Introduction
Hepatocellular carcinoma (HCC) is the one of the
most common malignancies and the second cause of cancer-related
deaths worldwide (1,2). More than 600,000 deaths are reported
annually (3). In the past few
years, early diagnosis and advances in therapeutic measures, such
as surgical resection and liver transplantation, have greatly
improved the outcome of HCC patients (4,5).
However, the general prognosis is still poor with overall survival
rates of 3–5% (6,7). In spite of well-established
surveillance programs in patients with chronic liver disease, more
than 85% of the cases are diagnosed at an intermediate-advanced
stage, which is not suitable for curative management (8,9). In
these cases, local surgical resection with chemotherapy is shown to
decrease the mortality rate of HCC (10). However, the option of chemotherapy
for HCC is limited and drug-resistance occurs in many of the HCC
patients. Novel candidate agents for use in chemotherapy for HCC
are urgently needed to control the development of HCC.
Pterostilbene (PTE),
trans-3,5-dimethoxy-4′-hydroxystilbene, is a natural
dimethylated analog of resveratrol. It is reported that PTE
possesses diverse pharmacological activities, including antitumor,
anti-inflammatory, antioxidant, antiproliferative and analgesic
activities (11–13). In recent years, much attention has
been given to the antitumor effect of PTE. Feng et al
(14) found that PTE is a potent
anticancer pharmaceutical against human esophageal cancer. Dhar
et al (15) discovered that
PTE is a promising natural agent for use as a chemopreventive and
therapeutic strategy to curb prostate cancer. PTE derivatives have
been found to suppress tumors, such as osteoclastogenesis (16) and colon cancer (17). Moreover, PTE was found to have an
inhibitory effect on the growth and invasion of HCC cells (12,18).
However, the mechanism involved in the inhibitory effect of PTE on
HCC is far from completely understood.
In the present study, we aimed to examine the effect
of PTE on tumor growth in mouse models of HCC and to elucidate the
possible molecular mechanism in vivo and in vitro. We
showed that PTE dose-dependently suppressed tumor growth in mice
induced by diethylnitrosamine (DEN) plus carbon tetrachloride
(CCl4) and inhibited cell viability and proliferation
in vitro. p53-mediated downregulation of superoxide
dismutase 2 (SOD2), generation of reactive oxygen species (ROS),
and the activation of mitochondrial apoptosis were involved in
PTE-induced inhibition of tumor growth in vivo and cancer
cell proliferation in vitro.
Materials and methods
Chemicals and reagents
p53 and β-actin antibodies were obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). PTE, DEN,
N-acetylcysteine (NAC), PFTα and most of the chemicals and
reagents used in this study were procured from Sigma-Aldrich (St.
Louis, MO, USA).
Animal treatment
Animals were treated in accordance with the
guidelines approved by the Animal Care and Use Committee of the
Fourth Military Medical University (Shaanxi, China). C57 mice were
purchased from the Experimental Animal Center of the Fourth
Military Medical University. Mice were kept in individual cages
with free access to food and water and the environment was at
constant temperature and humidity conditions on 12-h light/dark
cycles.
Mice were acclimated for 1 week and then randomly
divided into 7 groups (n=15): Ctrl, DEN, DEN+PTE (100 mg/kg),
DEN+PTE (200 mg/kg), DEN+PTE (200 mg/kg)+PFTα, DEN+PTE (200
mg/kg)+LV-SOD2 and DEN+PTE (200 mg/kg)+NAC. Mice were injected with
DEN plus CCl4 to construct the HCC model. In brief, mice
were intraperitonially injected with DEN (200 mg/kg, BW) once and 2
weeks later mice were given a CCl4 (3 ml/kg) injection
thrice a week for 6 consecutive weeks. In the groups of DEN+PTE
(100 mg/kg), DEN+PTE (200 mg/kg), DEN+PTE (200 mg/kg)+PFTα, DEN+PTE
(200 mg/kg)+LV-SOD2 and DEN+PTE (200 mg/kg)+NAC, mice were
intraperitonially injected with PTE (100 or 200 mg/kg daily) and/or
PFTα (10 mg/kg daily) or SOD2 lentivirus twice (on 2 consecutive
days) for 2 weeks throughout the experimental procedure. Mice in
the DEN+PTE (200 mg/kg)+LV-SOD group were treated with
0.4×108 TU LV-SOD2 through an intravenous injection in
the tail. Twenty weeks after the injection of DEN, mice were
sacrificed after overnight fasting. According to the accepted
criteria, the tumors were counted and measured (19). Blood samples were separated and
stored for further biochemical determination. A section of the
tumor tissue was fixed in 10% paraformaldehyde for TdT-mediated
dUTP nick end labeling (TUNEL) staining. Another section of the
tumor tissue was frozen and cut for ROS detection while another
area of tumor tissue was homogenized in saline for the
determination of caspase activity. The remaining tissues were
stored at −20°C for the determination of mRNA and protein
expression.
Cell culture and treatment
HepG2 cell lines were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with fetal bovine serum (FBS; 10%), 100 U/ml penicillin and 100
U/ml streptomycin. Cells were incubated at 37°C in a humidified
incubator containing 5% CO2. For the experimental
treatment, cells were incubated with 12.5–100 µM PTE in serum-free
medium for 24 h.
Transfection of lentivirus
The scramble or SOD2 lentivirus was transfected into
cells to build cells stably expressing SOD2 according to the
manufacturer's instructions. Subsequently, cells were treated with
100 µM PTE for 24 h.
Cell viability and proliferation
After the treatment, cell viability and
proliferation were measured. An MTT assay was conducted to evaluate
cell viability. A CCK-8 assay was performed to assess cell
proliferation according to the manufacturer's instructions
(Sigma-Aldrich). The absorbances at 570 and 450 nm were measured,
respectively. Results are presented as folds of the control.
Biochemical analysis
Serum levels of lactate dehydrogenase (LDH), alanine
aminotransferase (ALT), aspartate aminotransferase (AST) and
alkaline phosphatase (ALP) were determined using commercial assay
kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China)
according to the manufacturer's instructions.
Apoptosis
TUNEL staining was conducted to measure the
apoptosis in liver tissues and cells using the In Situ Cell
Death Detection kit (Roche Diagnostics, Basel, Switzerland)
according to the manufacturer's instructions. The total number of
cells and the number of TUNEL-positive stained cells were counted
by an independent researcher. At least 6 random fields were counted
for each slide. Results are expressed as the percentage of
apoptotic cells.
Determination of caspase activity
The activity of caspase-3 in homogenates of liver
tissues and cells was determined using commercial assay kits
(Beyotime Institute of Biotechnology, Haimen, China) according to
the manufacturer's instructions.
ROS determination
The ROS level was detected by DHE, a superoxide
sensitive probe. In brief, frozen liver sections and cells were
incubated with 10 µM DHE at 37°C for 30 min in the dark. After
being washed twice, slides or dishes were observed under a confocal
microscope (Olympus, Tokyo, Japan).
Real-time PCR
Total RNA was extracted from the tissue samples and
cells using TRIzol reagent according to the manufacturer's
instructions (Invitrogen). mRNA (500 ng) was reversely transcribed
into cDNA using the First Strand cDNA synthesis kit (Takara Bio,
Inc., Otsu, Japan). Target gene expression was quantified by a
real-time polymerase chain reaction (PCR) system using SYBR-Green
reagents (Takara Bio, Inc.) in a Bio-Rad Cycling Biosystem (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). β-actin was used as an
internal control. Amplification conditions were as follows: an
initial step at 94°C for 5 min, followed by 40 cycles of
denaturation at 94°C for 30 sec, annealing at 63°C for 30 sec and
then extension at 72°C for 10 sec. The relative amount of RNA was
quantified using the comparative threshold cycle (Ct)
(2−ΔΔCt) method.
Western blot analysis
Liver tissues and cells were lysed with cell lysis
buffer (50 Mm Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM
EDTA, 10 mM NaF, 1 mM Na3VO4, and a protease
inhibitor cocktail) on ice for 30 min. After centrifugation at
20,000 × g for 20 min at 4°C, the protein content of the
supernatants was determined by BCA assay kit (Pierce, Rockford, IL,
USA). Subsequently, equal volumes of supernatants and 2X SDS
loading buffer were mixed and boiled for 5 min. Samples containing
equal amounts of protein were subjected to SDS-PAGE and then
transferred onto an NC membrane. After blocking with non-fat milk
for 1 h at room temperature, the membranes were incubated with
indicated primary antibodies overnight at 4°C. After being washed
four times, the membranes were incubated in the appropriate
horseradish peroxidase-conjugated secondary antibodies at 37°C for
45 min. The protein bands were visualized using chemiluminescent
reagents according to the manufacturer's instructions (Thermo
Fisher Scientific) and quantified using an image analyzer Quantity
One system (Bio-Rad Laboratories, Inc.).
Statistical analysis
Results are expressed as the mean ± SEM. All the
experiments were performed at least 3 times. The results were
analyzed using GraphPad Prism software (GraphPad Software, Inc.,
San Diego, CA, USA) by one-way ANOVA followed by an SNK-q test for
multiple comparisons. P<0.05 was considered statistically
significant.
Results
PTE inhibits tumor growth in mice and
inhibits proliferation and promotes apoptosis in HepG2 cells
To investigate the effect of PTE on tumor growth, an
HCC mouse model was established using DEN/CCl4
administration. In Table I, we
showed that the incidence of tumorigenesis, the number of tumors
and the maximum size of the tumors in the DEN-treated mice were
100%, 18.6±2.9 and 9.7±0.8 mm, respectively. The administration of
PTE did not affect the incidence of tumorigenesis (Table I). However, PTE significantly
decreased the number of tumors and the maximum size of the tumors
in the DEN-treated mice. The results showed that in the 100 mg/kg
PTE group, the number of tumors and the maximum size of the tumors
were reduced to 13.2±2.3 and 5.8±0.7 mm, respectively (Table I). In the 200 mg/kg PTE group, the
number of tumors and the maximum size of the tumors were reduced to
6.4±3.7 and 4.0±0.6 mm, respectively (Table I). In addition, the activities of
LDH, ALT, AST and ALP were significantly increased by
DEN/CCl4 treatment which were inhibited by PTE in a
dose-dependent manner (Table II).
The results indicated that PTE administration protected against
tumor growth and liver injury in the DEN-treated mice. Moreover, we
examined the effect of PTE on cancer cell proliferation in HepG2
cell lines. Cells were incubated with 12.5–100 µM PTE for 24 h and
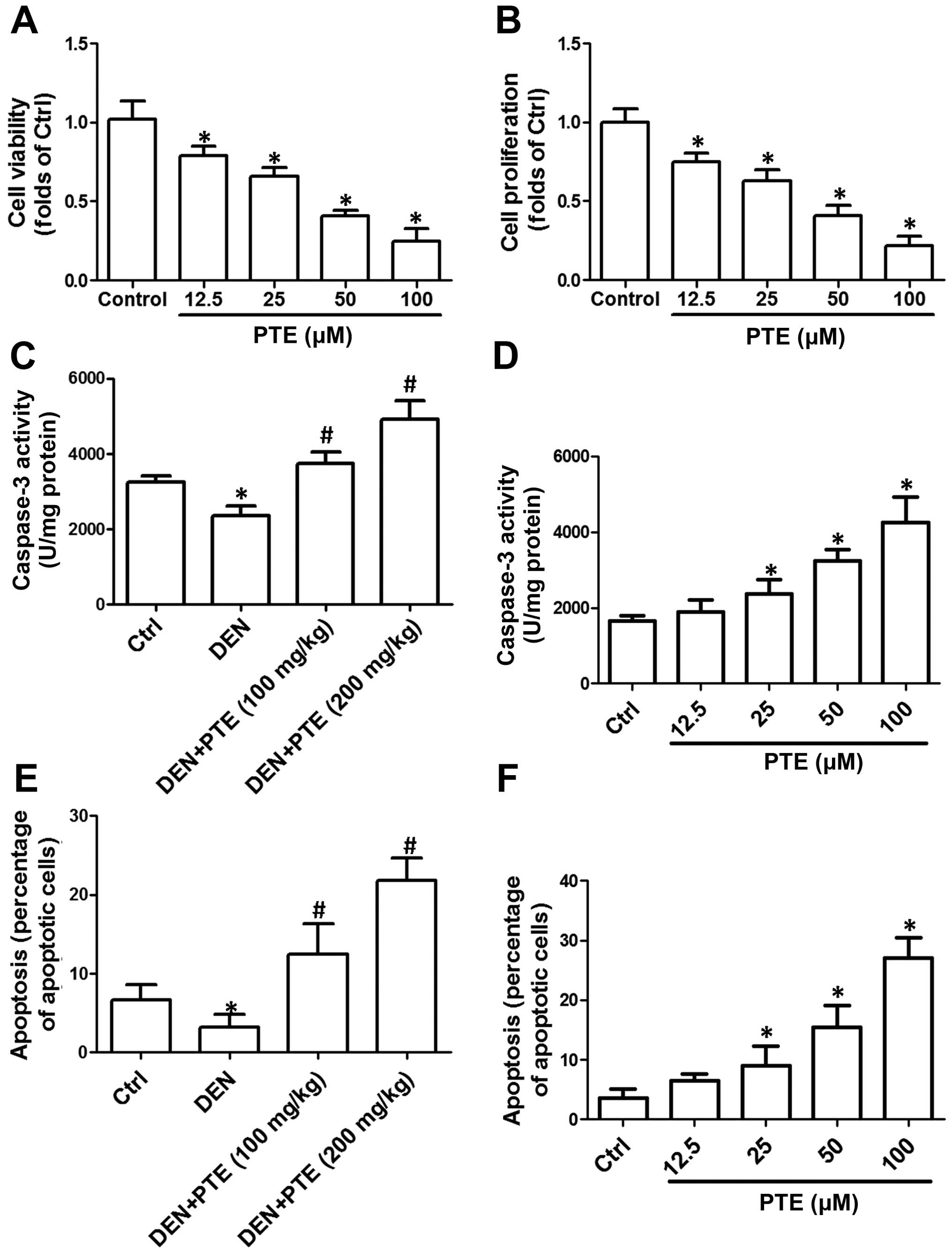
then cell viability and proliferation were measured. In Fig. 1A and B, we showed that 25–100 µM PTE
concentration-dependently decreased cell viability and
proliferation in HepG2 cells. The results indicated that PTE
inhibited the proliferation of HCC cells in vitro.
 | Table I.Effect of PTE on multiplicity, size
and incidence of tumors in DEN-treated mice. |
Table I.
Effect of PTE on multiplicity, size
and incidence of tumors in DEN-treated mice.
| Groups | No. | Max. size (mm) | Incidence (%) |
|---|
| Ctrl | 0 | 0 | 0 |
| DEN |
18.6±2.9a |
9.7±0.8a | 100 |
| DEN+PTE (100
mg/kg) |
13.2±2.3b |
5.8±0.7b | 100 |
| DEN+PTE (200
mg/kg) |
6.4±3.7b |
4.0±0.6b | 100 |
| DEN+PTE (200
mg/kg)+PFTα |
12.3±3.4c |
7.7±0.9c | 100 |
| DEN+PTE (200
mg/kg)+SOD2 KU |
10.6±3.1c |
7.3±1.1c | 100 |
| DEN+PTE (200
mg/kg)+NAC |
11.9±2.4c |
7.6±1.4c | 100 |
| Table II.Effect of PTE on the profiles of
liver enzymes in the DEN-treated mice. |
Table II.
Effect of PTE on the profiles of
liver enzymes in the DEN-treated mice.
| Groups | LDH (U/l) | ALT (U/l) | AST (U/l) | ALP (U/l) |
|---|
| Ctrl | 132±10.1 | 78±6.9 | 67±8.2 | 114±10.4 |
| DEN |
298±11.2a |
196±8.5a |
216±10.3a |
278±9.5a |
| DEN+PTE (100
mg/kg) |
235±10.8b |
151±9.7b |
166±5.5b |
232±6.7b |
| DEN+PTE (200
mg/kg) |
198±7.8b |
112±9.4b | 97±6.8b |
169±8.3b |
| DEN+PTE (200
mg/kg)+PFTα |
238±9.1c |
156±5.9c |
167±9.2c |
218±5.7c |
| DEN+PTE (200
mg/kg)+SOD2 KU |
242±8.1c |
152±7.3c |
159±4.8c |
229±10.8c |
| DEN+PTE (200
mg/kg)+NAC |
235±7.8c |
149±6.6c |
161±8.2c |
199±8.4c |
Furthermore, the effect of PTE on apoptosis in liver
tumor tissues and cells was evaluated. As shown in Fig. 1C, the activity of caspase-3 in the
DEN-treated mice was significantly increased with PTE
administration. In the HepG2 cells, 25–100 µM PTE
concentration-dependently increased caspase-3 activity (Fig. 1D). Compared with the DEN group, PTE
administration markedly increased the percentage of apoptotic cells
(Fig. 1E). PTE (25–100 µM)
concentration-dependently increased the percentage of apoptotic
cells in the HepG2 cells (Fig. 1F).
The results indicated that PTE activated the mitochondrial
apoptotic pathway and induced significant apoptosis in tumor
tissues and cells.
Upregulation of p53 is involved in the
PTE-induced inhibitory effect on HCC
In the next step, we examined the possible role of
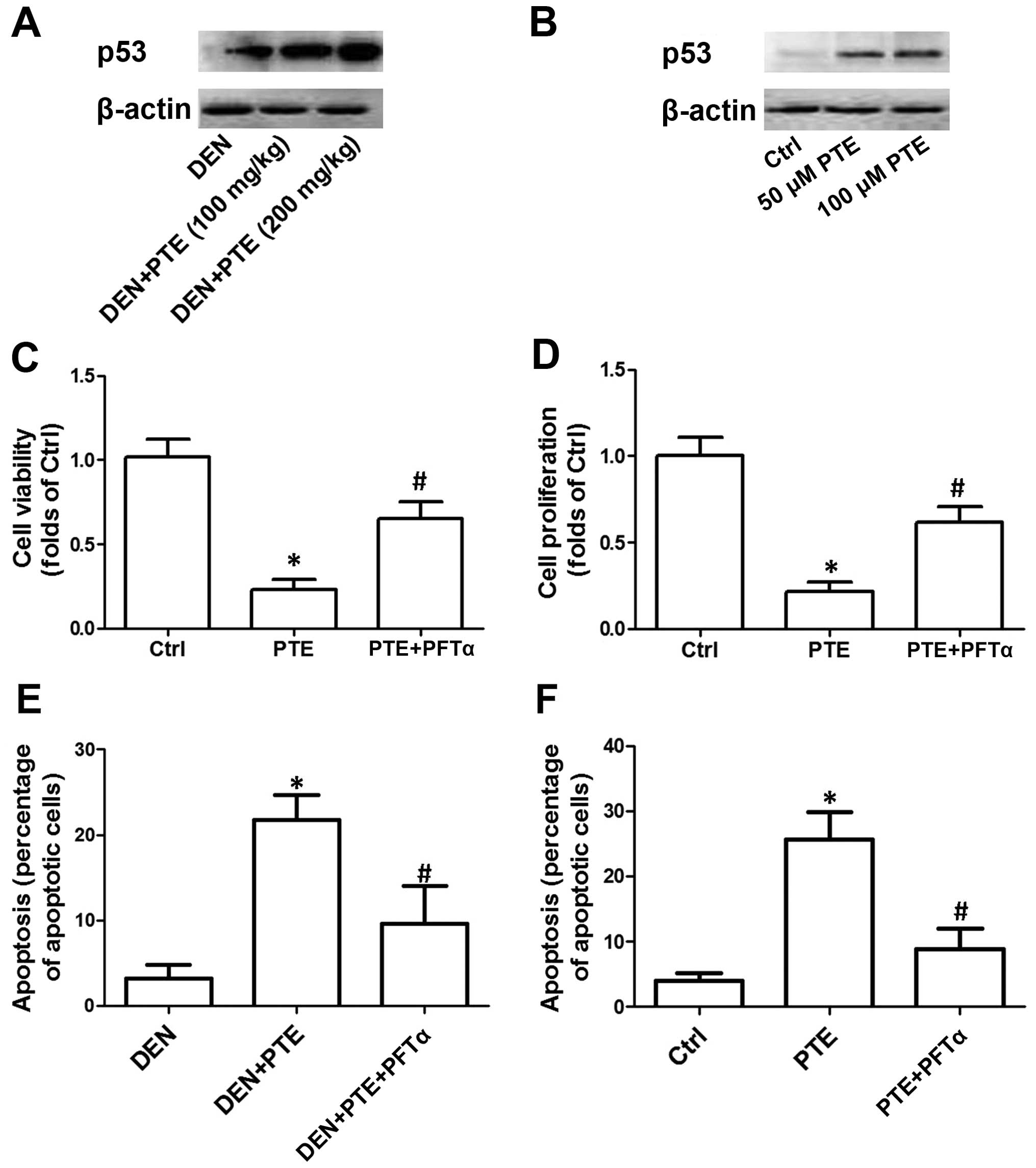
p53 in the inhibitory effect of PTE on HCC. As shown in Fig. 2A, in the DEN-treated mice, PTE
administration dose-dependently increased p53 expression. In
Fig. 2B, we showed that 50 and 100
µM PTE concentration-dependently increased the protein expression
of p53. To elucidate the role of upregulation of the p53
PTE-induced inhibitory effect on HCC in vivo and in
vitro, mice and cells were treated with PFTα, an inhibitor of
p53. The results showed that PFTα significantly inhibited the
decrease in the number of tumors and the maximum size of the tumors
as well as the LDH, ALT, AST and ALP activities induced by PTE in
the DEN-treated mice (Tables I and
II). In the presence of PFTα, the
PTE-induced decrease in cell viability and proliferation in HepG2
cells was significantly suppressed (Fig. 2C and D). Moreover, inhibition of p53
by PFTα suppressed the PTE-induced increase in apoptosis both in
mice in vivo and cells in vitro. These results
indicated that p53-mediated regulation of apoptosis was involved in
the inhibitory effect of PTE on HCC tumor growth in vivo and
on cell proliferation in vitro (Fig. 2E and F).
ROS generation mediates the inhibitory
effect of PTE on HCC
Next, we evaluated whether ROS generation mediated
apoptosis induced by PTE in vivo and in vitro. In
Fig. 3A, we showed that PTE
significantly increased DHE staining in liver sections, indicating
an increase in ROS production. PFTα, overexpression of SOD2 by
lentivirus infection and NAC, a potent antioxidant, significantly
inhibited PTE-induced ROS generation in the tumor tissues (Fig. 3A). Consistently, in the HepG2 cells,
PTE resulted in a significant increase in ROS generation which was
inhibited by PFTα, overexpression of SOD2 by lentivirus infection
and NAC (Fig. 3B). Moreover, the
role of ROS generation in the effect of PTE on tumor growth, cell
proliferation and apoptosis was examined. As shown in Tables I and II, the PTE-induced decrease in the number
of tumors and the maximum size of the tumors as well as LDH, ALT,
AST and ALP activities were inhibited by LV-SOD2 and NAC. LV-SOD2
and NAC also inhibited the PTE-induced decrease of cell viability
and proliferation in the HepG2 cells (Fig. 3C and D). Moreover, the PTE-mediated
increase in apoptosis in tumor tissues and cells was inhibited by
LV-SOD2 and NAC (Fig. 3E and F).
These results indicated that p53-mediated ROS generation was
involved in the inhibitory effect of PTE on HCC tumor growth in
vivo and on cell proliferation in vitro.
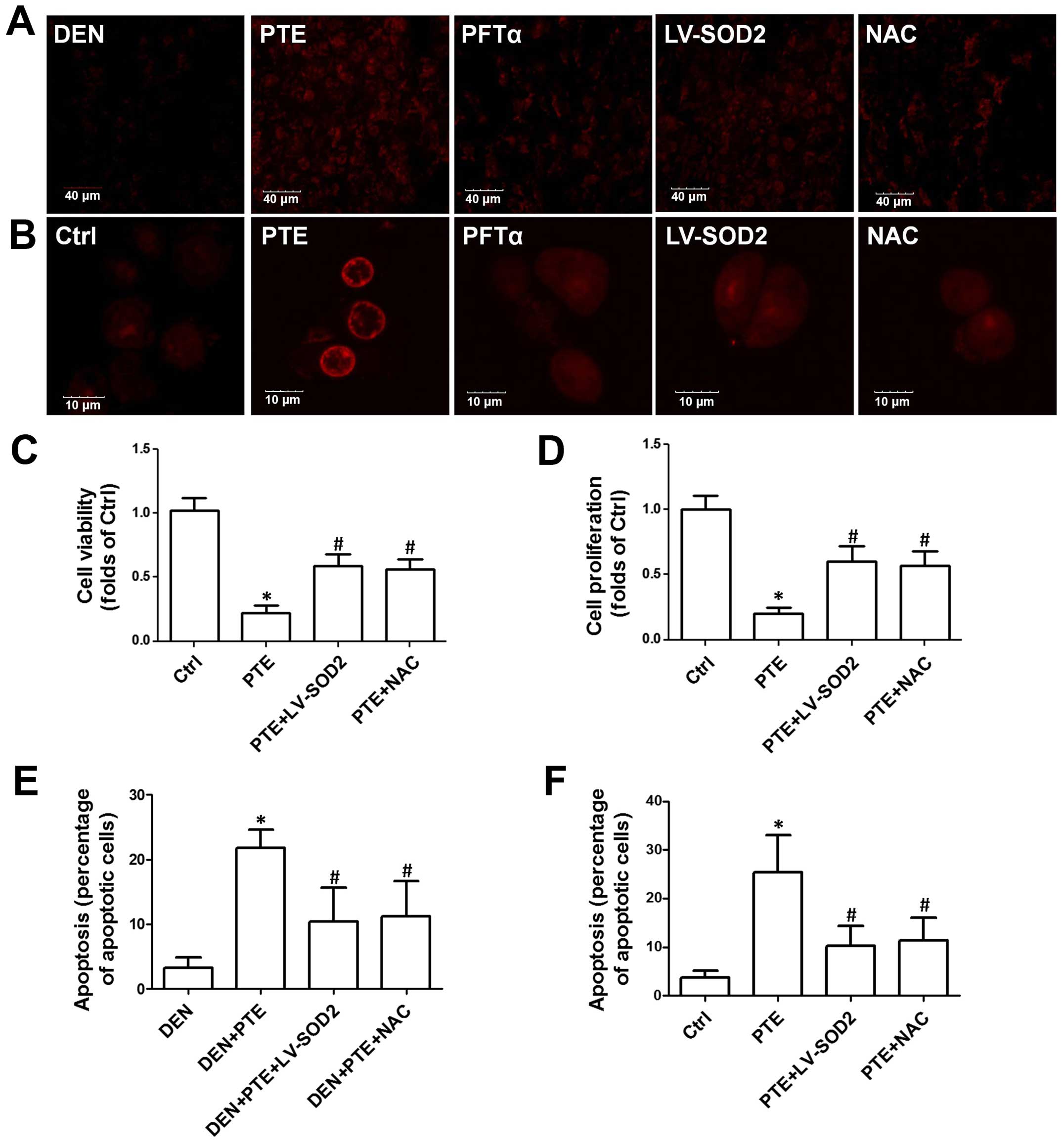 | Figure 3.Role of ROS generation in the effect
of PTE on cell viability, proliferation and apoptosis. DEN plus
CCl4-treated mice were injected with 200 mg/kg PTE with
or without 10 mg/kg PFTα, or LV-SOD2 or NAC. HepG2 cells were
transfected with scramble lentivirus or LV-SOD2 and then incubated
with 100 µM PTE in the presence or absence of 10 µM PFTα or NAC for
24 h. (A) ROS levels in liver tumor tissue and (B) HepG2 cells were
determined by DHE staining and representative images are shown. (C)
Cell viability was detected by an MTT assay and (D) cell
proliferation was detected with a CCK-8 kit. (E) Apoptosis in liver
tumor tissue and (F) HepG2 cells was determined by TUNEL assay and
the results are presented as the percentage of apoptotic cells.
*p<0.05, compared with that of the Ctrl or DEN.
#p<0.05, compared with that of the PTE group. ROS,
reactive oxygen species; PTE, pterostilbene; DEN,
diethylnitrosamine; CCl4, carbon tetrachloride; SOD2,
superoxide dismutase 2; NAC, N-acetylcysteine; Ctrl,
control. |
p53-mediated inhibition of SOD2 is
responsible for the inhibitory effect of PTE on HCC
Considering the important role of SOD2 in
counteracting ROS, we evaluated the role of SOD2 in p53-mediated
apoptosis induced by PTE. Since overexpression of SOD2
significantly inhibited PTE-induced suppression of tumor growth and
cell proliferation, it was indicated that a decrease in SOD2 was
involved in tumor growth and cell proliferation in HCC. However,
whether the expression of SOD2 was regulated by p53 was unknown. In
the next step, we evaluated the expression of SOD2 using real-time
PCR. As shown in Fig. 4A and B, in
both DEN-treated liver tissues and HepG2 cells, PTE resulted in a
significant decrease in SOD2 expression. LV-SOD2 enhanced the
expression of SOD2 to a level that was markedly higher than that of
the DEN group or control cells (Fig. 4A
and B). In the presence of PFTα, the PTE-induced decrease in
SOD2 was significantly inhibited both in vivo and in
vitro (Fig. 4A and B). However,
NAC had no significant effect on SOD2 expression, indicating that
downregulation of SOD2 was upstream of ROS generation (Fig. 4A and B). The results demonstrated
that downregulation of SOD2 was a link between upregulation of p53
and increase in the ROS level, which activated mitochondrial
apoptosis and resulted in inhibition of tumor growth and cell
proliferation.
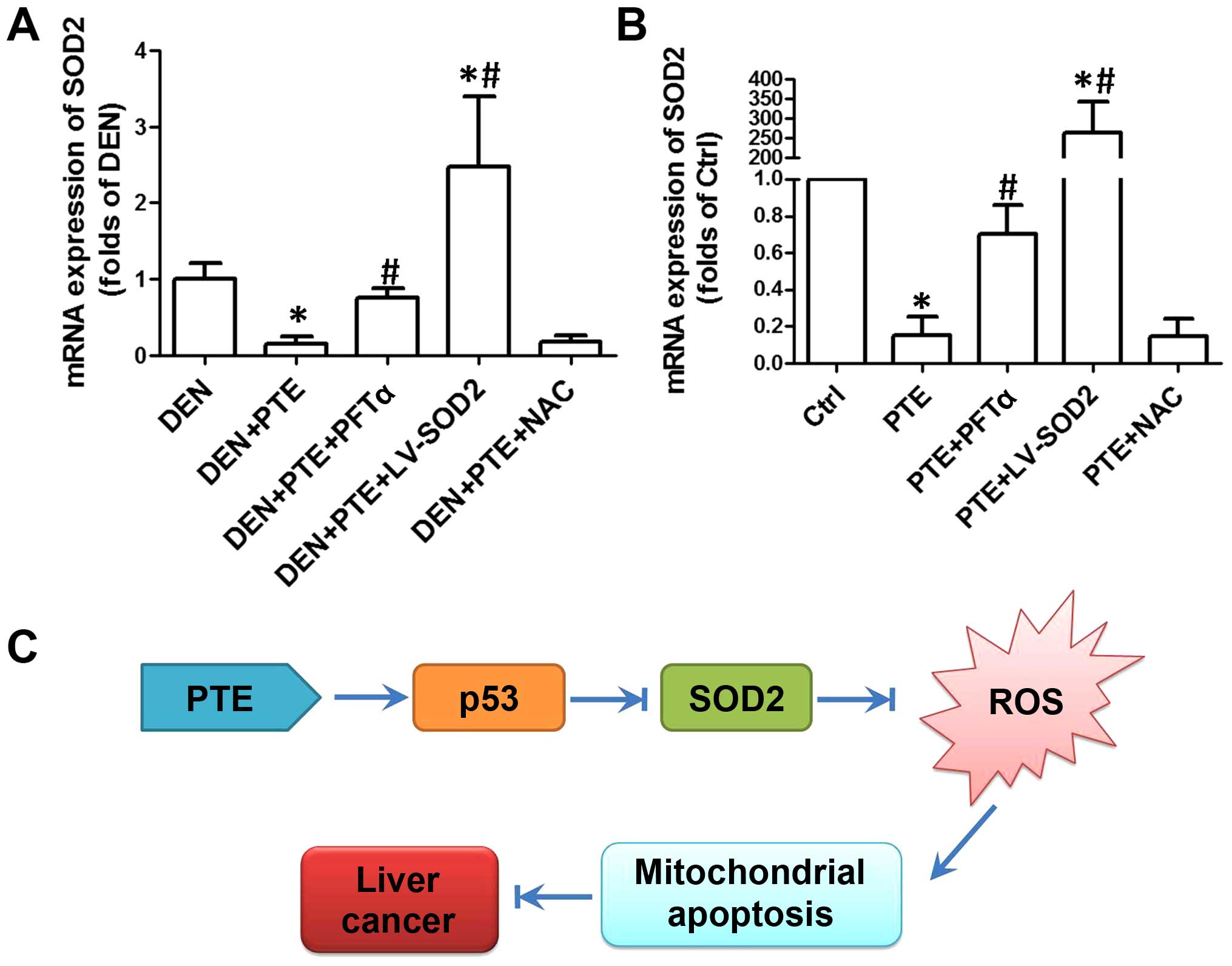 | Figure 4.Role of ROS generation in the effect
of PTE on cell viability, proliferation and apoptosis. DEN plus
CCl4-treated mice were injected with 200 mg/kg PTE with
or without 10 mg/kg PFTα, or LV-SOD2 or NAC. HepG2 cells were
transfected with scramble lentivirus or LV-SOD2 and then incubated
with 100 µM PTE in the presence or absence of 10 µM PFTα or NAC for
24 h. (A) mRNA expression of SOD2 in liver tumor tissue and (B) in
HepG2 cells was determined by real-time PCR and the results are
presented as a fold of Ctrl. *p<0.05, compared with that of the
Ctrl or DEN. #p<0.05, compared with that of the PTE
group. (C) The molecular mechanism of the antitumor effect of PTE
in HCC. ROS, reactive oxygen species; PTE, pterostilbene; DEN,
diethylnitrosamine; CCl4, carbon tetrachloride; SOD2,
superoxide dismutase 2; NAC, N-acetylcysteine; HCC,
hepatocellular carcinoma; Ctrl, control. |
Discussion
DEN a constituent of tobacco smoke, cheddar cheese,
curd and fried meals and a number of alcoholic beverages, is widely
used as a hepatocarcinogenic dialkylnitrosoamine to induce an in
vivo HCC model in its combination with CCl4
(20–23). In the present study, we investigated
the inhibitory effect of PTE on tumor growth in DEN-induced HCC in
mice and cancer cell proliferation in HepG2 cells. We found that
PTE resulted in significant inhibition of tumor growth and cancer
cell proliferation both in vivo and in vitro.
Malignant tumors are often characterized by
dysregulation of apoptotic cell death (24,25).
Moreover, induction of apoptosis is considered to be a potent
therapeutic strategy for the intervention of tumors (26–28).
Numerous literature has shown that PTE has pro-apoptotic
activities. Zhang et al found that PTE induced apoptosis in
HeLa cells (29). Hsiao et
al showed that PTE stimulated mitochondrial-derived apoptosis
in human acute myeloid leukemia cell lines (30). Pan et al showed that PTE
exhibited a pro-apoptotic and anti-proliferation effect in breast
cancer (31). In the present study,
we also examined the effect of PTE on apoptosis in vivo and
in vitro. Consistently, we demonstrated that PTE exhibited
pro-apoptotic effects both in liver tumor tissues of DEN-treated
mice and in HCC cells, as evidenced by the increase in caspase-3
activity and TUNEL-positive cells.
It is well-known that p53 is a tumor-suppressor
gene, playing key roles in cell cycle control and induction of
apoptosis through the regulation of a battery of target genes
(32,33). In response to death signals,
activated p53 regulates various genes of pro-apoptotic proteins,
which are transcription-dependent or -independent, leading to final
cell death (34,35). In our study, we also examined the
possible role of p53 in PTE-induced regulation of apoptosis, cell
proliferation and cell growth. We showed that p53 was responsible
for PTE-induced apoptosis and inhibition of cell proliferation and
tumor growth, as evidenced by PFTα-induced suppression of the
increase in apoptosis and decrease in cell proliferation and tumor
growth in the PTE-treated mice and cells.
p53 is also a redox-regulating transcription factor
via regulation of ROS production and the expression of various
antioxidant enzymes (36).
Increased ROS is believed to be able to activate the mitochondrial
apoptotic pathway, resulting in cell death (37–39).
In the present study, we found that PTE increased ROS generation
both in vivo and in vitro through a p53-dependent
manner. Moreover, PTE-induced ROS production was involved in the
antitumor effect. However, previous studies have shown that PTE
exhibits antioxidant activities (40,41).
The discrepancy is supported by the notion that the effect of PTE
on the redox state may be tissue/cell-specific and
concentration-dependent.
Among the antioxidant systems, SOD2 is an important
member which is located in the mitochondrial matrix where it
catalyzes the dismutation of a superoxide anion and plays pivotal
roles in protecting against mitochondrial and intracellular ROS
insult (42). In our study, we also
examined the possible role of SOD2 in the inhibitory effect of PTE
on tumor growth and cell proliferation. PTE resulted in a
significant decrease in SOD2 expression and overexpression of SOD2
by a lentivirus inhibited PTE-induced increase in apoptosis and
decrease in tumor growth and cell proliferation. The results
demonstrated that downregulation of SOD2 was an essential step in
the process of PTE-induced inhibition of tumor growth and HCC cell
proliferation.
In the next step, we examined the sequence of
upregulation of p53, downregulation of SOD2 and increase in ROS
generation in response to PTE treatment. We showed that PFTα, but
not NAC, inhibited PTE-induced decrease in SOD2 expression and
PFTα, LV-SOD2 and NAC inhibited PTE-induced ROS generation,
indicating that downregulation of SOD2 was required for
p53-mediated ROS production induced by PTE. Previous studies found
that p53-mediated downregulation of SOD2 was involved in the
antitumor effect of betulinic acid in HCC (43). In combination with the results, we
proposed that SOD2 may be a main target through which p53 regulates
redox status and apoptosis and the p53/SOD2/ROS pathway is a common
pathway that mediates the toxic effect of extracellular stimuli in
HCC.
In conclusion, in the present study, we examined the
effect of PTE on tumor growth in DEN-induced HCC in mice and HCC
cell proliferation in HepG2 cells. The results showed that PTE
inhibited tumor growth in vivo and HCC cell proliferation
in vitro. PTE increased p53 expression, decreased SOD2
expression, and resulted in an increase in the ROS level and the
activation of the mitochondrial apoptotic pathway, leading to
inhibition of tumor growth and cell proliferation. Collectively,
these data demonstrated that the p53/SOD2/ROS pathway is critical
for PTE-inhibited tumor growth and HCC cell proliferation.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeh YP, Hu TH, Cho PY, Chen HH, Yen AM,
Chen SL, Chiu SY, Fann JC, Su WW, Fang YJ, et al: Changhua
Community-Based Abdominal Ultrasonography Screening Group:
Evaluation of abdominal ultrasonography mass screening for
hepatocellular carcinoma in Taiwan. Hepatology. 59:1840–1849. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pascual S, Herrera I and Irurzun J: New
advances in hepatocellular carcinoma. World J Hepatol. 8:421–438.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cabibbo G, Enea M, Attanasio M, Bruix J,
Craxì A and Cammà C: A meta-analysis of survival rates of untreated
patients in randomized clinical trials of hepatocellular carcinoma.
Hepatology. 51:1274–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schmidt S, Follmann M, Malek N, Manns MP
and Greten TF: Critical appraisal of clinical practice guidelines
for diagnosis and treatment of hepatocellular carcinoma. J
Gastroenterol Hepatol. 26:1779–1786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roxburgh P and Evans TR: Systemic therapy
of hepatocellular carcinoma: Are we making progress? Adv Ther.
25:1089–1104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tejeda-Maldonado J, García-Juárez I,
Aguirre-Valadez J, González-Aguirre A, Vilatobá-Chapa M,
Armengol-Alonso A, Escobar-Penagos F, Torre A, Sánchez-Ávila JF and
Carrillo-Pérez DL: Diagnosis and treatment of hepatocellular
carcinoma: An update. World J Hepatol. 7:362–376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lombardi G, Vannini S, Blasi F,
Marcotullio MC, Dominici L, Villarini M, Cossignani L and Moretti
M: In vitro safety/protection assessment of resveratrol and
pterostilbene in a human hepatoma cell line (HepG2). Nat Prod
Commun. 10:1403–1408. 2015.PubMed/NCBI
|
|
12
|
Pan MH, Chiou YS, Chen WJ, Wang JM,
Badmaev V and Ho CT: Pterostilbene inhibited tumor invasion via
suppressing multiple signal transduction pathways in human
hepatocellular carcinoma cells. Carcinogenesis. 30:1234–1242. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Remsberg CM, Yáñez JA, Ohgami Y,
Vega-Villa KR, Rimando AM and Davies NM: Pharmacometrics of
pterostilbene: Preclinical pharmacokinetics and metabolism,
anticancer, antiinflammatory, antioxidant and analgesic activity.
Phytother Res. 22:169–179. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng Y, Yang Y, Fan C, Di S, Hu W, Jiang
S, Li T, Ma Z, Chao D, Feng X, et al: Pterostilbene inhibits the
growth of human esophageal cancer cells by regulating endoplasmic
reticulum stress. Cell Physiol Biochem. 38:1226–1244. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dhar S, Kumar A, Zhang L, Rimando AM, Lage
JM, Lewin JR, Atfi A, Zhang X and Levenson AS: Dietary
pterostilbene is a novel MTA1-targeted chemopreventive and
therapeutic agent in prostate cancer. Oncotarget. 7:18469–18484.
2016.PubMed/NCBI
|
|
16
|
Nikhil K, Sharan S and Roy P: A
pterostilbene derivative suppresses osteoclastogenesis by
regulating RANKL-mediated NFκB and MAPK signaling in RAW264.7
cells. Pharmacol Rep. 67:1264–1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Y, Wu X, Cai X, Song M, Zheng J, Pan
C, Qiu P, Zhang L, Zhou S, Tang Z, et al: Identification of
pinostilbene as a major colonic metabolite of pterostilbene and its
inhibitory effects on colon cancer cells. Mol Nutr Food Res.
60:1924–1932. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang CS, Ho CT, Tu SH, Pan MH, Chuang CH,
Chang HW, Chang CH, Wu CH and Ho YS: Long-term ethanol
exposure-induced hepatocellular carcinoma cell migration and
invasion through lysyl oxidase activation are attenuated by
combined treatment with pterostilbene and curcumin analogues. J
Agric Food Chem. 61:4326–4335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hacker HJ, Mtiro H, Bannasch P and
Vesselinovitch SD: Histochemical profile of mouse hepatocellular
adenomas and carcinomas induced by a single dose of
diethylnitrosamine. Cancer Res. 51:1952–1958. 1991.PubMed/NCBI
|
|
20
|
Fausto N and Campbell JS: Mouse models of
hepatocellular carcinoma. Semin Liver Dis. 30:87–98. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao X, Fu J, Xu A, Yu L, Zhu J, Dai R, Su
B, Luo T, Li N, Qin W, et al: Gankyrin drives malignant
transformation of chronic liver damage-mediated fibrosis via the
Rac1/JNK pathway. Cell Death Dis. 6:e17512015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qin XY, Tatsukawa H, Hitomi K, Shirakami
Y, Ishibashi N, Shimizu M, Moriwaki H and Kojima S: Metabolome
analyses uncovered a novel inhibitory effect of acyclic retinoid on
aberrant lipogenesis in a mouse diethylnitrosamine-induced hepatic
tumorigenesis model. Cancer Prev Res (Phila). 9:205–214. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shibata Y, Hara T, Nagano J, Nakamura N,
Ohno T, Ninomiya S, Ito H, Tanaka T, Saito K, Seishima M, et al:
The role of indoleamine 2,3-dioxygenase in
diethylnitrosamine-induced liver carcinogenesis. PLoS One.
11:e01462792016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kundu J, Chun KS, Aruoma OI and Kundu JK:
Mechanistic perspectives on cancer chemoprevention/chemotherapeutic
effects of thymoquinone. Mutat Res. 768:22–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang CF, Wen LZ, Yin C, Xu WP, Shi B,
Zhang X and Xie WF: Apoptosis signal-regulating kinase 1 mediates
the inhibitory effect of hepatocyte nuclear factor-4α on
hepatocellular carcinoma. Oncotarget. 7:27408–27421.
2016.PubMed/NCBI
|
|
27
|
Li J, Liang L, Liu Y, Luo Y, Liang X, Luo
D, Feng Z, Dang Y, Yang L and Chen G: Clinicopathological
significance of STAT4 in hepatocellular carcinoma and its effect on
cell growth and apoptosis. Onco Targets Ther. 9:1721–1734.
2016.PubMed/NCBI
|
|
28
|
Chen X, Tan M, Xie Z, Feng B, Zhao Z, Yang
K, Hu C, Liao N, Wang T, Chen D, et al: Inhibiting
ROS-STAT3-dependent autophagy enhanced capsaicin-induced apoptosis
in human hepatocellular carcinoma cells. Free Radic Res.
50:744–755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang B, Wang XQ, Chen HY and Liu BH:
Involvement of the Nrf2 pathway in the regulation of
pterostilbene-induced apoptosis in HeLa cells via ER stress. J
Pharmacol Sci. 126:216–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hsiao PC, Chou YE, Tan P, Lee WJ, Yang SF,
Chow JM, Chen HY, Lin CH, Lee LM and Chien MH: Pterostilbene
simultaneously induced G0/G1-phase arrest and MAPK-mediated
mitochondrial-derived apoptosis in human acute myeloid leukemia
cell lines. PLoS One. 9:e1053422014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pan C, Hu Y, Li J, Wang Z, Huang J, Zhang
S and Ding L: Estrogen receptor-α36 is involved in
pterostilbene-induced apoptosis and anti-proliferation in in vitro
and in vivo breast cancer. PLoS One. 9:e1044592014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakano K and Vousden KH: PUMA, a novel
proapoptotic gene, is induced by p53. Mol Cell. 7:683–694. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Zhang C, Hu W and Feng Z: Tumor
suppressor p53 and its mutants in cancer metabolism. Cancer Lett.
356:197–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chi SW: Structural insights into the
transcription-independent apoptotic pathway of p53. BMB Rep.
47:167–172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maillet A and Pervaiz S: Redox regulation
of p53, redox effectors regulated by p53: a subtle balance.
Antioxid Redox Signal. 16:1285–1294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Liu J, Jiang L, Wei X, Niu C, Wang
R, Zhang J, Meng D and Yao K: Bach1 induces endothelial cell
apoptosis and cell-cycle arrest through ROS generation. Oxid Med
Cell Longev. 2016:62340432016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang H, Sun N, Li X, Li K, Tian J and Li
J: Diallyl trisulfide induces osteosarcoma cell apoptosis through
reactive oxygen species-mediated downregulation of the PI3K/Akt
pathway. Oncol Rep. 35:3648–3658. 2016.PubMed/NCBI
|
|
39
|
Jia J, Qin Y, Zhang L, Guo C, Wang Y, Yue
X and Qian J: Artemisinin inhibits gallbladder cancer cell lines
through triggering cell cycle arrest and apoptosis. Mol Med Rep.
13:4461–4468. 2016.PubMed/NCBI
|
|
40
|
Bhakkiyalakshmi E, Sireesh D,
Sakthivadivel M, Sivasubramanian S, Gunasekaran P and Ramkumar KM:
Anti-hyperlipidemic and antiperoxidative role of pterostilbene via
Nrf2 signaling in experimental diabetes. Eur J Pharmacol. 777:9–16.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Elango B, Dornadula S, Paulmurugan R and
Ramkumar KM: Pterostilbene ameliorates streptozotocin-induced
diabetes through enhancing antioxidant signaling pathways mediated
by nrf2. Chem Res Toxicol. 29:47–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fridovich I: Superoxide radical and
superoxide dismutases. Annu Rev Biochem. 64:97–112. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang J, Qiu B, Li X, Zhang H and Liu W:
p53-p66shc/miR-21-Sod2 signaling is critical for the
inhibitory effect of betulinic acid on hepatocellular carcinoma.
Toxicol Lett. 238:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|