Introduction
Esophageal squamous cell carcinoma (ESCC) is one of
the most common and aggressive cancers in the world, particularly
with high incidence and morbidity in China (1,2).
Although treatment strategies have progressed in recent years, the
prognosis of ESCC is still poor. It is urgent to identify a new
target to improve patient outcomes. Signaling pathways have been
widely studied in cancers; however, the precise mechanisms
underlying ESCC are poorly understood.
Recently, cancer-related inflammation (CRI) has been
considered as the seventh hallmark of cancer (3). A close relationship has been revealed
between chronic inflammatory infection and cancer risk and
progression such as Helicobacter pylori and gastric cancer,
papilloma virus and cervical cancer, and hepatitis viruses and
liver carcinoma. Human papilloma virus (HPV) infection has also
been suggested as an etiology of ESCC, in particular types 16 and
18, although the conclusion is controversial (4,5). In
addition, overexpression of several inflammatory markers,
cyclooxygenase-2 (COX-2) and nuclear factor-κB (NF-κB), have also
been observed in ESCC with predictive prognostic value, indicating
the inflammatory mechanisms in ESCC development (6). However, the relationship between
chronic inflammation and ESCC is also poorly understood.
More and more evidence indicates that various
molecular and cellular pathways are involved in the link between
inflammation and cancer (7). Among
them, the signal transducer and activator of transcription 3
(STAT3) and NF-κB signaling pathways are considered to play a vital
role in regulating inflammation and cancer development (8). Persistent abnormal activation of STAT3
is oncogenic in many human cancers, including breast, prostate,
ovarian cancers, and pancreatic cancer (9–13).
Activated STAT3 promotes carcinogenesis through regulation of
downstream genes that encode cell apoptosis, cell cycle, metastasis
and angiogenesis (14,15). Janus kinase (JAK) is responsible for
STAT3 activation when stimulated by extracellular signals, and the
JAK2 inhibitor AG490 was found to block the constitutive activation
of STAT3 (16). Recently, a new
role of STAT3 in CRI has been reported by promoting pro-oncogenic
inflammatory pathways through the IL-6-GP130-JAK pathways, and also
by opposing antitumor immune responses while the underlying
mechanism is not fully clarified (17). Interleukin-6 (IL-6) belongs to a
large family of cytokines and activates JAKs through binding to its
receptor and dimerizing glycoprotein 130 (gp130) which is expressed
on many cells (18). It is involved
in many inflammatory processes and promotes oncogenesis. Recently,
a colitis-associated cancer model confirmed that IL-6 and STAT3 are
essential in the process from chronic inflammation to cancer.
Nevertheless, studies focusing on the effects of STAT3 on ESCC are
few compared with other cancers and their roles in ESCC CRI and
cancer growth are unknown (19).
NF-κB is another important regulator abnormally
activated in CRI in many types of cancers, and it also can be
triggered by IL-6 (20). When
activated, NF-κB induces the generation of many molecules
modulating inflammation, angiogenesis and adhesion. In addition, it
was found to participate in CRI to promote tumor initiation and
progression in a liver and gastrointestinal tract cancer model
(21,22). The NF-κB family includes RelA/p65,
RelB, c-Rel, NF-κB1/p50 and NF-κB2/p52. Among them, RelA/p65 is
reported to be closely correlated with inflammation, cell
proliferation, survival signals and cancer. However, the crosstalk
between the STAT3 and NF-κB signaling pathways in CRI and
oncogenesis has not been fully elucidated.
In the present study, expression levels of STAT3 and
its phosphorylated form in human ESCC cell lines were examined.
Additionally, the interrelationship between the STAT3 pathway and
inflammatory pathways NF-κB p65 and COX-2 were preliminarily
studied. Our objective was to demonstrate that the STAT3 signaling
pathway may be a key regulator linking cancer growth and CRI in
ESCC. Inhibition of this pathway may be a novel treatment and
preventative target for ESCC.
Materials and methods
Cell lines and reagents
The human ESCC cell line TE-1 (catalog no. TCHu 89)
was obtained from the Cell Bank of Shanghai Institute (Shanghai,
China). Cell lines EC-1 and K150 were provided by Professor Fenyong
Sun, Department of Central Laboratory, Shanghai Tenth People’s
Hospital. The cells were maintained in RPMI-1640 containing 10%
fetal bovine serum and 100 U/ml penicillin and 100 μg/ml
streptomycin. All cells were incubated at 37°C in a humidified
atmosphere containing 5% CO2. AG490 was purchased from
Merck (Whitehouse Station, NJ, USA), dissolved in dimethyl
sulfoxide and then diluted with the culture medium for experiments.
Recombinant IL-6 was purchased from PeproTech (Princeton, NJ, USA),
dissolved in acetic acid and then diluted with the culture medium.
Monoclonal antibodies against β-actin were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against
STAT3 and phospho-STAT3 (Tyr705), COX-2, NF-κB p65 for western
blotting were purchased from Cell Signaling Technology (Danvers,
MA, USA) and p-NF-κB p65 (Ser536) was from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Human IL-6 ELISA kit was purchased from
R&D Systems (Minneapolis, MN, USA).
Cell proliferation assay
Cell proliferation and viability were determined by
the Cell Counting Kit-8 (CCK-8) colorimetric assay (Dojindo,
Japan). TE-1 and EC-1 cells were seeded in a 96-well plate at
3×103 cells/well. When the cell density reached 80%,
fresh culture medium containing AG490 (5, 10, 20, 40 and 80 μmol/l)
was added to the cells after 12 h of starvation, respectively. Cell
viability was measured using the CCK-8 assay after culture for 24
and 48 h. Absorbance was measured at 450 nm with a microtiter plate
reader.
Flow cytometry for cell cycle
analysis
TE-1 cells treated with AG490 at different
concentrations for 48 h were washed and fixed with
phosphate-buffered saline (PBS) for two times and then fixed with
ethanol 95% and washed with cold PBS and then resuspended in 150 μl
hypotonic fluorochrome solution [50 μg/ml propidium iodide (PI), 10
μg/ml RNAse A in PBS]. The cells were incubated in the dark at 4°C
overnight before flow cytometric analysis was performed. The PI
fluorescence of individual nuclei was measured using a FACSCalibur
cytometer (BD Biosciences, Heidelberg, Germany). Data were analyzed
with the CellQuest Pro v 5.2.1 software (BD Biosciences). For each
condition, at least 3 independent experiments were performed.
Western blotting
TE-1 cells were treated with AG490 (5, 10, 20, 40
and 80 μmol/l) for 48 h, respectively. EC-1 cells were treated with
100 ng/ml IL-6 for 24 h. The harvested cells were washed with PBS
twice and lysed on ice for 30 min with whole cell extract lysis
buffer (Santa Cruz Biotechnology). Lysates were centrifugated at
12,000 rpm for 10 min at 4°C, and the protein concentration was
determined by an assay kit (Bio-Rad, Hercules, CA, USA). Cell
lysates were mixed with loading buffer and boiled for 5 min at
100°C. Protein samples were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes. The membranes were blocked in blocking
buffer (Tris-buffered saline, 0.1% Tween-20 and 5% non-fat dry
milk) for 1 h and then incubated overnight at 4°C with the specific
anti-STAT3 antibody (1:1,000 dilution), anti-p-STAT3 antibody
(1:1,000 dilution), anti-NF-κB p65 antibody (1:500 dilution),
anti-p-NF-κB p65 antibody (1:500 dilution), anti-vascular
endothelial growth factor (VEGF) antibody (1:1,000 dilution),
anti-COX-2 antibody (1:500 dilution) and anti-β-actin antibody
(1:2,000 dilution). Subsequently, the membranes were incubated with
a horseradish peroxidase-conjugated secondary antibody rabbit IgG
(1:2,000 dilution; Santa Cruz Biotechnology) for 1 h at room
temperature after being washed in TBS/0.1% Tween-20 for 3 times.
Then after 3 washes in TBS/0.1% Tween-20 again, the membranes were
detected by chemiluminescence using Western Blotting Luminol
Reagent (Santa Cruz Biotechnology).
RNA transfection and RT-PCR
Cells were transfected with a non-specific random
siRNA as a negative control and synthetic siRNA (GenePharma,
Shanghai, China) directed to STAT3 or NF-κB p65 (3 siRNAs for each
gene) at a final concentration of 100 nM for 48 h. Total RNA was
extracted by TRIzol LS (Invitrogen, Carlsbad, CA, USA). A
spectrophotometer was used to detect the concentration and purity
of the RNA. cDNA was synthesized with reverse transcriptase
(Takara, Japan). Quantitative real-time polymerase chain reaction
(RT-PCR) assays were carried out on the ABI Prism 7500 Fast System
(Applied Biosystems, Carlsbad, CA, USA) using the standard curve
method. The following primers were used: STAT3 sense strand,
5′-AGCATCCTGAAGCTGACCCAGGT-3′ and anti-sense strand,
5′-TCGGCAGGTCAATGGTATTGCTGC-3′;
NF-κBp65sensestrand,5′-TCTGCTTCCAGGTGACAGTG-3′ and antisense
strand, 5′-ATCTTGAGCTCGGCAGTGTT-3′; COX-2 sense strand,
5′-CCCCCACAGTCAAAGACACT-3′ and antisense strand,
5′-CTCATCACCCCACTCAGGAT-3′.
Results
STAT3 is phosphorylated in human ESCC
cell lines
STAT3 phosphorylation and COX-2 upregulation are
closely related to cancer development and CRI. In the present
study, we measured the protein levels of STAT3, phosphorylated
STAT3 as well as COX-2 by performing western blotting in 3 human
ESCC cell lines EC-1, TE-1 and K150. STAT3 was activated in the 3
cell lines at different levels. STAT3 phosphorylation was highest
in the TE-1 and lowest in the EC-1 cells. COX-2 was also expressed
in the TE-1 and K150 cells but very low in the EC-1 cells (Fig. 1).
JAK2 inhibitor attenuates the growth of
ESCC by regulating cell growth and cell cycle
A positive correlation between STAT3 activation and
cancer cell growth has been verified. In to the present study, cell
proliferation assay was conducted with TE-1 and EC-1 cells treated
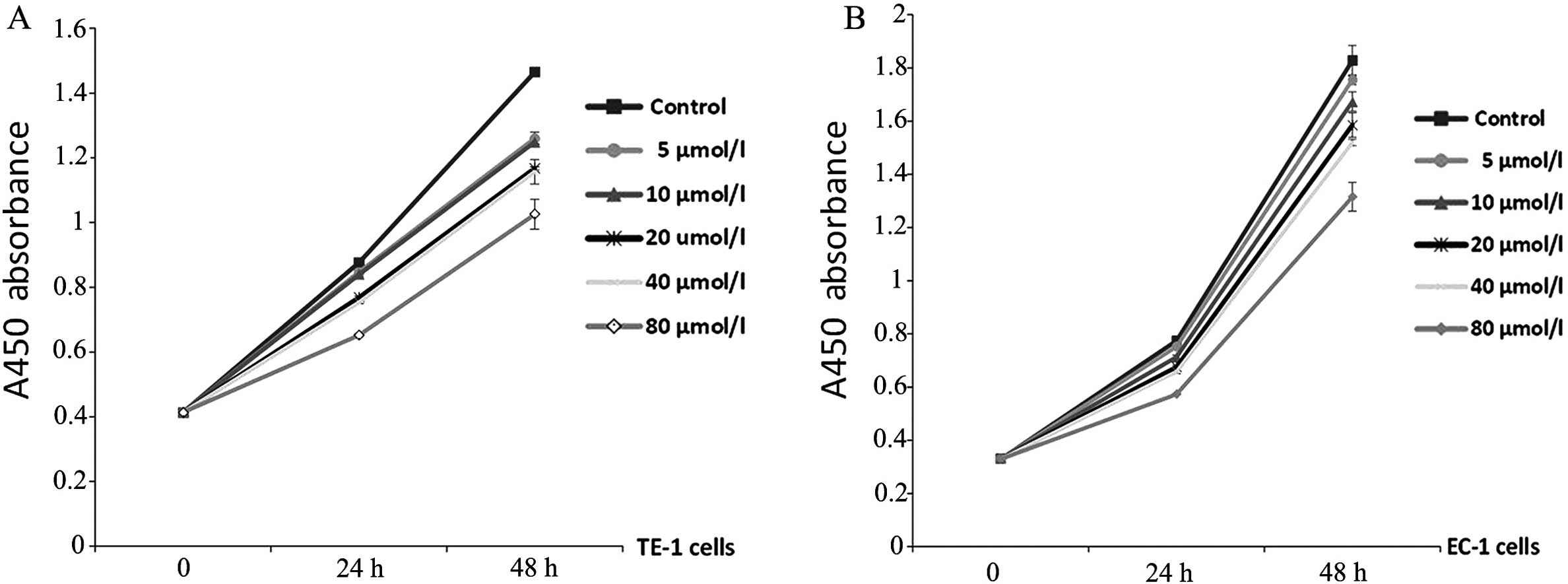
with AG490 at different concentrations for 24 and 48 h. As
expected, AG490 reduced the proliferation of both TE-1 and EC-1
cells dependent on time and concentration (P<0.05, Fig. 2A and B). In particular, AG490 at 80
μmol/l markedly inhibited cell proliferation compared with the
vehicle-treated cells (P<0.001, Tables I and II).
 | Table IInhibitory effect of AG490 on TE-1
cell growth. |
Table I
Inhibitory effect of AG490 on TE-1
cell growth.
| A450 (mean
± SD) | Inhibition rate
(%) |
|---|
|
|
|
|---|
| Group | 24 h | 48 h | 24 h | 48 h |
|---|
| Blank control | 0.876±0.004 | 1.465±0.010 | 0 | 0 |
| 5 μmol/l AG490 | 0.847±0.007 | 1.261±0.018 | 3.31 | 13.92b |
| 10 μmol/l AG490 | 0.839±0.006 | 1.249±0.008 | 4.22a | 14.74b |
| 20 μmol/l AG490 | 0.768±0.005 | 1.168±0.005 | 12.33b | 20.27b |
| 40 μmol/l AG490 | 0.753±0.007 | 1.157±0.038 | 14.04b | 21.02b |
| 80 μmol/l AG490 | 0.652±0.004 | 1.026±0.046 | 25.57b | 29.96b |
| Table IIInhibitory effect of AG490 on EC-1
cell growth. |
Table II
Inhibitory effect of AG490 on EC-1
cell growth.
| A450 (mean
± SD) | Inhibition rate
(%) |
|---|
|
|
|
|---|
| Group | 24 h | 48 h | 24 h | 48 h |
|---|
| Blank control | 0.774±0.007 | 1.827±0.057 | 0 | 0 |
| 5 μmol/l AG490 | 0.752±0.006 | 1.755±0.019 | 2.84 | 3.94a |
| 10 μmol/l AG490 | 0.713±0.008 | 1.673±0.036 | 7.88a | 8.43b |
| 20 μmol/l
AG490 | 0.675±0.007 | 1.585±0.046 | 12.79b | 13.25b |
| 40 μmol/l
AG490 | 0.657±0.006 | 1.519±0.012 | 15.11b | 16.86b |
| 80 μmol/l
AG490 | 0.573±0.002 | 1.315±0.054 | 25.97b | 28.02b |
Additionally, the JAK/STAT3 signaling pathway is
known for its regulatory role in the cell cycle. TE-1 cells
incubated in AG490 at different concentrations were detected for
cell cycle distribution. A decline in the percentage of cells in
the G0/G1 phase and an elevation in the percentage of cells in the
S phase were noted following treatment with increasing AG490
concentrations, although only cells treated with 80 μmol/l AG490
achieved statistical significance compared with the vehicle-treated
cells (P=0.007) (Fig. 3A and
B).
JAK2 inhibitor inhibits the growth of
ESCC by blocking the JAK/STAT3 signaling pathway
AG490, an important JAK2 inhibitor, has been found
to exhibit effective inhibition on cancer growth through abrogating
the JAK/STAT3 pathway in other cancers. In order to clarify whether
it also attenuates ESCC growth by inhibiting the JAK/STAT3 pathway
in ESCC, western blotting was conducted to evaluate the protein
levels of STAT3 and its phosphorylation levels in TE-1 cells. VEGF,
an important factor involved in tumor angiogenesis and metastases
regulated by STAT3, was also detected. As illustrated in Fig. 4, the phosphorylated levels of STAT3
and VEGF protein expression decreased in the AG490-treated groups
when compared with these levels in the control group; AG490 80
μmol/l group in particular (P<0.001). These results indicate
that AG490 inhibits ESCC growth and angiogenesis by blocking the
JAK/STAT3 pathway.
JAK2 inhibitor inhibits CRI in ESCC
through crosstalk between the JAK/STAT3 and NF-κB and COX-2
inflammatory pathways
A potential relationship between CRI and
carcinogenesis has been focused on in recent years since various
inflammatory factors have been discovered to be involved among
which NF-κB and COX-2 activations have been observed in many types
of cancers. However, how the STAT3 pathway interacts with them to
promote cancer growth, particularly STAT3-COX-2 crosstalk has been
poorly studied. In an attempt to shed light on the mechanism of
STAT3 signaling pathway in CRI, western blotting was performed to
study the crosstalk between STAT3 and NF-κB, and COX-2. Since all
subunits of NF-κB, RelA/p65 are closely associated with cancer
development and inflammation, NF-κB p65 was chosen for western
blotting. AG490 was also used to treat TE-1 cells to examine the
role of JAK/STAT3 in CRI. Protein expressions of NF-κB p65 and its
phosphorylated levels and COX-2 were detected following AG490
treatment for 48 h. Expectedly, AG490 efficiently downregulated
NF-κB p65, p-NF-κB p65 and COX-2 in a concentration-dependent
manner (Fig. 5). Our hypothesis
that JAK/STAT3 also plays an important role in CRI in ESCC through
crosstalk with the NF-κB pathway and COX-2 was confirmed by the
above results. Abrogation of the STAT3 signaling pathway also
inhibited CRI through downregulation of inflammatory factors NF-κB
and COX-2.
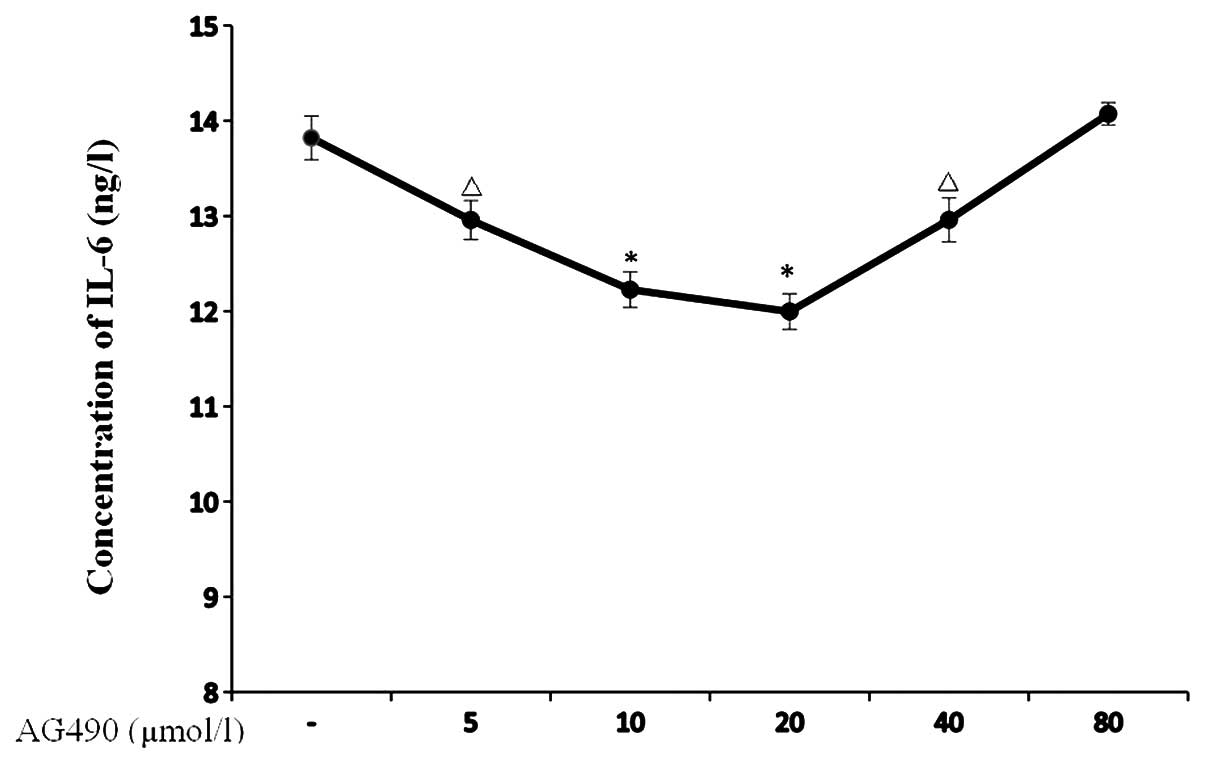
Moreover, we also validated the effects of AG490 on
IL-6 with an IL-6 ELISA kit. As a result, reduction in the IL-6
concentration was observed in the TE-1 cells after being treated
with AG490 at 5, 10 and 20 μmol/l for 24 h (P=0.009, P=0.001 and
P<0.001, respectively), and the inhibitory effect was strongest
in the AG490 20 μmol group. Notably, when the AG490 concentration
increased to 40 μmol/l, the inhibitory effect of AG490 on IL-6
decreased compared to the 20 μmol/l group, although it was still
stronger than the control group (P=0.01). The AG490 80 μmol/l group
did not show any inhibitory effects on IL-6 when compared with the
control group (Fig. 6).
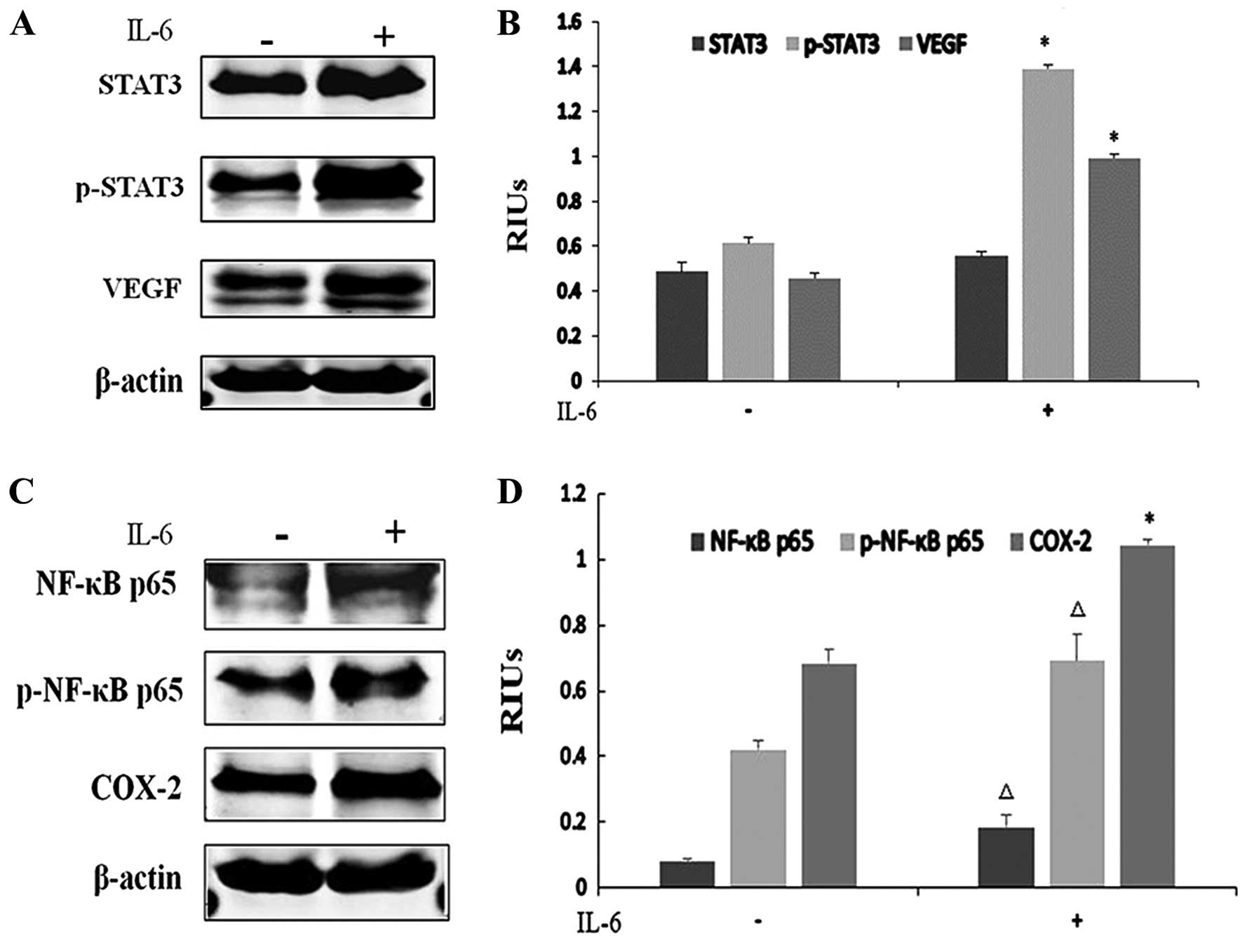
IL-6 promotes ESCC growth and CRI
As a downstream gene of IL-6, STAT3 activation in
tumors can be either IL-6-dependent or -independent. To determine
whether STAT3 was activated by IL-6 in ESCC, we performed western
blotting. Due to the low basal STAT3 phosphorylation levels noted
in our previous data, EC-1 cells were chosen for IL-6 stimulation.
IL-6 (100 ng/ml) treatment for 24 h increased the phosphorylated
STAT3 level and VEGF expression in the EC-1 cells, indicating that
the activation of the JAK/STAT3 signaling pathway is IL-6-dependent
in ESCC (Fig. 7A and B).
Particularly, marked elevation in the p-STAT3 and VEGF protein
levels confirmed that IL-6 activated STAT3 and promoted
angiogenesis (P<0.001). Similar results were also found in the
NF-κB and COX-2 pathways with the upregulation of NF-κB p65
phosphorylation levels and COX-2 protein levels in the EC-1 cells,
implying that IL-6 also induced CRI (P<0.05) (Fig. 7C and D).
Interaction of STAT3, NF-κB and COX-2 at
the mRNA level
To study the mechanism underlying the interaction of
STAT3, NF-κB and COX-2, siRNAs of STAT3 and NF-κB were transfected
in the TE-1 cells, respectively. Three siRNAs targeted at each gene
were designed. siRNAs with the highest inhibition rates were
selected for STAT3 and NF-κB inhibition (P=0.004 and P=0.027,
respectively) (Fig. 8A) to assess
the effects of siRNAs on the COX-2 mRNA level. The obvious
reduction in COX-2 mRNA expression by both STAT3 and NF-κB siRNA
suggested that both STAT3 and NF-κB regulate the transcription of
COX-2 (P=0.037 and P=0.014, respectively) (Fig. 8B).
Discussion
The JAK/STAT3 signaling pathway plays a vital role
in tumorigenesis by regulating downstream genes. Activation or
phosphorylation of several kinases such as JAK and Src lead to the
persistent activation of STAT3 in cancer (23). The availability of AG490, a JAK2
inhibitor, makes it possible to investigate the effect of JAK
inhibition on STAT3 activation and the role of the JAK/STAT3
pathway in tumors. In the present study, STAT3 was activated in 3
cell lines at different levels as in many other cancer types. STAT3
phosphorylation was highest in the TE-1 and lowest in the EC-1
cells. After treatment of AG490, cell proliferation was inhibited
dependent on AG490 concentration and time both in the TE-1 and EC-1
cells. Experiments focused on the cell cycle demonstrated a decline
in cells in the G0/G1 phase and an elevation of cells in the S and
G2/M phases. Although only the AG490 80 μmol/l group had
statistical difference compared to the control group, a regulatory
trend of AG490 on TE-1 cells was observed. Additionally, AG490
treatment led to a significant decrease in STAT3 phosphorylation
and VEGF protein levels in the TE-1 cells. These results suggest
that STAT3 accelerated tumor progression of ESCC by regulating cell
growth, cell cycle and angiogenesis. AG490 efficiently blocked
tumor growth and progression by suppressing STAT3 activity.
Clinical and epidemiological studies suggest a
strong association between chronic infection, inflammation and
cancer. Genetic studies reveal that there may be molecular
mechanisms linking inflammation and CRI (24). As research indicates the new role of
the STAT3 signaling pathway in CRI, we wanted to ascertain whether
the JAK/STAT3 pathway plays a central role in regulating cancer
growth and CRI in ESCC. NF-κB is known for the activation in many
human cancers and inflammatory processes. The finding that the
RelA, encoding p65 subunit of NF-κB is homologous to the viral
oncogene v-Rel indicates that NF-κB is involved in cancer.
Recently, the NF-κB family member, RelA/p65, has been found to
physically interact with STAT3 (20). In addition, STAT3 and NF-κB are both
transcriptional factors which play pivotal roles in various aspects
of the tumorigenic process by regulating downstream genes. Once
activated, NF-κB and STAT3 regulate downstream genes related to
cell apoptosis, proliferation and immune response, some of which
overlap and require a co-effect of the two factors (25). Recently, it has been found that
maintenance of NF-κB activation in tumors requires STAT3. In the
present study, we found that the JAK2 inhibitor also blocked
activation of NF-κB p65, implicating that there is an interaction
between STAT3 and NF-κB and that the STAT3 signaling pathway
regulates NF-κB through the p65 subunit. The STAT3 signaling
pathway inhibits CRI through interaction with NF-κB pathway.
In order to examine the role of the STAT3 signaling
pathway in CRI, it is essential to study other inflammatory factors
closely related to cancer which are also influenced by the STAT3
signaling pathway in addition to NF-κB. COX-2 is an important
enzyme which mediates inflammatory processes. Improper upregulation
of COX-2 leads to an increase in prostaglandin E2 (PGE2), resulting
in pathophysiology of certain types of human cancers as well as
inflammatory disorders (26). There
is also evidence that COX-2 is overexpressed in esophageal cancer
both in ESCC and adenocarcinoma and, inhibition of COX-2 suppresses
cancer growth and induces apoptosis (27,28).
Nevertheless, studies focused on the STAT3-COX-2 interaction are
rare compared to other inflammatory factors. In the present study,
STAT3 phosphorylation and COX-2 were co-expressed highly in the
ESCC cell lines. The expression levels of the two proteins were
consistent in the 3 cell lines, which indicated that the cell line
with higher p-STAT3 also had higher COX-2 protein levels. AG490 not
only efficiently suppressed the STAT3 activation but also decreased
COX-2 protein levels significantly. It appeared that COX-2 could
also be affected by the STAT3 pathway. To better clarify the
relationship between STAT3, NF-κB and COX-2, we silenced STAT3 and
NF-κB by siRNA, respectively. We found at the mRNA levels that both
silencing of STAT3 and NF-κB downregulated COX-2 mRNA expression,
indicating that both STAT3 and NF-κB regulate COX-2. COX-2 may be
one of the overlapping downstream genes regulated by STAT3 and
NF-κB.
IL-6 is an important cytokine participating in both
inflammation and oncogenesis. When stimulated by IL-6, gp130 is
phosphorylated and thereby activates JAK1 and JAK2, leading to the
activation of STAT3 (18). However,
STAT3 activation in tumors can be either dependent on or
independent of IL-6 signaling as mentioned before. To determine
whether STAT3 phosphorylation is IL-6-dependent in our ESCC cell
lines, we used IL-6 to stimulate EC-1 cells which expressed low
STAT3 phophorylation. Analysis of our results revealed that cells
treated with IL-6 had significantly increased expression of p-STAT3
and VEGF. Therefore, in ESCC, the aberrant activation of STAT3 was
IL-6-dependent, and IL-6 promoted cancer growth and angiogenesis by
activating the STAT3 pathway and its downstream genes such as VEGF.
In addition, we found that IL-6 also stimulated NF-κB p65
activation and upregulated COX-2, implicating that IL-6 could also
stimulate CRI by activating the NF-κB and COX-2 pathways.
Therefore, IL-6 is not only an important cytokine linking the STAT3
and NF-κB pathways but is also a bridge through which STAT3 links
cancer growth and CRI in ESCC. Moreover, it is necessary for us to
ascertain whether attenuating the STAT3 pathway influences the IL-6
concentration in turn. As a result, AG490 at 5, 10 and 20 μmol/l
efficiently reduced the IL-6 concentration in the TE-1 cells,
suggesting that blocking the JAK/STAT3 pathway can also decrease
IL-6; another way to inhibit CRI. However, notably, when the AG490
concentration increased to 40 and 80 μmol/l, the IL-6 concentration
increased compared with the 20 μmol/l group. The probable
explanation was that there was a negative feedback in the
IL-6-JAK-STAT3 pathway and another possibility was there may be
other signaling pathways activated to produce IL-6 when the STAT3
pathway is blocked.
Notably, the inhibitory effects of AG490 on cell
proliferation seemed modest compared with other chemotherapeutic
agents. It may be more appropriate to conduct AG490 treatment in
tumor initiation such as the process from CRI to cancer rather than
reducing tumor size based on its suppression of CRI. Animal models
are further needed to verify the role of AG490 in the prevention of
CRI and cancer growth in ESCC. In conclusion, our research showed
that the JAK/STAT3 pathway promoted tumor progression by regulating
cell growth, cell cycle and angiogenesis in ESCC. Moreover, STAT3
participated in CRI in ESCC through IL-6 and interacting with the
NF-κB p65 subunit and COX-2 which are important inflammatory
factors accelerating cancer growth (Fig. 9). The JAK/STAT3 pathway is an
important pathway which links CRI and cancer growth. The STAT3
pathway could be a novel target both for cancer treatment and
prevention in ESCC, and JAK2 inhibitor AG490 could be an
option.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 30872591 and 81372749).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei WQ, Yang J, Zhang SW, Chen WQ and Qiao
YL: Analysis of the esophageal cancer mortality in 2004–2005 and
its trends during last 30 years in China. Zhonghua Yu Fang Yi Xue
Za Zhi. 44:398–402. 2010.(In Chinese). PubMed/NCBI
|
|
3
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suzuk L, Noffsinger AE, Hui YZ and
Fenoglio-Preiser CM: Detection of human papillomavirus in
esophageal squamous cell carcinoma. Cancer. 78:704–710. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Antunes LC, Prolla JC, de Barros Lopes A,
da Rocha MP and Fagundes RB: No evidence of HPV DNA in esophageal
squamous cell carcinoma in a population of Southern Brazil. World J
Gastroenterol. 19:6598–6603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang GZ, Li L, Ding HY and Zhou JS:
Cyclooxygenase-2 is over-expressed in Chinese esophageal squamous
cell carcinoma, and correlated with NF-κB: an immunohistochemical
study. Exp Mol Pathol. 79:214–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bollrath J and Greten FR: IKK/NF-κB and
STAT3 pathways: central signalling hubs in inflammation-mediated
tumour promotion and metastasis. EMBO Rep. 10:1314–1319. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chung SS, Giehl N, Wu Y and Vadgama JV:
STAT3 activation in HER2-overexpressing breast cancer promotes
epithelial-mesenchymal transition and cancer stem cell traits. Int
J Oncol. 44:403–411. 2014.
|
|
11
|
Qu Y, Oyan AM, Liu R, et al: Generation of
prostate tumor-initiating cells is associated with elevation of
reactive oxygen species and IL-6/STAT3 signaling. Cancer Res.
73:7090–7100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burke WM, Jin X, Lin HJ, et al: Inhibition
of constitutively active Stat3 suppresses growth of human ovarian
and breast cancer cells. Oncogene. 20:7925–7934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang C, Cao J, Huang KJ, et al:
Inhibition of STAT3 activity with AG490 decreases the invasion of
human pancreatic cancer cells in vitro. Cancer Sci. 97:1417–1423.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong H, Zhang ZG, Tian XQ, et al:
Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle
arrest, and reduces tumor cell invasion in colorectal cancer cells.
Neoplasia. 10:287–297. 2008.PubMed/NCBI
|
|
15
|
Turkson J and Jove R: STAT proteins: novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar
|
|
16
|
Schindler C and Darnell JE Jr:
Transcriptional responses to polypeptide ligands: the JAK-STAT
pathway. Annu Rev Biochem. 64:621–652. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park EJ, Lee JH, Yu GY, et al: Dietary and
genetic obesity promote liver inflammation and tumorigenesis by
enhancing IL-6 and TNF expression. Cell. 140:197–208. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grivennikov S, Karin E, Terzic J, et al:
IL-6 and Stat3 are required for survival of intestinal epithelial
cells and development of colitis-associated cancer. Cancer Cell.
15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karin M: Nuclear factor-κB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Greten FR, Eckmann L, Greten TF, et al:
IKKβ links inflammation and tumorigenesis in a mouse model of
colitis-associated cancer. Cell. 118:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He G and Karin M: NF-κB and STAT3 - key
players in liver inflammation and cancer. Cell Res. 21:159–168.
2010. View Article : Google Scholar
|
|
23
|
Lo HW, Hsu SC, Xia W, et al: Epidermal
growth factor receptor cooperates with signal transducer and
activator of transcription 3 to induce epithelial-mesenchymal
transition in cancer cells via up-regulation of TWIST gene
expression. Cancer Res. 67:9066–9076. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grivennikov SI and Karin M: Dangerous
liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer.
Cytokine Growth Factor Rev. 21:11–19. 2010. View Article : Google Scholar :
|
|
26
|
Hao Q, Zhang C, Gao Y, et al: FOXP3
inhibits NF-κB activity and hence COX2 expression in gastric cancer
cells. Cell Signal. 26:564–569. 2014. View Article : Google Scholar
|
|
27
|
von Rahden BH, Stein HJ, Pühringer F, et
al: Coexpression of cyclooxygenases (COX-1, COX-2) and vascular
endothelial growth factors (VEGF-A, VEGF-C) in esophageal
adenocarcinoma. Cancer Res. 65:5038–5044. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhi H, Wang L, Zhang J, et al:
Significance of COX-2 expression in human esophageal squamous cell
carcinoma. Carcinogenesis. 27:1214–1221. 2006. View Article : Google Scholar
|