogenic driver mutations including EGFR,
EML4-ALK, ROS and RET have been identified in lung cancer.
Molecularly targeted therapies against the EGFR and ALK mutations
are showing success (3). Despite
these improvements in molecular diagnosis and targeted therapies,
the 5-year overall survival rate for all stages of lung cancer is
only 16% (American Cancer Society. Cancer Facts & Figures 2012.
Atlanta: American Cancer Society; 2012). The lack of substantial
improvement in cancer mortality is multifactorial. In part, this is
because the proportion of patients that carry the EGFR mutation or
ALK fusion gene are only 12 and 4% of all adenocarcinoma cases, and
the EGFR mutation is concentrated in female non-smokers. Clearly,
discovering novel oncogenic drivers in NSCLC is paramount for
improving current therapy. Recently, the fibroblast growth factor
receptor 1 (FGFR1) was identified to be frequently amplified in
squamous cell lung cancer (>20% of cases) (4,5) as
well as in small-cell lung cancer (6% of cases) (6). However, it is still unknown whether
these cells are completely addicted to FGFR1 signaling and whether
pharmacologically targeting FGFR1 is effective in inhibiting cancer
growth.
FGFR1 is a transmembrane tyrosine kinase receptor
that plays an important role in cell growth, differentiation and
survival among other functions (7).
FGFR1 activation leads to downstream signaling via the PI3K-AKT,
RAS-MEK-MAPK, STAT, PLCγ, as well as the Src signaling pathway
(8,9). Recently, ponatinib, also known as
AP24534, has been proven effective against FGFR1 kinase (10). Indeed, it proved to be a potent
inhibitor of leukemogenesis induced by the FGFR1 fusion kinases
(11,12). However, it is still not clear
whether ponatinib potently inhibits NSCLC cell growth or blocks
progression of tumors with FGFR1 overexpression. In the present
study, we found that approximately half (30 out of 59) of all NSCLC
cases showed a >2-fold increase in transcriptional activity of
FGFR1 compared with their adjacent ‘normal’ tissue counterparts by
using quantitative RT-PCR, indicating that not only genomic
amplification of FGFR1 but also transcriptional overexpression of
FGFR1 is a relative common molecular signature in NSCLC. We further
demonstrated that ponatinib can effectively and specifically
suppress cell growth in lung cancer cell lines as well as in
primary lung cancer cell cultures with overexpression of FGFR1.
This growth inhibition by ponatinib is associated with inactivation
of FGFR1 and its downstream targets. Our data suggest that
ponatinib is a potent drug for targeted therapy in lung cancer
patients with amplification of FGFR1, and provide a rationale for
further evaluation preclinically and clinically.
Materials and methods
Patient samples
Human tissue samples were obtained from the
biorepositories of the Georgia Health Sciences University Cancer
Center and Shanghai Pulmonary Hospital that collect anonymized
samples from cancer and non-cancer patients for research purposes
following protocols approved by the Georgia Health Sciences
University Human Assurance Committee (GHSUHAC) and the Tongjin
University Institution Review Board (TUIRB). All patients signed
written consents documenting donation of their tissue for research
purposes prior to tissue deposition. The 88 tumor samples together
with matched adjacent normal specimens were from lung cancer
patients who had undergone resection of lung cancer at the Shanghai
Pulmonary Hospital (Tongji University School of Medicine, Shanghai,
China) in 2009. All tumors and their paired normal tissue samples
were snap frozen and stored at −80°C until assayed after
histological confirmation. Human lung cancer tissue samples used
for isolation of primary lung cancer cell cultures were obtained
from the Georgia Health Sciences University, Cancer Center Tumor
Tissue and Serum Repository after de-identification.
Published microarray data set
Global gene expression profiles of lung cancer cell
lines (n=89) were downloaded and isolated from published microarray
data sets from the cancer cell line project (950 assays, E-MTAB-37;
European Bioinformatics Institute). The global gene expression
levels from these lung cancer cell lines compared with the normal
Wi38 human fetal lung fibroblast cells and normal human lung tissue
were re-analyzed using the GenePattern software program (http://www.broadinstitute.org) as described in detail
elsewhere (13).
Cell culture and proliferation
assays
All cell lines were purchased from the American Type
Culture Collection (ATCC) and cultured in RPMI-1640 (Invitrogen)
with 5% FBS (Hyclone), at 37°C in 10% CO2. For drug
treatments, 3,000–5,000 cells/well, dependent on the cell lines,
were seeded in 96-well plates and incubated overnight. Cells were
then treated with either DMSO (control) or ponatinib at various
concentrations. Cell viability was determined using the
CellTiter-Glo® luminescence cell viability kits
(Promega) and a SpectraMax® M5e (Molecular Probes)
luminescence plate reader.
Cell apoptosis assay and cell cycle
analysis
For analysis of apoptosis, cells were plated in
6-well plates and treated with ponatinib for 72 h. The cells were
then washed with PBS, stained with Annexin V-APC and 7-AAD (BD
Biosciences) following the manufacturer’s instructions. Appearance
of Annexin V and 7-AAD in the flow cytometric analysis indicated
onset of apoptosis. Cell cycle analysis was performed using
standard flow cytometric procedures following either propidium
iodide staining alone, or together with bromodeoxyuridine (BrdU)
incorporation. These cells were then stained with anti-BrdU-APC
(eBioscience), as described previously (14). Cells were analyzed using an LSRII
flow cytometer. All flow cytometric data were analyzed using FlowJo
software (Tree Star, Inc., Ashland, OR, USA).
Western blot analyses
Proteins were isolated using standard procedures.
Whole-cell lysates containing 50 μg of proteins, were separated by
SDS-PAGE and immunoblotted with the following specific antibodies:
anti-FGFR1 and GAPDH (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), anti-phosphoFGFR1 (Abcam), anti-Src and anti-pSrc (Cell
Signaling Technologies, Danvers, MA, USA) and β-actin (Sigma, St.
Louis, MO, USA) using standard protocols as described previously
(15).
Immunocytochemical staining
Immunocytochemical analysis was performed as
described previously (16).
Briefly, the cells were grown on sterile glass coated coverslips,
and treated with or without 1.0 μM ponatinib for 1 h, fixed and
permeablized. Cells were first blocked with 3% ovalbumin for 30 min
at 25°C. They were then stained with 1:50 primary rabbit polyclonal
anti-phospho-FGFR1 (Y653/654; Abcam) antibody at 4°C overnight. The
cells were then washed with PBS and incubated further with the
Cy2-conjugated goat anti-rabbit IgG (Abcam) secondary antibody
(1:200) for 30 min at 37°C. After two more washings, the slides
were counterstained with Hoechst 33342 DNA dye before mounting. The
images were captured using a Zeiss fluorescent microscope.
Molecular analyses
Total RNA was isolated using TRIzol (Invitrogen)
reagent, and digested with RNA-free DNase to eliminate genomic DNA
contamination. Reverse-transcription was achieved using a
SuperScript first-strand synthesis system (Invitrogen), and
amplified by PCR or quantitated by real-time RT-PCR using
conditions described previously (13).
Clonogenicity assay
Cells (500–1,000/well) were seeded in 6-well plates,
allowed to attach overnight to the plastic substrate before
treatment with ponatinib (1.0 μM) or vehicles (controls). After 48
h treatment, the media were replaced with drug-free media for the
desired length of time. As the colonies became visible (usually 2–3
weeks), cells were fixed with methanol, stained with Giemsa (1:10
in distilled water), and counted.
shRNA knockdown of FGFR1
Retroviral shRNAs for FGFR1 were purchased from
Thermo Scientific (catalog nos. RHS3979-9568791, 3979-98488862 and
3979-9568788, targeting different exons of FGFR1). shRNA-GFP was
used as a control. Retroviral supernatants (≥5×106
CFU/μl) were generated and introduced into cells as described
previously (17), and puromycin was
used for selection of clones where needed.
Statistical analysis
Data from the cell proliferation assay and
clonogenicity assay are presented as means ± SD. Differences
between groups were analyzed using the Student’s t-test for
independent samples. The level of significance was set at
p<0.05.
Results
Transcriptional overexpression of FGFR1
is frequently observed in both lung cancer cell lines and human
NSCLC tumor samples
To initiate the study, we first evaluated published
global gene expression array datasets from the transcriptomics of
cancer cell line project (950 assays, E-MTAB-37, European
Bioinformatics Institute). This dataset contains triplicate
expression profiles of various cancer cell lines. Using the
GenePattern software program (http://www.broadinstitute.org) we re-analyzed data for
lung cancer cell lines and found that FGFR1 was overexpressed in
38% (35 out of 89) of lung cancer cell lines compared with Wi38
normal human fetal lung fibroblast cells and normal human lung
tissue (Fig. 1A). To determine
whether overexpression of FGFR1 also occurs in primary human lung
cancers, we identified 88 NSCLC samples with matched normal lung
tissues resected from lung cancer patients at the Shanghai
Pulmonary Hospital. After total RNA isolation, we identified 59
pairs of high quality matched RNA samples suitable for further
analysis. The clinical features of these 59 NSCLC patients are
presented in Table I. We used
real-time RT-PCR to quantify human FGFR1 mRNA levels and
demonstrated that 30 out of 59 (50.8%) tumors showed a >2-fold
increase in FGFR1 mRNA expression compared with their adjacent
normal counterparts (Fig. 1B). Five
(8.5%) tumors showed lower expression levels compared with the
matched normal lung tissues (Fig.
1B). We did not observe a significant difference between
smokers and non-smokers (Table
I).
 | Table IClinical characteristics of
patients. |
Table I
Clinical characteristics of
patients.
| No. of
specimen | Gender | Age | Smoker | TNM stage | Location,
histology, differentiation | FGFR1 level |
|---|
| 9 | M | 67 | N | Ib | LLL, SQC,
moderate | Down |
| 10 | M | 74 | Y | Ia | RUL, SQC,
moderate | UNC |
| 11 | M | 57 | Y | IIIa | RLL, SQC,
moderate | UNC |
| 14 | M | 59 | N | Ib | LLL, ADC, well | UNC |
| 15 | M | 53 | Y | IIIa | RML, SQC,
moderate | Down |
| 16 | M | 57 | N | IIIA | RLL, ADC, poor to
moderate | UNC |
| 18 | M | 55 | Y | IIb | RUL, ADC, well | UNC |
| 20 | M | 55 | Y | Ib | LLL, SQC,
moderate | UNC |
| 21 | M | 58 | Y | IIa | LLL, SQC,
moderate | UNC |
| 22 | M | 72 | Y | Ib | RUL, SQC | Up |
| 23 | M | 71 | N | Ib | RUL, ADC, moderate
to well | UNC |
| 26 | F | 55 | N | IIb | LLL, ADC,
moderate | Up |
| 27 | M | 74 | Y | Ib | LUL, SQC, well | Up |
| 28 | M | 75 | Y | IIb | RUL, SQC, poor | UNC |
| 29 | F | 57 | N | Ib | RLL, ADC,
moderate | Up |
| 30 | M | 78 | N | IIb | RLL, ADC, well | UNC |
| 31 | F | 74 | N | Ib | RML, ADC, well | Down |
| 32 | M | 59 | N | Ib | RUL, ADC, poor to
moderate | Up |
| 34 | M | 59 | N | IIIb | LUL, ADC | Up |
| 35 | F | 63 | N | Ib | LUL,
adenosquamous | UNC |
| 37 | M | 65 | Y | IIIa | LLL, ADC, poor to
moderate | Up |
| 38 | M | 47 | N | N/A | LUL, TB | Up |
| 39 | M | 67 | Y | IIIb | LUL, SQC, well | Up |
| 40 | M | 63 | Y | Ib | LUL, ADC, poor to
moderate | Up |
| 49 | F | 63 | N | IIIa | RUL, ADC,
moderate | Down |
| 51 | M | 77 | Y | IIIa | LLL, ADC, moderate
to well | Up |
| 52 | M | 74 | Y | IIIa | RML, RLL,
adenosquamous | UNC |
| 58 | M | 45 | Y | IIb | LUL, ADC, partially
mucinous | UNC |
| 59 | M | 59 | N | IIIa | RUL, ADC, poor to
moderate | UNC |
| 61 | F | 62 | N | IIIa | RLL, ADC, poor to
moderate | Up |
| 62 | M | 71 | Y | IIIb | LUL, AQC,
moderate | UNC |
| 63 | F | 66 | N | Ib | RLL, mucinous | Up |
| 64 | F | 38 | N | IIb | LLL, mucinous with
bronchioalveolar | UNC |
| 65 | M | 69 | Y | IIIa | RML, RLL, SQC,
moderate to well | Up |
| 66 | M | 63 | N | Ib | LUL, SQC, poor to
moderate | UNC |
| 67 | M | 73 | Y | Ib | LUL,
adenosquamous | Up |
| 68 | F | 64 | N | Ib | RUL, sarcoma | Up |
| 69 | M | 68 | Y | IIb | LUL, SQC,
moderate | Up |
| 74 | M | 63 | Y | N/A | RML, nasopharyngeal
SQC | Up |
| 75 | M | 49 | Y | IIa | LLL, ADC, poor to
moderate | Up |
| 76 | M | 53 | N | IIb | RLL, ADC, poor to
moderate | Up |
| 77 | F | 54 | N | IIb | LUL, SQC,
moderate | Up |
| 78 | M | 78 | N | Ib | RLL, SQC, poor | Up |
| 79 | F | 52 | N | IIIa | RML, large
cell | Up |
| 81 | M | 62 | N | N/A | RUL, TB | UNC |
| 82 | F | 75 | Y | IIIa | LUL, SQC,
moderate | UNC |
| 83 | M | 45 | Y | IIIb | LUL, SQC,
moderate | Up |
| 84 | M | 59 | Y | IV | RLL, atypical
carcinoid | Up |
| 85 | F | 38 | N | N/A | Mediastinal mass,
lymphoma | UNC |
| 86 | M | 61 | Y | IIIa | RLL, SQC, poor to
moderate | UNC |
| 87 | M | 63 | Y | Ib | LLL, ADC, moderate
to well | Up |
| 88 | M | 78 | N | Ib | LLL, SQC,
moderate | UNC |
| 89 | F | 53 | N | Ib | LUL, SQC,
moderate | Up |
| 90 | F | 59 | N | N/A | RUL, TB | UNC |
| 91 | M | 62 | Y | IIIa | RML, SQC,
moderate | UNC |
| 92 | M | 63 | N | Ib | RLL, SQC, well | UNC |
| 93 | M | 70 | Y | Ib | RML,
sarcomatoid | Up |
| 94 | M | 39 | Y | Ib | RML, SQC, poor to
moderate | Up |
| 97 | M | 63 | N | IIIa | LUL, ADC, moderate
to well | UNC |
Ponatinib effectively inhibits cell
growth and colony formation in lung cancer cell lines
overexpressing FGFR1
The frequent upregulation of FGFR in primary lung
cancers suggests that targeting the FGFR1 signaling pathway using
pharmacological inhibitors may be an effective way to restrict
NSCLC growth. Our previous research targeting FGRF1 fusion kinases
in stem cell leukemia/lymphoma syndrome, demonstrated that
ponatinib, a multiple tyrosine kinase inhibitor, can effectively
suppress FGFR1 fusion kinase activity and subsequently inhibit cell
growth in vitro and leukemogenesis in mouse models in
vivo(12). This study
demonstrated that ponatinib was more potent than several other
available FGFR1 inhibitors, such as TKI258 and PD17142. To
determine whether this was also the case for NSCLC, we analyzed
FGFR1 expression levels in 12 NSCLC cell lines using RT-PCR. Three
of these cell lines H1299, A549 and H520, showed FGFR1
overexpression compared to H3122, which was confirmed using western
blot analysis (Fig. 2A). H520 is a
squamous cell line that carries FGFR1 amplification (18). H1299 and A549 (both
undifferentiated), and H3122 (adenocarcinoma, EML4-ALK fusion
positive) do not carry FGFR1 amplification or mutations (Cancer
Cell Line Encyclopedia, http://www.broadinstitute.org/ccle/home). These cell
lines were used to determine whether ponatinib affects cell growth
using a range of concentrations. Ponatinib showed no effect on the
growth of H3122 cells which express low levels of FGFR1 when used
at concentrations up to 1000 nM (Fig.
2B). In contrast, the three cell lines overexpressing FGFR1
showed a dramatic reduction in cell growth rate at various
concentrations, ranging from 200 to 1000 nM. We further used a
colony formation assay to evaluate the effect of ponatinib
treatment. Similar to the cell proliferation effects, ponatinib
significantly inhibited colony formation in cells overexpressing
FGFR1 but not in H3122 cells which express low levels of FGFR1
(Fig. 2C).
Ponatinib suppresses activation of FGFR1
and downstream targets and leads to inhibition of cell
division
Having shown that ponatinib inhibits cell growth of
NSCLC cells overexpressing FGFR1, we then determined whether this
cell growth inhibition is associated with decreased levels of
phospho-FGFR1. Ponatinib dramatically decreased the tyrosine
phosphorylation levels in all three cell lines expressing FGFR1
(Fig. 3A). Western blot analyses
with phospho-specific antibodies showed that ponatinib treatment
also led to inactivation of several direct targets of FGFR1, such
as Src and PLCγ (Fig. 3B). Since
ponatinib directly inhibits Src activation (10), the level of phospho-Src in H3122 was
also diminished (Fig. 3A).
Inactivation of Src, however, did not lead to growth inhibition of
the H3122 cells (Fig. 2B).
Consistently, immunocytochemical analysis using a specific
anti-phospho-FGFR1 antibody further confirmed that ponatinib (1.0
μM) effectively reduced the membrane phospho-FGFR1 staining levels
in both H1299 and A549 cells (Fig.
3B) after only 1 h. In parallel with these analyses, we also
determined whether downregulation of FGFR1 activation could lead to
cell cycle arrest or/and cell apoptosis. Ponatinib markedly
inhibited the cell cycle in these FGFR1-positive cells but not in
FGFR1-negative H3122 cells (Fig.
3C). No dramatic increase in the apoptotic cell population was
noted in the ponatinib-treated group (data not shown). To further
investigate whether cell growth inhibition is related to
inactivation of FGFR1 kinase by ponotinib, we treated the H520 and
H1581 cells with ponatinib in step-wise increasing doses. The H1581
cell line has been shown to overexpress FGFR1 (18), FGFR2 and FGFR4 (Fig. 6). The H520 cells, however, mainly
overexpress FGFR1 with only low levels of FGFR2-4 compared with the
H1581 cells. Other NSCLC cell lines showed a similar expression
pattern as the H520 cell line (Fig.
6). Immunoblotting, with the anti-phospho-FGFR1 antibody,
demonstrated that the FGFR1 activity in the H1581 cells was
inhibited by a lower dose of ponatinib than that used for the H520
cell line (Fig. 3D). Consistently,
the cell proliferation assay showed that the H1581 cells were more
sensitive to ponatinib when compared with the H520 cells. The
IC50 values for the H1581 and H520 cells were 30 and 50
nM, respectively (Fig. 3E).
Collectively, these data suggest that cell growth inhibition by
ponatinib correlates with inhibition of FGFR1 activation.
Knockdown of FGFR1 expression induces
cell growth inhibition in NSCLC cell lines overexpressing
FGFR1
Ponatinib was designed to act against BCR-ABL kinase
and its mutants, but it also effectively inhibits FGFR1, SRC,
VEGFR, PDGFR and KIT (10). To
determine whether the growth inhibition noted in the three NSCLC
cell lines was the direct result of inactivation of FGFR1, we used
shRNAs to specifically knockdown FGFR1 expression in these cells.
shGFP was used as a negative control. Seventy-two hours post
transfection, cells from independently transfected wells were
pooled for analysis. The pooled cells were coincidently analyzed
for FGFR1 expression, activation, cell cycle analysis and cell
proliferation. Real-time RT-PCR analysis showed that shFGFR1
markedly decreased the transcription levels of FGFR1 in all three
NSCLC cell lines compared with the vector control carrying shGFP.
The FGFR1-negative H3122 cells showed no alterations in expression
as predicted (Fig. 4A).
Consistently, western blotting with an anti-FGFR1 antibody also
showed the effect of the knockdown in the shFGFR1 but not in the
shGFP-infected cells (Fig. 4B). To
determine whether knockdown of FGFR1 can decrease cell
proliferation, we performed a standard cell cycle analysis which
showed that knockdown of FGFR1 dramatically decreased the
percentage of S/G2 phase cells compared with that in the
shGFP-infected cells (Fig. 4C).
Consistent with the cell cycle analysis, cell proliferation assay
showed that shFGFR1 transduction significantly induced cell growth
inhibition in the FGFR1-positive NSCLC cells compared with that in
the shGFP-infected cells. We did not observe any differences
between the two groups in the FGFR1-negative H3122 cells (Fig. 4D).
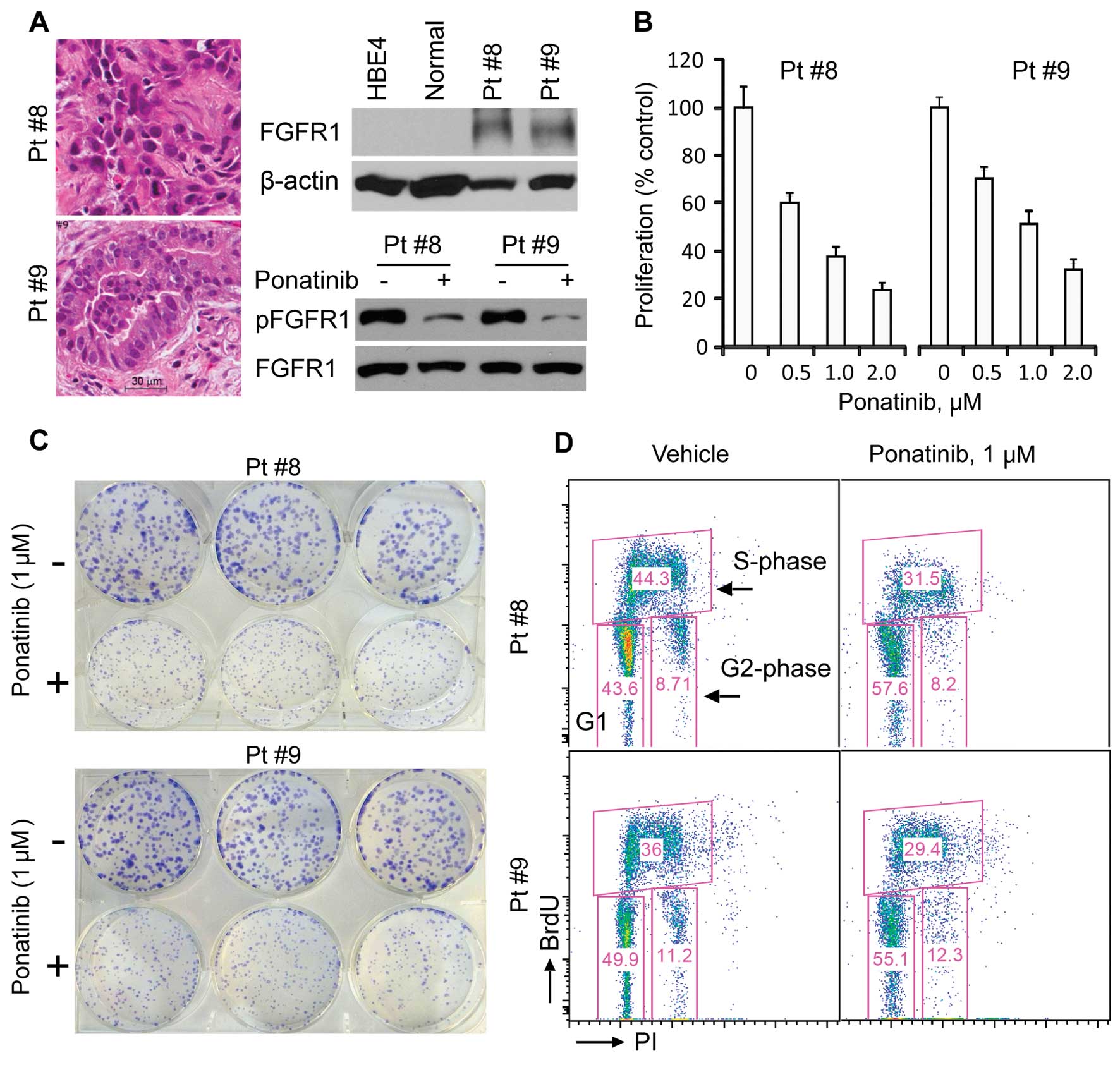
Ponatinib effectively inhibits human
primary lung cancer cell culture growth
To investigate whether ponatinib can decrease the
growth of primary lung cancer cells, we seeded freshly resected
human non-small cell lung cancers into culture from 12 patients.
Two of these cultures were successfully established and assayed.
One was derived from a squamous-cell carcinoma (Pt #8) and another
was from an adenocarcinoma (Pt #9) based on their histological
features (Fig. 5A, left panel).
These two primary lung cancer cell cultures were routinely grown in
RPMI-1640 medium plus 10% FBS without addition of any growth
factors. In the absence of ponatinib these cells overexpressed many
oncogenes frequently upregulated in cancer e.g. Plk1, survivin and
RSK2, compared to normal lung tissue (data not shown). Western blot
analysis with FGFR1 antibodies showed high levels of expression of
FGFR1 in these two tumor cell cultures compared to normal lung
tissues and HBE4 cells (an immortalized human bronchial epithelial
cell line) (Fig. 5A, right panel).
Consistently, both primary cell cultures were sensitive to
ponatinib treatment and showed a dramatic reduction in the cell
growth rate at various IC50 values, ranging from 0.5 to
1.0 μM (Fig. 5B). Further analysis
suggested that this cell growth inhibition was associated with a
decrease in phospho-FGFR1 levels in the presence of ponatinib
(Fig. 5A, right bottom panel). To
confirm the cell proliferation effect of ponatinib, we also
performed colony formation assays, which demonstrated that
ponatinib significantly inhibited colony formation of both primary
lung cancer cell cultures (Fig.
5C). Subsequently, cell cycle analysis, in combination with
BrdU uptake, showed that the percentage of cells in the S phase was
dramatically decreased. Apoptotic cells were not observed (Fig. 5D). These results indicate that
ponatinib has a profound effect on human lung cancer cells
expressing increased levels of FGFR1.
Discussion
Cancer is driven by different types of genetic
mutations acquired during tumorigenesis and tumor growth (19). Identifying these mutations that are
critical for tumor growth (oncogenic drivers) is the first step.
Once the oncogenic drivers are identified, they can then be
targeted accordingly. This therapeutic strategy is becoming
increasingly more possible due to advances in second generation
sequencing technology and other profiling platforms (20) which facilitate the identification of
driver mutations together with the increasing availability of drugs
that target these molecular changes (21). To date, EGFR (22,23),
KRAS (24), EML4-ALK (25), ROS1 (26,27),
RET (28,29), ERBB2 and PIK3CA (30) and MET (31), among others, are found to drive
tumorigenesis in varying proportions of NSCLC, mostly
adenocarcinomas. Targeted therapies have materialized in these
tumors carrying EGFR, EML4-ALK, ROS1 as well as RET. In the case of
FGFR1, however, there is no mutation(s) or translocation found to
be involved, to date. Instead, copy number increase appears to be
responsible for the activation of this pathway particularly in the
squamous cell subtype (18,32). Other important mechanisms for
over-activation of FGFR1 may be caused by increasing availability
of FGFR1 through increased transcription and/or autocrine/paracrine
mechanisms as part of the epithelial to mesenchymal transition
(EMT) (33). In the present study,
we showed that ~50% (30 out of 59) of the NSCLC patients
overexpressed FGFR1, a percentage that is much higher than that of
FGFR1 amplification. Together these studies indicate that i) copy
number increase of FGFR1 contributes to the overexpression of FGFR1
in these tumor cells and ii) overexpression of FGFR1 in a
proportion of patients is due to transcriptional upregulation of
FGFR1. Given that aberrant FGFR pathway activation is also involved
in drug resistance (34), FGFR
targeting is an important strategy in containing cancer growth and
possibly in overcoming resistance. Importantly, in two previous
studies, while the FGFR1 inhibitor PD173074 was shown to be
effective against cells with overactivated FGFR1, this was not
uniformly effective suggesting other mutation(s) may affect
sensitivity to PD173074.
In the present study, we demonstrated that targeting
the FGFR1 signaling pathway using a novel FGFR1 inhibitor in NSCLC
cell lines as well as primary NSCLC samples overexpressing FGFR1
significantly inhibited cancer cell growth and clonogenicity,
suggesting the potential clinical benefit of ponatinib. It is
noteworthy that two NSCLC cell lines, H1581 and H520, both
harboring the FGFR1 gene amplification [4 copies (18)], have much higher levels of FGFR1
transcription than other cell lines (Figs. 2A). Consequently these two cell
lines were more sensitive to ponatinib (Fig. 3D and E; IC50 <50 nM),
confirming the results from a recent study (35). At the recommended clinical trial
dose of 45 mg, the trough level of ponatinib reached 40 nM. The
peak level was several fold higher (35). This suggests that ponatinib at a
clinically achievable concentration markedly reduces the growth of
NSCLC in which FGFR1 is amplified. However, further studies in
animal models are necessary in order to determine the efficacy
before clinical trials are initiated in lung cancer patients whose
tumors overexpress FGFR1.
It is noteworthy that an increasing number of
studies have demonstrated that overexpression of FGFR1 is not only
observed in NSCLC, but also in other types of cancers, such as
breast cancer (36–38), prostate cancer (39,40)
and ovarian cancer (41). Recent
large scale analyses of somatic copy-number alterations from 3,131
cancer specimens, spanning 26 different cancer types, found
amplification of the FGFR1 gene in 10% of cases. This is the most
significantly reported focal amplification (42), indicating that FGFR1 signaling is
commonly involved in tumorigenesis and/or tumor progression and may
represent a promising therapeutic target in many cancer types. The
precise role of FGFR1 signaling in the pathogenesis and progression
of these tumors, however, is still unclear. In rearranged FGFR1
fusion kinase-induced leukemia and lymphoma, we and others
(17,43) have demonstrated that constitutive
activation of the chimeric FGFR1 kinase alone is not sufficient to
cause overt leukemia/lymphoma development in mouse models. Using
immunohistochemical staining, Behrens et al(4) studied 321 NSCLC tissue samples and 426
adjacent bronchial epithelial specimens, and found a significant
increase in bFGF and FGFR proteins in respiratory epithelium with
squamous dysplasia, compared with metaplastic bronchial epithelia.
Using SNP array analysis, Dutt et al(32) also observed that 60% (11 out of 19)
of primary NSCLC patients were in stage I. Together these
observations suggest that the activation of the FGFR signaling
pathway is an early event in the pathogenesis of NSCLC. If so, FGFR
inhibitors may prove to be effective against early stage
tumors.
In summary, our study provides convincing evidence
that abnormal expression of FGFR1 occurs in almost 50% of all NSCLC
patients presented with both major histological types. FGFR1,
therefore, may serve as a novel target in molecularly targeted
therapies against lung cancer. Targeting FGFR1 kinases with the
novel FGFR1 inhibitor ponatinib is a potentially successful
approach for the treatment of NSCLC patients carrying aberrant
activation of FGFR1. It is conceivable that ponatinib may be used
where appropriate either alone or in combination with other drugs
in the future.
Acknowledgements
We thank Scott Antonia of the Lee Moffitt Cancer
Center and Matthew Meyerson of Dana-Farber Cancer Institute,
Harvard Medical School, for sharing their non-small cell cancer
cell lines. We are grateful to Ms. Haiyan Qin for technical support
in the molecular analyses and Michael Boyd for assistance with
micrograph preparation from the pathology slides. Ponatinib was
kindly provided by Ariad Pharmaceuticals, Inc.
References
|
1
|
Stinchcombe TE, Bogart J, Wigle DA and
Govindan R: Annual review of advances in lung cancer clinical
research: a report for the year 2009. J Thorac Oncol. 5:935–939.
2010.PubMed/NCBI
|
|
2
|
Perez-Moreno P, Brambilla E, Thomas R and
Soria JC: Squamous cell carcinoma of the lung: molecular subtypes
and therapeutic opportunities. Clin Cancer Res. 18:2443–2451. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Subramanian J, Corrales L, Soulieres D,
Morgensztern D and Govindan R: Summary of presentations from the
46th Annual Meeting of the American Society of Clinical Oncology
(2010) focus on tumor biology and biomarkers related to lung
cancer. J Thorac Oncol. 6:399–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Behrens C, Lin HY, Lee JJ, et al:
Immunohistochemical expression of basic fibroblast growth factor
and fibroblast growth factor receptors 1 and 2 in the pathogenesis
of lung cancer. Clin Cancer Res. 14:6014–6022. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sasaki H, Shitara M, Yokota K, et al:
Increased FGFR1 copy number in lung squamous cell carcinomas. Mol
Med Rep. 5:725–728. 2012.PubMed/NCBI
|
|
6
|
Peifer M, Fernandez-Cuesta L, Sos ML, et
al: Integrative genome analyses identify key somatic driver
mutations of small-cell lung cancer. Nat Genet. 44:1104–1110. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turner N and Grose R: Fibroblast growth
factor signalling: from development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Larsson H, Klint P, Landgren E and
Claesson-Welsh L: Fibroblast growth factor receptor-1-mediated
endothelial cell proliferation is dependent on the Src homology
(SH) 2/SH3 domain-containing adaptor protein Crk. J Biol Chem.
274:25726–25734. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sandilands E, Akbarzadeh S, Vecchione A,
McEwan DG, Frame MC and Heath JK: Src kinase modulates the
activation, transport and signalling dynamics of fibroblast growth
factor receptors. EMBO Rep. 8:1162–1169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O’Hare T, Shakespeare WC, Zhu X, et al:
AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia,
potently inhibits the T315I mutant and overcomes mutation-based
resistance. Cancer Cell. 16:401–412. 2009.PubMed/NCBI
|
|
11
|
Chase A, Bryant C, Score J and Cross NC:
Ponatinib as targeted therapy for FGFR1 fusions associated with the
8p11 myeloproliferative syndrome. Haematologica. 98:103–106. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ren M, Qin H, Ren R and Cowell JK:
Ponatinib suppresses the development of myeloid and lymphoid
malignancies associated with FGFR1 abnormalities. Leukemia.
27:32–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren M and Cowell JK: Constitutive Notch
pathway activation in murine ZMYM2-FGFR1-induced T-cell lymphomas
associated with atypical myeloproliferative disease. Blood.
117:6837–6847. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Darzynkiewicz Z and Juan G: Analysis of
DNA content and BrdU incorporation. Curr Protoc Cytom. 7(unit
7.7)2001. View Article : Google Scholar
|
|
15
|
Ren M, Qin H, Ren R, Tidwell J and Cowell
JK: Src activation plays an important key role in lymphomagenesis
induced by FGFR1 fusion kinases. Cancer Res. 71:7312–7322. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Silva J, Wang G and Cowell JK: The
temporal and spatial expression pattern of the LGI1 epilepsy
predisposition gene during mouse embryonic cranial development. BMC
Neurosci. 12:432011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren M, Li X and Cowell JK: Genetic
fingerprinting of the development and progression of T-cell
lymphoma in a murine model of atypical myeloproliferative disorder
initiated by the ZNF198-fibroblast growth factor receptor-1
chimeric tyrosine kinase. Blood. 114:1576–1584. 2009. View Article : Google Scholar
|
|
18
|
Weiss J, Sos ML, Seidel D, et al: Frequent
and focal FGFR1 amplification associates with therapeutically
tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl
Med. 2:62ra932010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stratton MR: Exploring the genomes of
cancer cells: progress and promise. Science. 331:1553–1558. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garnett MJ, Edelman EJ, Heidorn SJ, et al:
Systematic identification of genomic markers of drug sensitivity in
cancer cells. Nature. 483:570–575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ashworth A, Lord CJ and Reis-Filho JS:
Genetic interactions in cancer progression and treatment. Cell.
145:30–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
III study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rosell R, Li S, Skacel Z, et al:
Prognostic impact of mutated K-ras gene in surgically resected
non-small cell lung cancer patients. Oncogene. 8:2407–2412.
1993.PubMed/NCBI
|
|
25
|
Kwak EL, Bang YJ, Camidge DR, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bergethon K, Shaw AT, Ou SH, et al: ROS1
rearrangements define a unique molecular class of lung cancers. J
Clin Oncol. 30:863–870. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Janne PA and Meyerson M: ROS1
rearrangements in lung cancer: a new genomic subset of lung
adenocarcinoma. J Clin Oncol. 30:878–879. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kohno T, Ichikawa H, Totoki Y, et al:
KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 18:375–377.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takeuchi K, Soda M, Togashi Y, et al: RET,
ROS1 and ALK fusions in lung cancer. Nat Med. 18:378–381. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samuels Y and Velculescu VE: Oncogenic
mutations of PIK3CA in human cancers. Cell Cycle. 3:1221–1224.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim ES and Salgia R: MET pathway as a
therapeutic target. J Thorac Oncol. 4:444–447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dutt A, Ramos AH, Hammerman PS, et al:
Inhibitor-sensitive FGFR1 amplification in human non-small cell
lung cancer. PLoS One. 6:e203512011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marek L, Ware KE, Fritzsche A, et al:
Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine
signaling in non-small-cell lung cancer cells. Mol Pharmacol.
75:196–207. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ware KE, Marshall ME, Heasley LR, et al:
Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in
NSCLC cell lines through de-repression of FGFR2 and FGFR3
expression. PLoS One. 5:e141172010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gozgit JM, Wong MJ, Moran L, et al:
Ponatinib (AP24534), a multi-targeted pan-FGFR inhibitor with
activity in multiple FGFR-amplified or mutated cancer models. Mol
Cancer Ther. 11:690–699. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kwek SS, Roy R, Zhou H, et al:
Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate
with two major pathways in oncogenesis. Oncogene. 28:1892–1903.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shiang CY, Qi Y, Wang B, et al:
Amplification of fibroblast growth factor receptor-1 in breast
cancer and the effects of brivanib alaninate. Breast Cancer Res
Treat. 123:747–755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Turner N, Pearson A, Sharpe R, et al:
FGFR1 amplification drives endocrine therapy resistance and is a
therapeutic target in breast cancer. Cancer Res. 70:2085–2094.
2010. View Article : Google Scholar
|
|
39
|
Freeman KW, Welm BE, Gangula RD, et al:
Inducible prostate intraepithelial neoplasia with reversible
hyperplasia in conditional FGFR1-expressing mice. Cancer Res.
63:8256–8263. 2003.PubMed/NCBI
|
|
40
|
Freeman KW, Gangula RD, Welm BE, et al:
Conditional activation of fibroblast growth factor receptor (FGFR)
1, but not FGFR2, in prostate cancer cells leads to increased
osteopontin induction, extracellular signal-regulated kinase
activation, and in vivo proliferation. Cancer Res. 63:6237–6243.
2003.
|
|
41
|
Mayr D, Kanitz V, Anderegg B, et al:
Analysis of gene amplification and prognostic markers in ovarian
cancer using comparative genomic hybridization for microarrays and
immunohistochemical analysis for tissue microarrays. J Clin Pathol.
126:101–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Beroukhim R, Mermel CH, Porter D, et al:
The landscape of somatic copy-number alteration across human
cancers. Nature. 463:899–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Roumiantsev S, Krause DS, Neumann CA, et
al: Distinct stem cell myeloproliferative/T lymphoma syndromes
induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11
translocations. Cancer Cell. 5:287–298. 2004. View Article : Google Scholar : PubMed/NCBI
|