Introduction
Glioblastoma multiforme (GBM) is common and is an
aggressive brain tumor in adults. Despite the introduction of
radiation therapy and chemotherapy with temozolomide in addition to
surgery, general survival has been increased from 12 to 14 months
only (1,2). Specific genetic changes have been
classified and related to characteristic molecular pathways
involved in the control of tumor development. With an increasing
understanding of the molecular behaviour of tumors, specifically of
GBM, variations in pharmacogenomic profiles in combination with
radiation therapy potentially improve the outcome of therapy in
this lethal disease. Schiffer et al reported that in
glioblastoma, the highest quantity of apoptosis was detected in the
area with the highest mitotic rate suggesting that, during mitosis,
some glioblastomas are exposed to cell death (3).
Uncommon regulation of cyclins, cyclin-dependent
kinases (CDKs) and endogenous CDK inhibitors has been described in
gliomas, demonstrating that CDK modulators may be important for
treatment of GBM (4–9). Roscovitine, a CDK modulator is a
potent and selective small molecule inhibitor of the
cyclin-dependent kinases CDK2/cyclin B, CDK2/cyclin A, CDK2/cyclin
E and CDK5, and leads to induction of p53 (10,11).
Furthermore, roscovitine inhibits the growth of several human
cancer cell lines including breast, ovarian, pancreatic, colon,
renal, hepatocellular, pituitary carcinoma and neuroblastoma
(12–18). Roscovitine has been found to inhibit
different solid and hematological tumor cell lines including acute
lymphoblastic leukemia (ALL), which occurs frequently in children
and is correlated with the central nervous system (CNS) (19,20).
The drug inhibits the G1/S and G2/M transition in a concentration
dependent manner. Other studies reported an inhibition of the DNA
synthesis in the cerebral cortex, an effect that appears to be
independent from its ability to inhibit CDKs or the replication
licensing factor (RLF) (10,11,21).
The concentration and half-life of roscovitine are almost similar
in plasma and in brain tissues. Roscovitine is metabolized in
humans mainly by CYP3A4 and CYP2B6 enzymes, and its elimination is
rapid via the bile within the first 24 h (22,23).
Most chemotherapeutic agents do not cross the blood-brain barrier
and do not reach the CNS in adequate concentrations to eliminate
tumor cells, while roscovitine is highly distributed over the
blood-brain barrier (24).
Roscovitine is a potential inhibitor of CDK5, which has a
significant function in the developing brain, such as neuronal
migration (23). A functional p53
protein level was suggested as an efficient enhancer in
roscovitine-induced apoptosis in cancer. Roscovitine-induced
apoptosis was shown to be p53-dependent in MCF-7 cells, whereas
roscovitine may induce apoptosis in B-CLL cells, irrespective of
the functional status of the p53 pathway, and may be considered a
therapeutic agent able to improve the outcome of B-CLL-resistant
tumors. Therefore, the possible relationship between roscovitine
and p53 regulation remains to be elucidated (25–27).
Furthermore, roscovitine decreased production of the cell cycle
inhibitor p21 and induced apoptosis. This effect was observed as
the most efficient in cell lines expressing p53 protein with a full
activity. The cells expressing partially and conditionally active
p53wt mutants responded to roscovitine less efficiently. This
observation suggests that patients with tumors exhibiting p53 can
benefit from roscovitine therapy (28).
In the present study, we investigated the effects of
roscovitine on proliferation, apoptosis and cell cycle regulation
in glioblastoma cell cultures. Due to selective inhibition of CDKs
by roscovitine, particularly CDK1, 2, 5, 7 and 9, which are
involved in the cell cycle regulation, we assessed the interaction
between roscovitine and CDKs. Furthermore, we investigated CDK
binding partners including cyclin A, D and E and tumor-suppressor
proteins p21 and p53. The results identified the influence of
roscovitine in apoptosis in glioblastoma cell line as a promising
therapeutic agent.
Materials and methods
Cell culture
The A172 and NCE-G28 cell lines were obtained from
the American Type Culture Collection (ATCC, Manassas, VA, USA). The
two cell lines were routinely cultured in plastic flasks (75
cm2) or 6-well-plates in Dulbecco's modified eagle's
medium (DMEM; Biochrom GmbH, Berlin, Germany) supplemented with 10%
fetal bovine serum (FBS; Biochrom) and 1 ppm antibiotics
(penicillin and streptomycin; Biochrom). The cells were incubated
at 37°C, under 95% humidity and 5% CO2 saturation. At
70–80% confluence, the cells were passaged by being washed with PBS
and trypsinized with trypsin/EDTA (both from Biochrom) for 3–7 min.
PBS and trypsin/EDTA were preheated at 37°C in a water bath. The
procedures were performed under sterile conditions in a laminar
flow cabinet.
Drugs and treatments
Roscovitine was prepared as a 1M stock solution in
dimethyl sulfoxide (DMSO; Sigma Aldrich, Hamburg, Germany). Stock
solution was diluted in culture medium in order to obtain the final
concentrations of 1, 10, 25, 50 and 100 µM for the cell
treatment. Aliquots of the roscovitine stock and the final
concentrations were stored until use at-20°C. The cells were
treated with roscovitine 24 h after seeding to guarantee adherence.
In the presence of the drug, the cells were incubated for 24–96 h.
Controls were treated with complete medium as described above while
omitting the drug.
xCELLigence
Impedance-based measurement of cell proliferation
and IC50 were performed with the xCELLigence Real-Time
Cell Analyzer (RTCA) in 96-well plates (E-plates) (all from Roche
Applied Science, Penzberg, Germany) under standard culture
conditions as described above. The readout recorded by the RTCA is
a dimensionless cell index (CI) value that correlates with cell
number. The setup of the experiment and recording of data included
background measurement with 100 µl of cell-free medium,
followed by the addition of a drug-free cell suspension
(104 cells/50 µl/well). The cells were
continuously measured every 15 min and allowed to adhere and
proliferate for 24 h in drug-free medium. For the treatment, the
medium was completely replaced with roscovitine-containing medium
in serial concentrations of 1, 10, 25, 50 and 100 µM or
drug-free DMSO-containing medium for the controls. Readings were
performed for at least 96 h after the treatment. Experiments were
performed in FBS-deficient-medium (0.125% FBS) to assess the drug
response under starvation. The samples were analyzed at least in
triplicate.
The RTCA-software version 1.2.2 was used to analyze
the data. Curves were normalized to the time-point just prior to
adding the drug, to which a CI of 1 was assigned.
Flow cytometry (FACS)
Cell cycle analysis was performed using flow
cytometry. Cells were seeded in triplicates in 6-well plates
(3×105/well) and allowed to attach for 24 h.
Subsequently, the cells were treated with roscovitine at the final
concentrations of 10, 25, 50 and 100 µM. After an additional
incubation period of 24–96 h, the cells were washed with PBS
(Biochrom) and collected by standard trypsinization. The collected
medium, the PBS and the cell-trypsin suspension were centrifuged
for 10 min at 1,000 rpm. Cell pellets were resuspended with
propidium iodide (PI; Sigma Aldrich) at a ratio of 105
cells/ml and incubated for 60 min on ice in the dark. Analysis was
immediately performed. For the evaluation of the assays and the
data, the BD FACSCalibur flow cytometer and the corresponding
CellQuest™ Pro Software (BD Biosciences, San Jose, CA, USA) were
used.
RNA isolation, cDNA synthesis and
real-time-quantitative PCR (RT-qPCR)
Cells were collected using standard trypsinization
as described above and lysed using QIAzol Lysis reagent (Qiagen,
Hilden, Germany), followed by RNA isolation with the RNeasy Mini
kit (Qiagen), as per the manufacturer's instructions. RNA
concentration was measured photometrically using the nanoDrop
nD-1000 Sprectrophotometer (Thermo Fisher Scientific, Wilmington,
DE, USA). Five-hundred nanograms of total RNA were used for cDNA
reverse transcription by using the iScript™ cDNA synthesis kit
(Bio-Rad Laboratories GmbH, Munich, Germany). RT-qPCR was performed
using the SsoFast evaGreen Supermix kit (Bio-Rad) and the primers:
CDK2 (cat. no. QT00005586), CDK7 (QT00028700), CDK9 (QT00208523),
cyclin A (QT00014798), cyclin D (QT00057575), cyclin E
(QT00063511), p21 (QT00062090), p53 (QT00060235) and GAPDH
(QT01192646) (all from Qiagen). RT-qPCR was performed on a CFX96
Real-Time PCR system (Bio-Rad).
Protein isolation and western blot
analysis
Cells were trypsinized as described before,
centrifuged for 12 min at 4°C and 1,000 rpm, resuspended and washed
in 1 ml PBS and pelleted at 1,000 rpm for 12 min. The pellet was
resuspended in 80–200 µl Jie's Buffer and allowed to
dissolve on ice for 30 min with intermittent vortexing. After
centrifugation at 13,000 rpm for 30 min the protein-enriched
suspension was transferred into a new tube and stored at -80°C
until further processing.
Protein quantification was performed using the
Pierce BCA Protein Assay kit (Thermo Scientific) as per the
manufacturer's instructions. The readout was carried out on a
SoftMax microplate reader using the Softmax Pro 3.11 software
(Molecular Devices, Sunnyvale, USA). Gel electrophoresis was
performed using a 30 µg protein solution. Antibodies were
incubated using the Snap i.d. protein detection system (Millipore,
Billerica, MA, USA) according to the manufacturer's instructions.
Chemiluminescence detection was performed with the Fusion-SL4
system and the Fusion-Capt software 15.15 by using the Pierce™ ECL
Western Blotting Substrate (Thermo) and quantified using the Bio-1D
package v.15.01 software (all from Vilber Lourmat, Eberhardzell,
Germany). β-actin was used as the loading control. The antibodies
used were: anti-Cdk2 (rabbit polyclonal, 1:300, cat no. ab6538) and
anti-p21 (mouse monoclonal EA10, 1:100, ab16767; both from Abcam,
Cambridge, UK), anti-β-actin (mouse monoclonal, clone AC-15,
1:3,000), and anti-mouse and anti-rabbit secondary antibodies
(1:3,000, all from Sigma-Aldrich, St. Louis, USA).
Statistics
Statistic analysis and scientific graphing were
performed using SPSS 20 (IBM software; Ehningen, Germany) and Prism
6 (GraphPad Software, Inc., La Jolla, CA, USA). The flow cytometric
results were analyzed using the ANOVA test and corrected using the
Dunnett's multiple comparison test for multiple comparisons.
P<0.05 was considered statistically significant.
Results
Proliferation analysis
The effects of roscovitine on proliferation were
examined in A172 and G28 glioma cell lines.
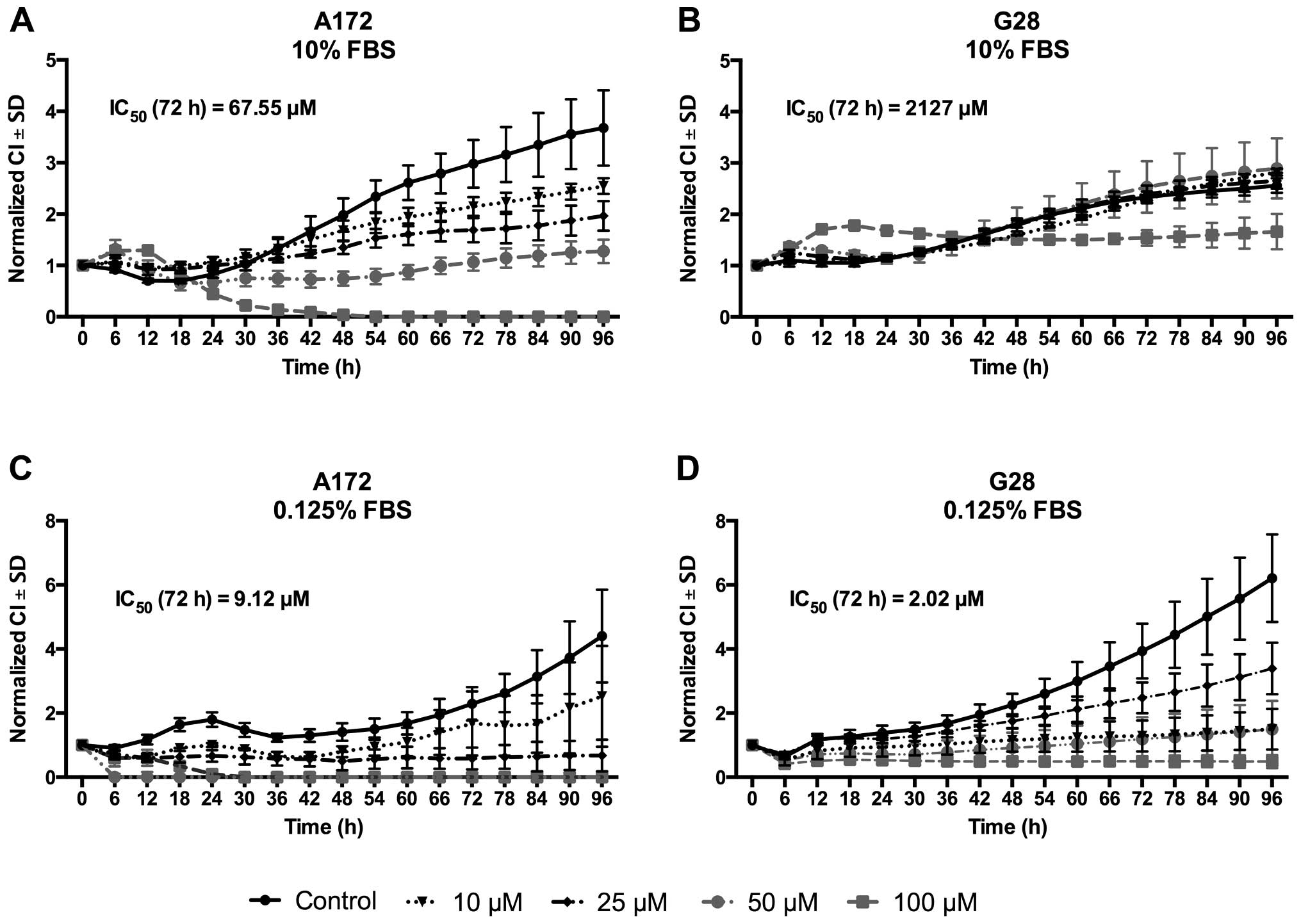
Roscovitine induced a dose-dependent growth
inhibition in A172 cells up to 50 µM and a distinct
reduction of cell viability at 100 µM already after 24 h of
treatment. IC50 at 72 h was 67.55 µM. G28 cells
were less sensitive and cells proliferated in a similar manner as
the untreated controls at concentrations of up to 50 µM. One
hundred micromoles roscovitine exerted only a cytostatic effect,
resulting in an IC50 of 2,127 µM (Fig. 1).
When the same experiment was performed with
FBS-deficient medium (0.125% FBS), IC50 at 72 h was
decreased to 9.12 µM in the A172 cells and to 2.02 µM
in G28 cells. Under these conditions, there was an evident
dose-dependent effect in G28 cells, in contrast to culturing with
10% FBS. Proliferation was similar under both conditions. The cells
were monitored for up to 96 h to demonstrate continued
proliferation even under nutrition-deficient conditions. The A172
cells reached a mean normalized cell index (CI) in the untreated
controls of 4.4 after 96 h (vs. 3.6 when grown with 10% FBS).
However, the G28 cells proliferated stronger with a mean CI after
96 h of 6.2 (vs. 2.5 when grown with 10% FBS). For comparable
results with other studies on these cell lines, all subsequent
experiments were performed using 10% FBS.
Cell cycle analysis by flow
cytometry
To determine by which mechanism roscovitine
inhibited cell proliferation, we analyzed cell cycle distribution
by flow cytometry. After 72 h incubation, there was a clear
dose-dependent increase of the pre-G1 cell fraction in the A172
cells from 2.5% in the controls, to 4.4, 3.9, 8.7 and 16.7% at 10,
25, 50 and 100 µM, respectively (Fig. 2A). At the same time-point, there was
an increase of the G2/M fraction from 26.9 in the controls to 29.7,
37.8, 48.2 and 54.5% at these concentrations. The G1/S percentage
was reduced from 70.6 in the controls to 28.8% in cells treated
with 100 µM roscovitine. After 96 h, the percentage of
apoptotic cells reached 54.2 and 55.1% at 50 and 100 µM,
respectively, vs. 3.6 in the untreated controls (Fig. 2B). The G1/S phase decreased from
80.8 in the controls to 17.9% at 100 µM and at the same time
the G2/M percentage increased from 15.6 in the untreated controls
to 26.9% at 100 µM.
In G28 cells, a significant increase of the
apoptotic cell fraction was only observed at 100 µM
roscovitine at the 72 h time-point and levels ranged from 0.9% in
the controls to 1.2, 1.0, 0.6 and 20.5% at 10, 25, 50 and 100
µM, respectively (Fig. 2C).
However, the G1/S fraction decreased from 41.5 in the controls to
38.0, 42.3, 31.0 and 24.1% at 10, 25, 50 and 100 µM,
respectively, and cells in the G2/M phase amounted to 57.6 in the
controls and 61.1, 56.7, 68.3 and 55.4% at the same concentration
range, respectively. A further increase of the apoptotic fraction
was observed after 96-h incubation with roscovitine, reaching 12.1%
already at 50 µM and 49.9% at 100 µM (vs. 2.6% in the
controls). At this time-point, 50 and 100 µM roscovitine
further reduced G1/S cells to 12.9 and 15.2%, respectively. The
G2/M cell fraction fluctuated at 61.2, 64.2 and 75.0% at 10, 25 and
50 µM, and only at 100 µM roscovitine was the level
reduced to 34.9% (vs. 69.6 in the untreated controls).
Expression analysis of cell cycle
regulator genes
A qPCR analysis for cell cycle regulators cyclins A,
D and E, CDK2, CDK7, CDK9, p21 and p53 was performed in the two
cell lines after 24, 48 and 72 h of treatment with roscovitine
(Fig. 3). Under incubation with
roscovitine at 10, 25, 50 and 100 µM for 72 h, we measured
dose-and time-dependent changes in all the genes investigated. In
A172 cells, roscovitine induced the expression of CDK2 and cyclin D
and suppressed p21, p53, CDK7, CDK9, cyclins A and E at 72 h. At
the growth-inhibiting concentrations of 10, 25 and 50 µM,
CDK2 expression was increased 1.1-, 1.3- and 1.5-fold,
respectively. At 100 µM, the concentration leading to cell
death, decreased to 0.3-fold of the untreated controls. The
expression of p21 was steadily induced over the dosing range,
reaching a peak of 4.6-fold at 100 µM roscovitine vs. The
untreated controls. The expression of p53, CDK7 and 9 and cyclins A
and E was gradually suppressed with increasing roscovitine
concentrations and at 100 µM reached values of 0.3-, 0.02-,
0.04- and 0.01-fold of the untreated controls, respectively. Cyclin
D was not detected using qPCR in these cells.
In the less roscovitine-sensitive G28 cells, the
gene expression patterns followed the trend identified in A172
cells, reaching expression levels of 0.3 for p53, 0.5-fold for
CDK7, 0.2-fold for CDK 9, 0.03-fold for cyclin A and 0.1-fold for
cyclin E. CDK 2 also increased slightly and steadily at 10, 25 and
50 µM (1.3, 1.4 and 1.4-fold) and at 100 µM showed
similar decreases as identified in A172 cells (0.3-fold). In
contrast to the A172 cells, G28 cells expressed cyclin D, which was
induced by the lower roscovitine concentrations of 10 and 25
µM (2.1- and 2.8-fold, respectively) and decreased at 50 and
100 µM to 1.0- and 0.5-fold, respectively. The expression of
p21 was induced to 1.3-, 1.3- and 1.6-fold at 10, 25 and 50
µM and decreased to 0.2-fold at 100 µM. The clear
dose-dependent effects observed in the proliferation and cell cycle
analyses of the A172 cells were reflected in the gene expression
and cell cycle studies, while the roscovitine effects of the G28
cells was less definite in all the analyses at 10% FBS.
Analysis of CDK2 and p21 expression by
western blot analysis
To confirm whether the expression changes observed
at the mRNA level as a consequence of roscovitine treatment
translate to the protein level, we analyzed the protein expression
of CDK2 and p21 by western blot analysis. In the A172 cells,
roscovitine treatment induced CDK2 expression already after 24 h,
with the strongest upregulation evident at 50 µM
(2.44-fold). At the longer incubation period of 48 h, CDK2 was
upregulated 3.50-fold at 50 and 100 µM but only 1.66-fold at
25 µM. The protein expression of p21 was strongly induced
with increasing concentrations already after 24 h to values of
4.06-, 10.41- and 43.93-fold at 25, 50 and 100 µM,
respectively. A less notable upregulation was observed after 48 h
with values reaching 0.86-, 1.17- and 2.30-fold of untreated
controls for the same concentration range.
The concentration of 50 µM appeared to be
most efficient in upregulating CDK2 in G28 cells. After 24 h, CDK2
was upregulated at the protein level to 1.50-fold of the untreated
controls and 25 and 100 µM concentrations only resulted in
values of 1.38- and 0.85-fold, respectively. In these cells, 72 h
of incubation resulted in a suppression of CDK2 protein to 0.65-,
0.39- and 0.67-fold at 25, 50 and 100 µM, respectively.
Levels of p21 at 24 h were reduced at 25 and 100 µM in these
cells (0.22- and 0.39-fold, respectively) or were not altered at 50
µM (1.01-fold). After the longer incubation of 72 h, higher
concentrations of roscovitine led to a strong downregulation of p21
to 0.19,- 0.18- and 0.13-fold at 25, 50 and 100 µM. Of note,
the two cell lines behaved in an opposite manner with respect to
CDK2 and p21 expression, i.e., while A172 cells upregulated CDK2
and p21 when challenged with roscovitine, G28 cells suppressed the
two proteins.
Discussion
GBM is the most common and most aggressive primary
brain tumor in adults. Despite intensive therapeutic efforts, the
prognosis remains poor (1). A
successful multimodal therapy in this cancer needs to target
multiple pathways, achieve sufficient concentration at the site of
action within the brain and the treatment administered should have
few side effects.
In the present study, we investigated the effect on
the expression of CDK2, CDK7, CDK9 and cyclin A, D and E by using
roscovitine in glioblastoma cells. We demonstrated that roscovitine
induced cell cycle arrest in the G28 and A172 cell lines, which was
paralleled by a decreased expression of CDK7, CDK9 and cyclin A, D
and E. Furthermore, roscovitine induced apoptosis and decreased
cell proliferation of the GBM cells.
Lim et al suggested that several modes of
action can be postulated by the gene expression patterns observed
after treatment with roscovitine. One of these modes of action
leads to a reduced expression of CDK7, CDK 9 and cyclin D, the
release inhibition of E2F and therefore suppression of
e2F-dependent transcription, as well as suppression of the
transcription by inhibiting RNA polymerase II-dependent
transcription (29).
Previous findings have shown that cyclin E is often
over-expressed in human tumors and the expression of the p21 and
p27 inhibitors is often suppressed during tumor growth. This result
demonstrates an involvement of CDK2 in human cancer (18). Other studies have shown that
roscovitine induces cell cycle arrest by inhibiting CDK2 through
competition for ATP-binding (10,30).
Our results show distinct expression patterns in the two
investigated cell lines, suggesting that different modes of action
may cause the cell cycle effects observed, i.e., while the CDK2
expression was suppressed only at the highest concentrations, we
observed a clearer and dose-dependent effect for p21, p53, CDK7 and
cyclin A. Our data suggest that the observed effects of roscovitine
on proliferation and cell survival depend on the inhibition of
various kinases, as shown by Bach et al (31). On the other hand, a potent
inhibition of the CDK2/cyclin E complex by roscovitine has already
been shown in clinical trials (32). Therefore, roscovitine affects more
pathways simultaneously and its specific inhibition of CDKs is not
its only effect.
Roscovitine also inhibits CDK7, forming an enzyme
complex with cyclin H. These data provide a direct connection
between cell cycle regulation and transcription, CDK7 and cyclin H
which form CAK, and a constituent of the basal transcription factor
TFIIF, which phosphorylates serine residues within the heptapeptide
repeat of the carboxyterminal domain (CTD) of RNA polymerase II
(33).
Furthermore, roscovitine inhibits cyclin H,
cyclinD/CDK4 and cyclinD/CDK6 complexes by inhibiting CDK7 and
reducing the cyclin D expression. This is due to the fact that CDK
activation requires cyclin binding for conformational changes in
the tertiary structure as well as phosphorylation at a conserved
threonine residue. Cyclin binding leads to conformational changes
in the tertiary structure of the CDKs, including the ATP-binding
side, which allows subsequent phosphorylation that is required for
complete CDK activation. Phosphorylation is catalyzed by the CAK
(34,35). CAK comprises a CDK7 complex with
cyclin H and Mat1 (33). In
quiescent cells and at the beginning of the G1 phase, E2F is
controlled by pRB (35,36). As soon as pRB is phosphorylated by
cyclinD/CDK4 and cyclinD/CDK6 and subsequently by cycline/CDK2, E2F
is released and transcription initiated (35,37).
Collectively, CDK2, CDK4, CDK6 and cyclin D are necessary for
transcription. Most probably, roscovitine inhibits the activation
of cyclinD/CDK4 and cyclinD/CDK6 by suppressing the expression of
CDK7 and cyclin D, leading to inhibition of the transcription.
Thus, E2F-dependent transcription of cycle relevant proteins,
including cyclin A and E, is suppressed, leading to cell cycle
arrest at different stages. RNA-polymerase II performs the actual
transcription. For activation it requires phosphorylation of its
C-terminal domain (CDT). CDK7/cyclinH/Mat1 CAK, which are
components of the TFIIH complex, and CDK9/cyclinT (pTEFb)
phosphorylate the CDT of RNA polymerase II (38). Thus, roscovitine may also inhibit
transcription by impairing RNA-polymerase II activation.
Our results are not consistent with those of other
studies which showed an increased mRNA expression of cyclin A, B, D
(32,39) and CDK7 and 9 (26,39) in
various types of cancer, but not glioma cells.
Similar to our gene expression results in the
glioblastoma cell lines, Whittaker et al showed reduced
cyclin A and D expression at the protein level by western blot
analysis in human colon cancer cells (32). This decrease in phosphorylation may
contribute to or even cause the decreased cyclin expression. This
increased phosphorylation may most likely be due to an inhibition
of CDK7 and 9 (32,39). Similar mechanisms likely caused the
observed effects in the present study, although functional analyses
to confirm these results were not performed.
Previous findings showed that roscovitine induced
cell cycle arrest and concomitantly apoptosis in tumor cells of
breast and colon cancer (14,35)
mostly due to mitochondrial-mediated apoptosis activation of
caspase-3, -8 (40) and -9
(34). Our results are in agreement
with observations by Wȩsierska-Gądek et al (26,30),
showing that roscovitine induces G2/M-phase cell cycle arrest in
tumor cells. In our study we observed progressive G2/M arrest
accompanied by decreasing G1/S fraction in A172 and G28 cell lines.
The A172 cells showed a time-and concentration-dependent G2/M
arrest in almost all the samples, whereas G28 cells showed G2/M
arrest after treatment with highest concentrations only. As the
G2/M phase is known as the most radio-sensitive phase in the cell
cycle (41), roscovitine may be
useful in combination with radiotherapy, which is also part of the
current therapeutic standard for glioblastoma.
McClue et al (22) identified that the major effect of
roscovitine is not by cell cycle arrest in a specific phase but
rather an induction of apoptosis in different cell cycle phases.
However, findings by those authors cannot be supported with the
data of the present study.
Regarding our FACS results, we identified a
concentration-and time-dependent induction of apoptosis with
roscovitine treatment, accompanied by a decreased CDK7 and 9
expression. Previous results have shown that roscovitine induces
apoptosis by inhibiting RNA polymerase II-dependent transcription
of the anti-apoptotic Bcl-2-family member Mcl-1 (myeloid cell
leukemia 1) (38,40). Those studies showed a decreased CDK7
and 9 expression due to roscovitine treatment with a consecutive
decrease of RNA polymerase II phosphorylation leading to its
depression. Using qPCR we determined a decreased expression of CDK7
and 9. Even in this tumor entity, roscovitine was capable of
inducing apoptosis by disabling the RNA polymerase II-dependent
transcription of Mcl-1 due to a lack of CDK7 and 9 expression.
Previous studies reported a p53 increase in
glioblastoma cells following roscovitine treatment (16,26).
There are p53-dependent and-independent pathways described in the
literature (10,16,18,26)
with respect to apoptosis. However, there is a greater potency
described against p53 wild-type cells than against mutant cells
(10,16,18,26).
In the present study, the expression response of p53 under
roscovitine incubation was different in the two cell lines. This is
explained by the fact that one of the most important differences
between the two cell lines is the p53 status. A172 cells bear
wild-type p53, while a mutated p53 is present in the G28
gliosarcoma cell line (42,43). It is likely that the stronger
roscovitine influence identified in the A172 cells is likely due to
the wild-type p53.
Previous findings have shown that roscovitine
decreased the expression of p21 and p21 cleavage was involved in
roscovitine induced apoptosis. Even in earlier studies on
roscovitine it was presumed that a high expression of p21 inhibited
the induction of apoptosis (28,44–46).
That finding is similar to our results for the G28 cells. The
inhibition of p21 was time-and concentration-dependent. The
investigation of A172 showed similar results until 48 h. The
expression of p21 after application of 100 µM of roscovitine
after 72 h was higher. This result can be interpreted as a
regulatory mechanism to escape apoptosis.
In conclusion, our pilot study demonstrates an
anti-proliferative and pro-apoptotic effect of roscovitine on two
human glioblastoma cell lines in vitro, accompanied by
distinct changes in gene expression of CDKs and cyclins, along with
p53 and p21. Given the good oral bioavailability of roscovitine and
its effect on the G2/M cell cycle phase, which is most sensitive to
radiotherapy, this drug should be investigated in depth in further
pre-clinical and clinical studies as it shows to be a promising
agent against glioblastoma alone or in combination therapy
(24,47).
Abbreviations:
|
ALL
|
acute lymphatic leukemia
|
|
CAK
|
CDK activating kinase
|
|
CDK
|
cyclin-dependent kinase
|
|
CNS
|
central nervous system
|
|
CTD
|
c-terminal-domain
|
|
DMSO
|
dimethyl sulfoxide
|
|
FACS
|
flow cytometry
|
|
FBS
|
fetal bovine serum
|
|
GBM
|
glioblastoma
|
|
RLF
|
replication licensing factor
|
|
RTCA
|
real-time cell analyzer
|
References
|
1
|
Maher EA, Furnari FB, Bachoo RM, Rowitch
DH, Louis DN, Cavenee WK and DePinho RA: Malignant glioma: Genetics
and biology of a grave matter. Genes Dev. 15:1311–1333. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hengstschläger M, Hölzl G and
Hengstschläger-Ottnad E: Different regulation of c-Myc- and
E2F-1-induced apoptosis during the ongoing cell cycle. Oncogene.
18:843–848. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiffer D, Cavalla P, Migheli A, Chiò A,
Giordana MT, Marino S and Attanasio A: Apoptosis and cell
proliferation in human neuroepithelial tumors. Neurosci Lett.
195:81–84. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Costello JF, Plass C, Arap W, Chapman VM,
held WA, Berger MS, Su Huang HJ and Cavenee WK: Cyclin-dependent
kinase 6 (CDK6) amplification in human gliomas identified using
two-dimensional separation of genomic DNA. Cancer Res.
57:1250–1254. 1997.PubMed/NCBI
|
|
5
|
Dirks PB, Hubbard SL, Murakami M and Rutka
JT: Cyclin and cyclin-dependent kinase expression in human
astrocytoma cell lines. J Neuropathol Exp Neurol. 56:291–300. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olson JJ, Barnett D, Yang J, Assietti R,
Cotsonis G and James CD: Gene amplification as a prognostic factor
in primary brain tumors. Clin Cancer Res. 4:215–222.
1998.PubMed/NCBI
|
|
7
|
Ono Y, Tamiya T, Ichikawa T, Kunishio K,
Matsumoto K, Furuta T, Ohmoto T, Ueki K and Louis DN: Malignant
astrocytomas with homozygous CDKN2/p16 gene deletions have higher
Ki-67 proliferation indices. J Neuropathol Exp Neurol.
55:1026–1031. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rollbrocker B, Waha A, Louis DN, Wiestler
OD and von Deimling A: Amplification of the cyclin-dependent kinase
4 (CDK4) gene is associated with high cdk4 protein levels in
glioblastoma multiforme. Acta Neuropathol. 92:70–74. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Srivenugopal KS and Ali-Osman F: Deletions
and rearrangements inactivate the p16INK4 gene in human glioma
cells. Oncogene. 12:2029–2034. 1996.PubMed/NCBI
|
|
10
|
Meijer L, Borgne A, Mulner O, Chong JP,
Blow JJ, Inagaki N, Inagaki M, Delcros JG and Moulinoux JP:
Biochemical and cellular effects of roscovitine, a potent and
selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and
cdk5. Eur J Biochem. 243:527–536. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schutte B, Nieland L, Van Engeland M,
Henfling ME, Meijer L and Ramaekers FC: The effect of the
cyclin-dependent kinase inhibitor olomoucine on cell cycle
kinetics. Exp Cell Res. 236:4–15. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nair BC, Vallabhaneni S, Tekmal RR and
Vadlamudi RK: Roscovitine confers tumor suppressive effect on
therapy-resistant breast tumor cells. Breast Cancer Res.
13:R802011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pizarro JG, Folch J, Junyent F, Verdaguer
E, Auladell C, Beas-Zarate C, Pallàs M and Camins A: Antiapoptotic
effects of roscovitine on camptothecin-induced DNA damage in
neuro-blastoma cells. Apoptosis. 16:536–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arısan ED, Coker A and Palavan-Ünsal N:
Polyamine depletion enhances the roscovitine-induced apoptosis
through the activation of mitochondria in HCT116 colon carcinoma
cells. Amino Acids. 42:655–665. 2012. View Article : Google Scholar
|
|
15
|
Cho SJ, Kim YJ, Surh YJ, Kim BM and Lee
SK: Ibulocydine is a novel prodrug Cdk inhibitor that effectively
induces apoptosis in hepatocellular carcinoma cells. J Biol Chem.
286:19662–19671. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu NA, Jiang H, Ben-Shlomo A, Wawrowsky
K, Fan XM, Lin S and Melmed S: Targeting zebrafish and murine
pituitary corti-cotroph tumors with a cyclin-dependent kinase (CDK)
inhibitor. Proc Natl Acad Sci USA. 108:8414–8419. 2011. View Article : Google Scholar
|
|
17
|
Coley HM, Safuwan NA, Chivers P,
Papacharalbous E, Giannopoulos T, Butler-Manuel S, Madhuri K,
Lovell DP and Crook T: The cyclin-dependent kinase inhibitor
p57(Kip2) is epigenetically regulated in carboplatin resistance and
results in collateral sensitivity to the CDK inhibitor seliciclib
in ovarian cancer. Br J Cancer. 106:482–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wellwood J and Taylor K: Central nervous
system prophylaxis in haematological malignancies. Intern Med J.
32:252–258. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sallam H, Jimenez P, Song H, Vita M,
Cedazo-Minguez A and Hassan M: Age-dependent pharmacokinetics and
effect of rosco-vitine on Cdk5 and Erk1/2 in the rat brain.
Pharmacol Res. 58:32–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yakisich JS, Sidén A, Idoyaga Vargas V,
Eneroth P and Cruz M: Early inhibition of DNA synthesis in the
developing rat cerebral cortex by the purine analogues olomoucine
and roscovitine. Biochem Biophys Res Commun. 243:674–677. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McClue SJ and Stuart I: Metabolism of the
trisubstituted purine cyclin-dependent kinase inhibitor seliciclib
(R-roscovitine) in vitro and in vivo. Drug Metab Dispos.
36:561–570. 2008. View Article : Google Scholar
|
|
23
|
Dhavan R and Tsai LH: A decade of CDK5.
Nat Rev Mol Cell Biol. 2:749–759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hassan M, Sallam H and Hassan Z: The role
of pharmaco kinetics and pharmacodynamics in early drug development
with reference to the cyclin-dependent kinase (Cdk)
inhibitor-roscovitine. Sultan Qaboos Univ Med J. 11:165–178.
2011.PubMed/NCBI
|
|
25
|
Paprskárová M, Krystof V, Jorda R, Dzubák
P, Hajdúch M, Wesierska-Gadek J and Strnad M: Functional p53 in
cells contributes to the anticancer effect of the cyclin-dependent
kinase inhibitor roscovitine. J Cell Biochem. 107:428–437. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wesierska-Gadek J, Borza A, Komina O and
Maurer M: Impact of roscovitine, a selective CDK inhibitor, on
cancer cells: Bi-functionality increases its therapeutic potential.
Acta Biochim Pol. 56:495–501. 2009.PubMed/NCBI
|
|
27
|
Alvi AJ, Austen B, Weston VJ, Fegan C,
MacCallum D, Gianella-Borradori A, Lane DP, Hubank M, Powell JE,
Wei W, et al: A novel CDK inhibitor, CYC202 (R-roscovitine),
overcomes the defect in p53-dependent apoptosis in B-CLL by
down-regulation of genes involved in transcription regulation and
survival. Blood. 105:4484–4491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Slovackova J, Smarda J and Smardova J:
Roscovitine-induced apoptosis of H1299 cells depends on functional
status of p53. Neoplasma. 59:606–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim YM, Yamasaki Y and Tsuda L: Ebi
alleviates excessive growth signaling through multiple epigenetic
functions in Drosophila. Genes Cells. 18:909–920. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Węsierska-Gądek J, Gritsch D, Zulehner N,
Komina O and Maurer M: Roscovitine, a selective CDK inhibitor,
reduces the basal and estrogen-induced phosphorylation of ER-α in
human ER-positive breast cancer cells. J Cell Biochem. 112:761–772.
2011. View Article : Google Scholar
|
|
31
|
Bach S, Knockaert M, Reinhardt J, Lozach
O, Schmitt S, Baratte B, Koken M, Coburn SP, Tang L, Jiang T, et
al: Roscovitine targets, protein kinases and pyridoxal kinase. J
Biol Chem. 280:31208–31219. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Whittaker SR, Walton MI, Garrett MD and
Workman P: The cyclin-dependent kinase inhibitor CYC202
(R-roscovitine) inhibits retinoblastoma protein phosphorylation,
causes loss of cyclin D1, and activates the mitogen-activated
protein kinase pathway. Cancer Res. 64:262–272. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bertoli C, Klier S, McGowan C, Wittenberg
C and de Bruin RA: Chk1 inhibits e2F6 repressor function in
response to replication stress to maintain cell-cycle
transcription. Curr Biol. 23:1629–1637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fischer PM and Lane DP: inhibitors of
cyclin-dependent kinases as anti-cancer therapeutics. Curr Med
Chem. 7:1213–1245. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
MacCallum DE, Melville J, Frame S, Watt K,
Anderson S, Gianella-Borradori A, Lane DP and Green SR: Seliciclib
(CYC202, R-Roscovitine) induces cell death in multiple myeloma
cells by inhibition of RNA polymerase II-dependent transcription
and down-regulation of Mcl-1. Cancer Res. 65:5399–5407. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Erguven M, Bilir A, Yazihan N, Korkmaz S,
Aktas E, Ovalioglu C, Dundar T and Seyithanoglu H: Imatinib
mesylate decreases the cytotoxic effect of roscovitine on human
glioblastoma cells in vitro and the role of midkine. Oncol Lett.
3:200–208. 2012.PubMed/NCBI
|
|
39
|
Whittaker SR, Te Poele RH, Chan F,
Linardopoulos S, Walton MI, Garrett MD and Workman P: The
cyclin-dependent kinase inhibitor seliciclib (R-roscovitine;
CYC202) decreases the expression of mitotic control genes and
prevents entry into mitosis. Cell Cycle. 6:3114–3131. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Murphy ÁC, Weyhenmeyer B, Noonan J,
Kilbride SM, Schimansky S, Loh KP, Kögel D, Letai AG, Prehn JH and
Murphy BM: Modulation of Mcl-1 sensitizes glioblastoma to
TRAIL-induced apoptosis. Apoptosis. 19:629–642. 2014. View Article : Google Scholar :
|
|
41
|
Pawlik TM and Keyomarsi K: Role of cell
cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol
Biol Phys. 59:928–942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sato Y, Kurose A, Ogawa A, Ogasawara K,
Traganos F, Darzynkiewicz Z and Sawai T: Diversity of DNA damage
response of astrocytes and glioblastoma cell lines with various p53
status to treatment with etoposide and temozolomide. Cancer Biol
Ther. 8:452–457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akudugu J, Gäde G and Böhm L: Cytotoxicity
of azadirachtin A in human glioblastoma cell lines. Life Sci.
68:1153–1160. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang T, Jiang T, Zhang F, Li C, Zhou YA,
Zhu YF and Li XF: Involvement of p21Waf1/Cip1 cleavage
during roscovitine-induced apoptosis in non-small cell lung cancer
cells. Oncol Rep. 23:239–245. 2010. View Article : Google Scholar
|
|
45
|
Schmidt M and Fan z: Protection against
chemotherapy-induced cytotoxicity by cyclin-dependent kinase
inhibitors (CKI) in CKI-responsive cells compared with
CKI-unresponsive cells. Oncogene. 20:6164–6171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
47
|
Neyns B, D'haeseleer M, Rogiers A, Van de
Cauter J, Chaskis C, Michotte A and Strik H: The role of cytotoxic
drugs in the treatment of central nervous system gliomas. Acta
Neurol Belg. 110:1–14. 2010.PubMed/NCBI
|