Introduction
Melanoma is the most aggressive skin malignancy,
accounting for more than 80% of skin cancer-related deaths
(1). An increasing incidence of
melanoma has been observed globally despite early detection,
appropriate resection and adjuvant therapy (2). The prognosis of advanced melanoma is
poor, with a median survival of 6–9 months. Currently, treatment
options for advanced melanoma are limited. Mutations in BRAF
(mostly V600E) are commonly present in 40–60% of cutaneous melanoma
(3) and are associated with brain
and lung metastases (4), therefore
representing a promising therapeutic target for melanoma. Patients
harboring BRAF mutations indeed had shown clinical response
to BRAF inhibitors such as vemurafenib. However, most of them had
only a short-lived response with a progression-free survival of
only 5 months, and resistance occurred eventually in most cases
(5). Thus, development of novel
effective chemotherapeutic agents used as a monotherapy or in
combination with BRAF inhibitors is in urgent demand.
Diphenhydramine (DPH) is a first-generation
antihistamine (H1 histamine receptor antagonist) developed in the
late 1940s. It has been widely used for many different purposes in
clinics due to its various pharmacological effects. Because of the
relative safety of this drug, DPH was approved by the FDA for
non-prescription use to treat allergies, insomnia, cough, motion
sickness and itchiness (6). It can
also be used to relieve extrapyramidal symptoms and treat
chemotherapy-induced nausea/vomiting (7). Furthermore, as a sodium channel
blocker, DPH can act as a local anesthetic (8,9). In
normal clinical dosage, the side effects of DPH are mild and well
tolerated. The common side effects are drowsiness, dry mouth and
increased heart rate.
Previous studies by Jangi et al have explored
the anti-melanoma potential of DPH by showing its ability to induce
in vitro cytotoxicity against four human melanoma cell lines
(10). However, the mechanisms of
action underlying DPH-induced cytotoxicity and the in vivo
anti-melanoma effect of this drug remain unknown. In light of this,
we herein provide the first evidence to demonstrate the selective
proapoptotic action of DPH on melanoma cells while sparing normal
melanocytes, establishing DPH as a drug targeting the STAT3/MCL-1
survival signaling pathway to induce apoptosis, and validate the
in vivo anti-melanoma effect of DPH in a B16-F10 melanoma
mouse model. Our discovery therefore suggests the potential to
repurpose DPH as an anti-melanoma therapeutic agent.
Materials and methods
Chemicals
DPH was obtained from Sigma-Aldrich (St. Louis, MO,
USA). The pan-caspase inhibitor z-VAD.fmk was purchased from Cayman
Chemical (Ann Arbor, MI, USA).
Cell culture
Human melanoma cell lines A2058 (no. CRL-11147),
A375.S2 (no. CRL-1872), MDA-MB-435S (no. HTB-129) and murine
B16-F10 melanoma cell line [no. CRL-6475; all from American Type
Culture Collection (ATCC), Manassas, VA, USA] were grown at 37°C
and 5% CO2 in the culture media recommended by ATTC.
Primary human melanocytes were obtained from Invitrogen Life
Technologies (Carlsbad, CA, USA) and grown in culture media
according to the manufacturers instructions. All culture media and
supplements were purchased from Invitrogen Life Technologies.
Cell viability assay
DPH-elicited cytotoxicity was evaluated by the
levels of cell viability after drug treatment using an MTS assay
(Promega Corp., Madison, WI, USA), which was performed according to
our established protocol (11). In
the present study, cells were seeded onto 96-well plates at a
density of 8×103 cells/well, followed by drug treatment
and subsequent MTS assay.
Annexin V/propidium iodide (PI) dual
staining assay using flow cytometry
Apoptosis induced by DPH was determined by Annexin
V/PI dual staining assay using an ApoTarget™ Annexin V-FITC
Apoptosis kit (BioSource International, Inc., Camarillo, CA, USA)
following our reported protocol (11). Data acquisition and analysis were
performed on FACScan flow cytometer (BD Biosciences, USA). Results
are expressed as the mean ± SEM of at least three independent
experiments. Cells with Annexin V-positive staining are recognized
as cells undergoing apoptosis.
Construction of the retroviral vector
pBabe-based MCL-1 or STAT3-expressing plasmid for stable clone
generation
The open reading frames (ORF) of human MCL-1
(Genbank accession no. NM_021960) were PCR-amplified from the
first-strand cDNA pools of human colon adenocarcinoma cell line
HCT116 using the following primer pair: MCL-1 forward,
5-ACCggTTTTggCCTCAAAAgAAAC-3 and MCL-1 reverse,
5-CAgTAAggCTATCTTATTAgATATg-3. The ORF of the constitutive STAT3
mutant (A661C/N663C) was PCR-amplified using the plasmid
pMXs-Stat3-C (plasmid #13373; Addgene, Inc., Cambridge, MA, USA) as
the template. The PCR-amplified ORFs were then subcloned to the
retroviral vector pBabe.puro engineered to encode an in-frame
N-terminal hemagglutinin (HA) epitope (pBabe-HA). The resultant
expression plasmids for MCL-1 (pBabe-HA-MCL-1) or STAT3
(pBabe-HA-STAT3-CA) were then subjected to production of retroviral
particles for subsequent cell infection and puromycin selection for
stable infectants according to our published protocol (11).
Immunoblot analysis
Immunoblotting was performed as previously described
(11). Polyclonal antibodies
against MCL-1, BCL-xL, BAX, HA epitope, total ERK, phospho-ERK
(Thr202/Tyr204), total STAT3, phospho-STAT3 (Tyr705), and the
cleaved forms of poly(ADP-ribose) polymerase (PARP), caspase-3,
caspase-8 and caspase-9 were all purchased from Cell Signaling
Technology (Beverly, MA, USA). α-tubulin antibody was purchased
from GeneTex (Irvine, CA, USA). The signals were detected with an
enhanced SuperSignal West Pico chemiluminescence kit (Pierce,
Rockford, IL, USA).
In vivo anti-melanoma activity
assay
The in vivo anti-melanoma effect of DPH was
evaluated using the murine B16-F10 cell line-based preclinical
melanoma model. Briefly, 3×105 B16-F10 cells were
subcutaneously injected into the ventral flank of C57BL/6 mice to
allow tumor growth for 8 days until reaching the size of 10
mm3. Tumor-bearing mice were then randomized into 2
groups (n=6) and received an i.p injection of 100 µl PBS or DPH (20
mg/kg). Mice were subjected to an i.p. injection every other day.
Tumor volumes were calculated with a digital caliper as length ×
width × thickness × 0.5 and expressed in mm3 before each
treatment. Mice were euthanized when the tumor size reached
<2,000 mm3 in mean diameter. All treatments were
performed according to the guidelines approved by IACUC of the
National Chung Hsing University.
Statistical analysis
All data from in vitro experiments were
expressed as means ± standard error of the mean (SEM) from at least
three independent experiments. Differences between groups were
evaluated for statistical significance using a Student's t-test. A
p-value <0.05 was regarded as the minimum criteria for
statistical significance. As for the in vivo studies, the
Kaplan-Meier method and log-rank test using GraphPad Prism software
package version 5.0 (GraphPad Software, Inc., San Diego, CA, USA)
were employed to compare the survival rates between vehicle
controls and DPH-treated mice. Differences were considered
significant at p<0.05.
Results
DPH induces apoptosis-dependent
cytotoxicity selectively in malignant melanoma cells while sparing
normal melanocytes
To comprehensively explore the anti-melanoma
activity of DPH, we started by testing the in vitro
cytotoxic effect of DPH on a panel of melanoma cell lines,
including murine B16-F10 cells (BRAF wild-type) and human A2058,
A375.S2 and MDA-MB-435S cells (all carrying the
BRAFV600E mutation) in addition to normal human
melanocytes. As shown in Fig. 1A,
DPH was cytotoxic against all melanoma cell lines tested; in
contrast, normal human melanocytes were refractory to DPH-elicited
cell death. Additionally, we found that DPH treatment led to a
marked increase in Annexin V-positive (apoptotic) cells in both the
A2058 (from 8.96±0.49 to 33.10±2.12%, p<0.001) and A375.S2 (from
8.65±0.49 to 30.36±2.68%, p<0.001) cell lines (Fig. 1B), illustrating the induction of
apoptosis. Immunoblotting further revealed that DPH induced a
dose-dependent increase in the levels of caspase-dependent cleavage
of PARP in addition to the proteolytic processing and thus
activation of caspase-9, caspase-8 and caspase-3 (Fig. 1C). These results thus confirmed the
proapoptotic effect of DPH on melanoma cells. Noteworthy,
pre-treatment with the pan-caspase inhibitor z-VAD.fmk
significantly rescued A2058 and A375.S2 cells from DPH-induced cell
death (Fig. 1D). Altogether, our
results established the anti-melanoma effect of DPH through the
induction of apoptosis-dependent cytotoxicity.
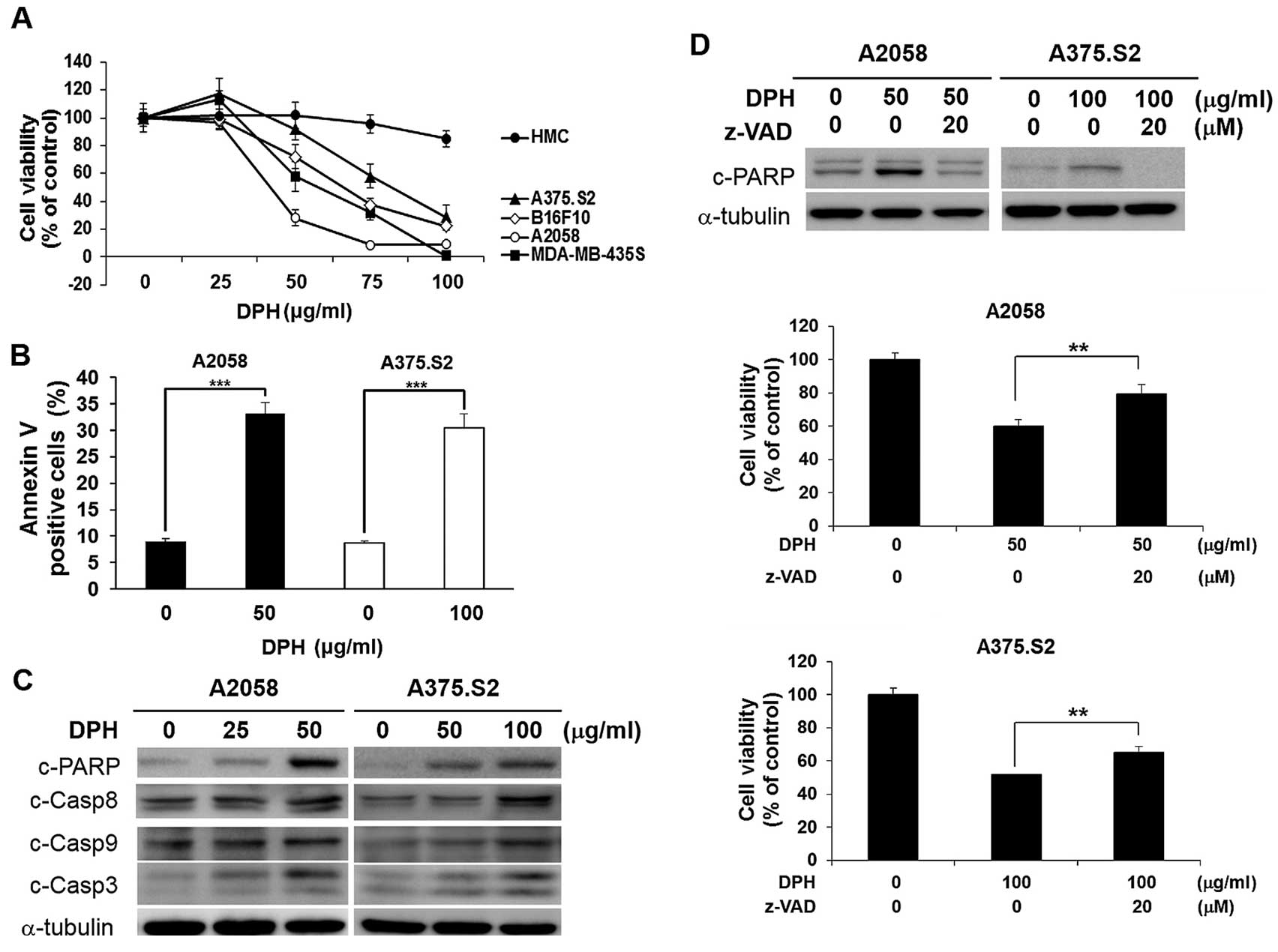 | Figure 1.Diphenhydramine (DPH) induces
apoptosis-dependent cytotoxicity selectively in melanoma cells. (A)
Selective cytotoxic effect of DPH on melanoma cells with limited
toxicity to normal melanocytes. Normal human melanocytes (HMC) (●),
human melanoma cell lines A2058 (○), A375.S2 (▲) and MDA-MB-435S
(■), and murine melanoma cell line B16-F10 (◇) were subjected to a
48-h treatment with DPH (0–100 µg/ml), followed by cell viability
determination using an MTS assay. (B) Diphenhydramine enhances the
Annexin V-positive (apoptotic) cell population. A2058 and A375.S2
cells were exposed to 50 and 100 µg/ml of diphenhydramine,
respectively, followed by Annexin V/propidium iodide (PI) dual
staining analyzed by flow cytometry. Shown here are the
quantitative results of the levels of Annexin V-positive cell
population after diphenhydramine stimulation. (C) DPH treatment
leads to caspase activation. A2058 and A375.S2 cells treated with
indicated concentrations of DPH were subjected to immunoblotting
for the levels of cleaved forms of poly(ADP-ribose) polymerase
(c-PARP), caspase-9 (c-Casp9), caspase-8 (c-Casp8) and caspase-3
(c-Casp3). α-tubulin levels were used as the loading control. (D)
Blockade of caspase activity attenuated DPH-induced cell death.
A2058 and A375.S2 cells were pre-treated for 1 h with 20 µM of
z-VAD.fmk (Z-VAD), a pan-caspase inhibitor, followed by 24-h
treatment with DPH. Immunoblotting revealed a marked reduction in
DPH-elicited PARP cleavage by z-VAD.fmk, confirming the blockade of
caspase activity. α-tubulin levels were used as the control for
equal loading (upper panel). z-VAD.fmk pre-treatment significantly
decreased DPH-induced cell death (middle and lower panels).
**p<0.01, ***p<0.001. |
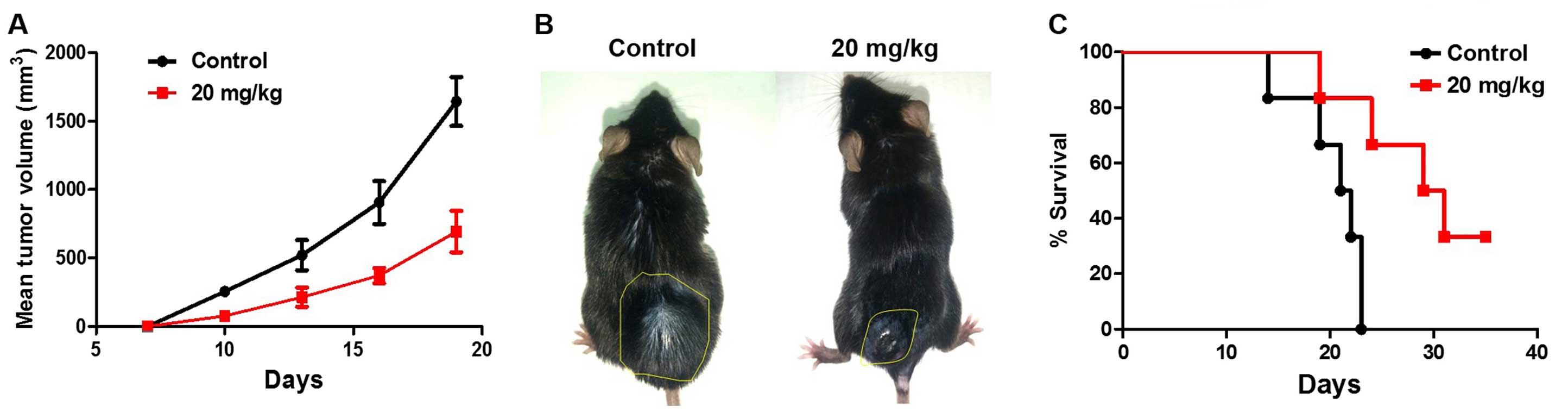
DPH suppresses the growth of B16-F10
melanoma in vivo
We next addressed whether the in vitro
cytotoxicity of DPH accounts for its in vivo anti-melanoma
effect. Using the well-established B16-F10 melanoma mouse model, we
found that i.p. administration of DPH (20 mg/kg) evidently
suppressed tumor growth compared to the vehicle control (Fig. 2A and B). Furthermore, the tumor
volume was decreased to nearly 30% of the vehicle control 19 days
after DPH administration (from 1750±176 to 610±209 mm3,
p<0.001). It is also noteworthy that DPH markedly prolonged the
survival of the B16-F10-bearing mice (Fig. 2C). Overall, these results validated
the in vivo anti-melanoma effect of DPH.
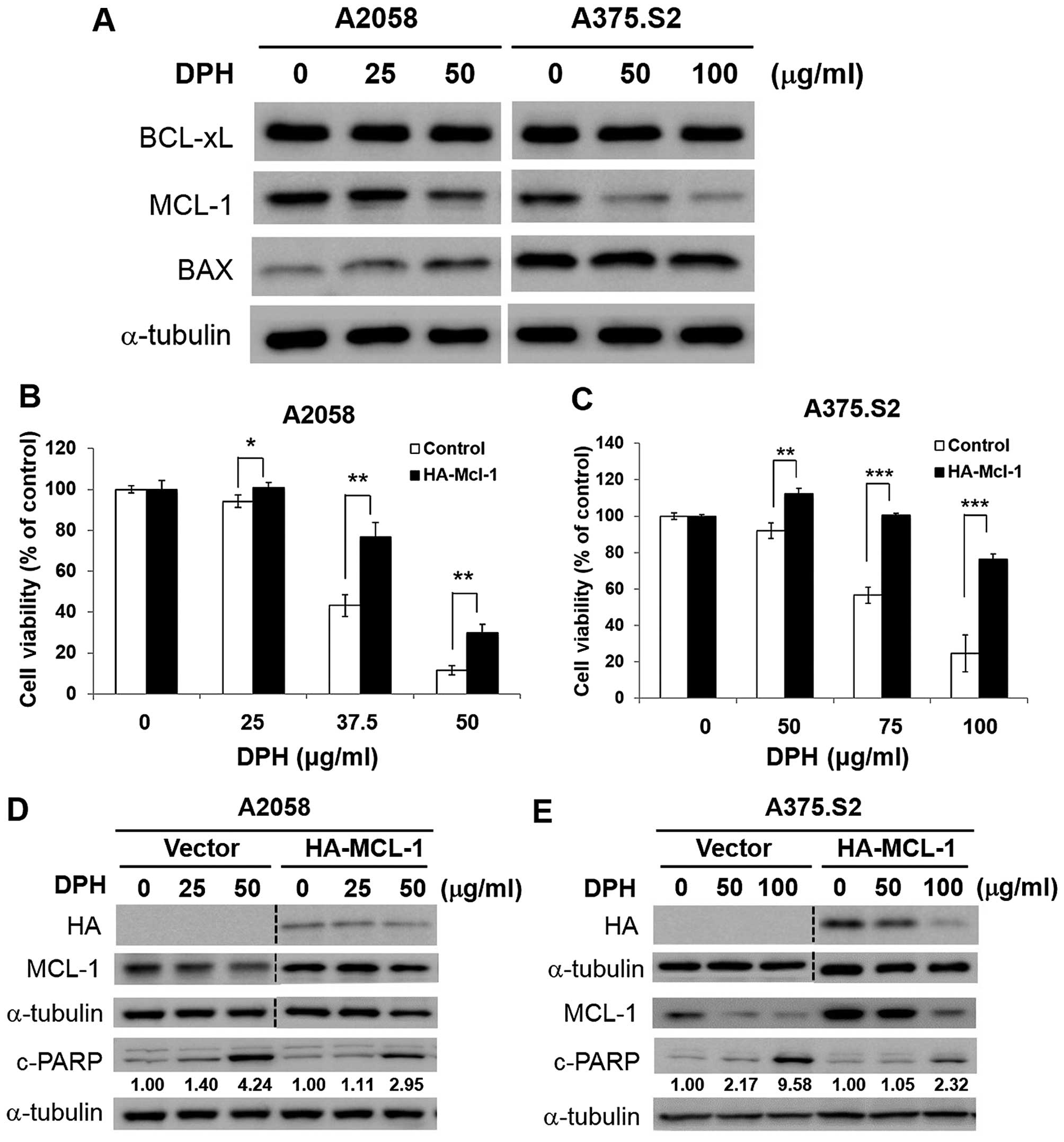
MCL-1 downregulation is vital to
DPH-induced melanoma cell apoptosis
The mechanism underlying the proapoptotic action of
DPH was further investigated. We started by testing the effect of
DPH on the expression of proteins responsible for the control of
apoptosis initiation, including proapoptotic BAX and antiapoptotic
BCL-xL and MCL-1. It is noteworthy that only MCL-1 was markedly
reduced by DPH in both the A2058 and A375.S2 cells (Fig. 3A). Given that MCL-1 is fundamental
for melanoma progression, relapse and drug resistance (12–14),
the role of MCL-1 downregulation in DPH-elicited anti-melanoma
activity was further examined in the A2058 and A375.S2 clones
stably overexpressing MCL-1. We found that MCL-1 overexpression
potently rescued cells from DPH-evoked cell death (Fig. 3B and C). Immunoblotting further
revealed noticeable reduction in DPH-induced PARP cleavage in the
MCL-1 stable clones (Fig. 3D and
E), suggesting that MCL-1 overexpression blunts DPH-elicited
apoptosis. Collectively, we conclude that downregulation of MCL-1
is integral to the proapoptotic action of DPH.
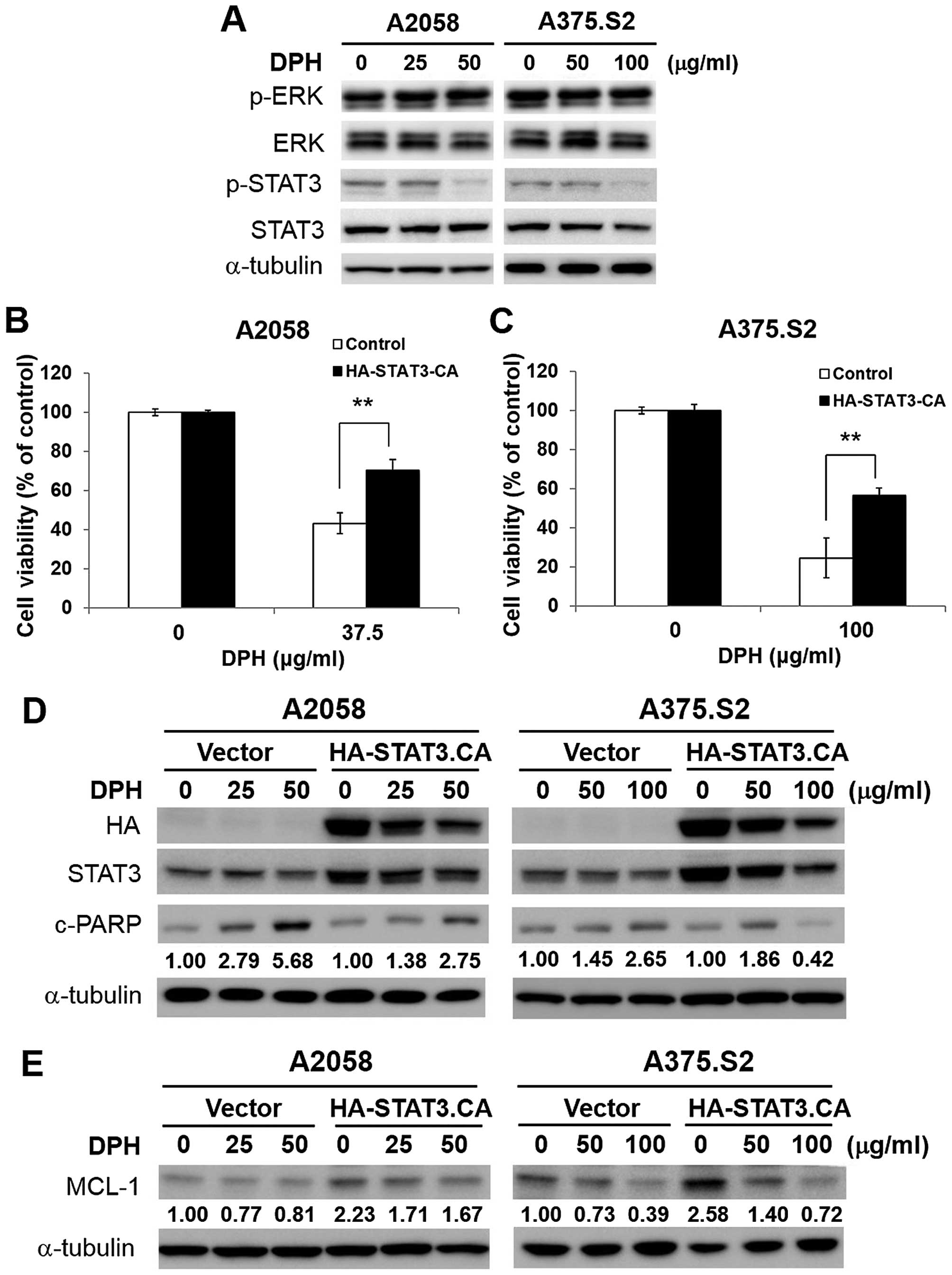
DPH impairs STAT3/MCL-1 survival
signaling in melanoma cells
We next explored how DPH downregulates MCL-1. It has
been shown that oncogenic BRAFV600E-initiated persistent
ERK signaling upregulates MCL-1 (15). However, immunoblotting showed
limited alteration in the levels of phospho-ERK (Thr202/Tyr204)
following DPH treatment (Fig. 4A),
suggesting that DPH failed to inhibit ERK signaling to decrease
MCL-1. Alternatively, we tested the effect of DPH on the activation
of STAT3 [revealed by phosphorylation of tyrosine 705
(p-Y705-STAT3)], considering that MCL-1 is a known transcriptional
target of STAT3 (16). In addition,
constitutive activation of STAT3 is associated with melanoma
tumorigenesis, progression and chemoresistance through the
upregulation of the transcription of genes promoting cell
proliferation, survival, angiogenesis and metastasis (12,17,18).
Our results indicated that DPH impeded constitutive STAT3
activation, as evidenced by the decrease in the levels of
p-Y705-STAT3 in both the A2058 and A375.S2 cells (Fig. 4A). Furthermore, overexpression of a
constitutively active STAT3 mutant (HA-STAT3-CA) effectively
protected cells from DPH-induced cell death (Fig. 4B and C), likely through the
reduction of apoptosis (Fig. 4D),
thus illustrating an essential role of STAT3 blockage in the
proapoptotic action of DPH. Moreover, HA-STAT3-CA-expressing cells
displayed an increase in the basal levels of MCL-1 but also
markedly restored MCL-1 expression levels following DPH treatment
(Fig. 4E), indicating that DPH
downregulates MCL-1 by impairing STAT3-mediated induction of MCL-1.
Overall, we conclude that DPH likely blocks the STAT3-MCL-1
survival signaling pathway to induce apoptosis in melanoma
cells.
Discussion
In this study, we present clear evidence supporting
the in vitro and in vivo anti-melanoma activities of
DPH. Particularly, we revealed that DPH induced apoptosis-dependent
cytotoxicity against a panel of melanoma cell lines while sparing
normal melanocytes (Fig. 1). We
also verified a marked suppression of in vivo B16-F10
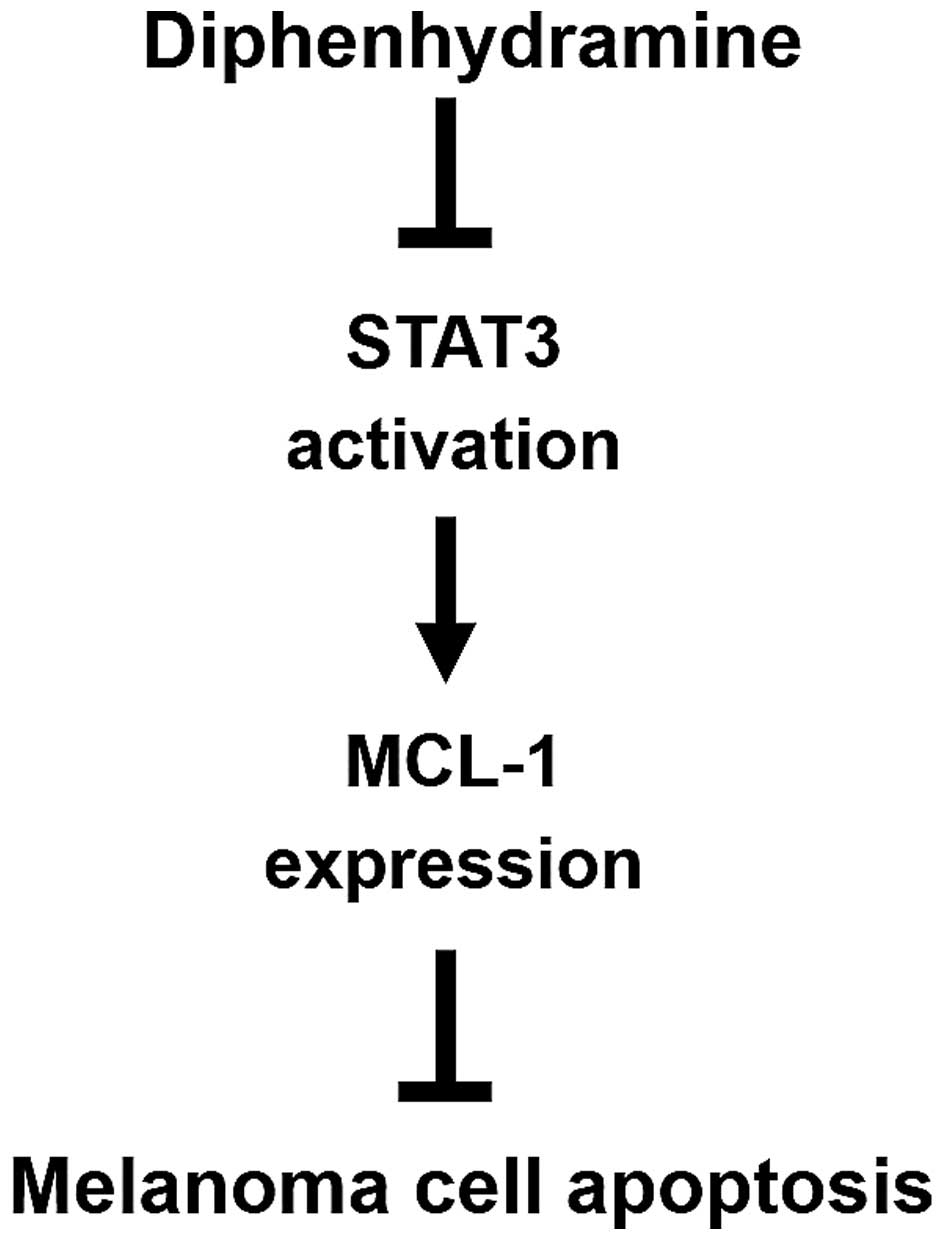
melanoma growth after DPH administration (Fig. 2). Mechanistically, DPH thwarts STAT3
activation to downregulate MCL-1, leading to the induction of
apoptosis in melanoma cells (Fig.
5). To the best of our knowledge, this is the first report
validating the in vivo anti-melanoma activity of DPH in
addition to the role of the STAT3/MCL-1 survival signaling pathway
in DPH-induced melanoma cell apoptosis.
Data presented here for the first time confirm that
DPH is selectively cytotoxic in malignant melanoma cell lines
(Fig. 1), hence highlighting its
advantage as an anticancer agent. The molecular basis underlying
this selective cytotoxicity is currently unknown. Notably, recent
studies have revealed higher expression levels of MCL-1 in melanoma
cell lines and also in tissue samples from melanoma patients
compared to their normal counterparts (19). It is thus reasonable to presume that
melanoma cells are more sensitive to the decrease in MCL-1-mediated
survival signals than normal melanocytes. Along this line,
DPH-elicited MCL-1 downregulation likely accounts for its selective
cytotoxicity against melanoma cells.
We found that the anti-melanoma activity of DPH is
attributed to the induction of apoptosis (Fig. 1). DPH was previously shown to evoke
apoptosis in T-cell acute lymphoblastic leukemia cell lines
(20), suggesting that apoptosis
induction is a general mode of action underlying the anticancer
effect of DPH. It has also been shown that additional H1 histamine
receptor antagonists such as terfenadine and astemizol were proven
to induce apoptosis in melanoma cells (10). Surprisingly, terfenadine provokes
melanoma cell apoptosis through an H1 histamine
receptor-independent mechanism (21); likewise, the anticancer effect of
astemizol is not entirely ascribed to its antihistamine activity
(22). Given that DPH is both an H1
antihistaminic and a sodium channel blocker, further investigation
is warranted in order to ascertain whether DPH induces apoptosis
through inhibition of the H1 receptor activity or blockage of
sodium channels, whose promoting roles in cancer cell proliferation
and metastasis have been recently uncovered (23).
Our mechanistic exploration identified MCL-1
downregulation as a pivotal mode of action underlying DPH-elicited
melanoma cell apoptosis (Fig. 3).
Notably, high MCL-1 expression is fundamental for the progression,
relapse and chemoresistance of multiple types of human cancer,
including melanoma, therefore making MCL-1 a promising therapeutic
target (12–14). Indeed, recent studies have validated
MCL-1 targeting as an effective strategy to eradicate cancer cells
which are dependent on MCL-1 for survival or chemoresistance
(19,24), leading to intensive efforts toward
the discovery of novel MCL-1-targeted therapeutic agents. Our
finding that DPH decreases MCL-1 expression therefore adds DPH to
the growing list of novel MCL-1-targeted small molecules with great
translation potential as anticancer agents.
We also confirmed the essential role of STAT3
activation blockage in the anti-melanoma action of DPH (Fig. 4). Constitutive STAT3 activation is
highly correlated with tumorigenesis, progression and drug
resistance in a variety of human tumors, including melanoma
(16–18). In contrast, ablating STAT3
activation has been shown to inhibit tumor growth, retard
metastasis and overcome resistance to drugs such as vemurafenib
(25), highlighting that
STAT3-targeted drugs are promising cancer therapeutics. Along this
line, DPH acting as an inhibitor of STAT3 activation holds great
potential as an anticancer agent. The mechanisms whereby DPH
supresses STAT3 activation remain elusive and are currently under
investigation in our laboratory.
In the present study, we confirmed that DPH impairs
STAT3 activation thus blocking STAT3-mediated induction of MCL-1
(Fig. 4), indicating that DPH
targets the STAT3/MCL-1-mediated survival signaling pathway
triggering melanoma cell apoptosis. Several lines of evidence have
underscored the central role of the STAT3/MCL-1 signaling axis in
the progression and survival of melanoma cells. In melanoma tumor
samples, the levels of p-Y705-STAT3 and MCL-1 were shown to be
increased together in association with melanoma progression
(12). Furthermore, blocking STAT3
activation led to apoptosis in melanoma cells accompanied by
downregulation of MCL-1 (16).
Moreover, recent studies have validated that oncogenic
BRAFV600E-initiated survival signaling in melanoma cells
actually depends on STAT3-mediated MCL-1 upregulation, as blockade
of STAT3 activation impairing the induction of MCL-1 by
BRAFV600E and consequently leading to massive apoptosis
(15). Thus, drugs such as DPH
targeting the STAT3/MCL-1 survival signaling pathway should hold
great potential as novel therapeutic agents for the treatment of
melanoma.
In conclusion, we herein establish DPH as a
selective apoptosis inducer of melanoma cells through targeted
suppression of the STAT3/MCL-1 survival signaling pathway. The
in vivo anti-melanoma activity of DPH was also clearly
validated. Our discovery therefore suggests the possible clinical
application to repurpose DPH as a novel cancer therapeutic agent in
the treatment of melanoma.
Acknowledgements
This study was supported by the Ministry of
Education, Taiwan, R.O.C. under the ATU plan. The authors are
indebted to Professor Chi-Chen Lin (National Chung Hsing
University) for his generous assistance in the in vivo
B16-F10 melanoma growth study.
References
|
1
|
Friedman RJ and Heilman ER: The pathology
of malignant melanoma. Dermatol Clin. 20:659–676. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lens MB and Dawes M: Global perspectives
of contemporary epidemiological trends of cutaneous malignant
melanoma. Br J Dermatol. 150:179–185. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Houghton AN and Polsky D: Focus on
melanoma. Cancer Cell. 2:275–278. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Diepgen TL and Mahler V: The epidemiology
of skin cancer. Br J Dermatol. 146:(Suppl 61). 1–6. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Linos E, Swetter SM, Cockburn MG, Colditz
GA and Clarke CA: Increasing burden of melanoma in the United
States. J Invest Dermatol. 129:1666–1674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Antihistamine Drugs, . Drug information.
85. American Society of Hospital Pharmacists; Bethesda, MD:
1985
|
|
7
|
Dupuis LL and Nathan PC: Optimizing emetic
control in children receiving antineoplastic therapy: Beyond the
guidelines. Paediatr Drugs. 12:51–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuo CC, Huang RC and Lou BS: Inhibition of
Na+ current by diphenhydramine and other diphenyl
compounds: Molecular determinants of selective binding to the
inactivated channels. Mol Pharmacol. 57:135–143. 2000.PubMed/NCBI
|
|
9
|
Green SM, Rothrock SG and Gorchynski J:
Validation of diphenhydramine as a dermal local anesthetic. Ann
Emerg Med. 23:1284–1289. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jangi SM, Díaz-Pérez JL, Ochoa-Lizarralde
B, Martín-Ruiz I, Asumendi A, Pérez-Yarza G, Gardeazabal J,
Díaz-Ramón JL and Boyano MD: H1 histamine receptor antagonists
induce genotoxic and caspase-2-dependent apoptosis in human
melanoma cells. Carcinogenesis. 27:1787–1796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsieh HY, Shieh JJ, Chen CJ, Pan MY, Yang
SY, Lin SC, Chang JS, Lee AYL and Chang CC: Prodigiosin
down-regulates SKP2 to induce p27KIP1 stabilization and
antiproliferation in human lung adenocarcinoma cells. Br J
Pharmacol. 166:2095–2108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhuang L, Lee CS, Scolyer RA, McCarthy SW,
Zhang XD, Thompson JF and Hersey P: Mcl-1, Bcl-XL and Stat3
expression are associated with progression of melanoma whereas
Bcl-2, AP-2 and MITF levels decrease during progression of
melanoma. Mod Pathol. 20:416–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perciavalle RM and Opferman JT: Delving
deeper: MCL-1s contributions to normal and cancer biology. Trends
Cell Biol. 23:22–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Belmar J and Fesik SW: Small molecule
Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther.
145:76–84. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Becker TM, Boyd SC, Mijatov B,
Gowrishankar K, Snoyman S, Pupo GM, Scolyer RA, Mann GJ, Kefford
RF, Zhang XD, et al: Mutant B-RAF-Mcl-1 survival signaling depends
on the STAT3 transcription factor. Oncogene. 33:1158–1166. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Niu G, Bowman T, Huang M, Shivers S,
Reintgen D, Daud A, Chang A, Kraker A, Jove R and Yu H: Roles of
activated Src and Stat3 signaling in melanoma tumor cell growth.
Oncogene. 21:7001–7010. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siveen KS, Sikka S, Surana R, Dai X, Zhang
J, Kumar AP, Tan BKH, Sethi G and Bishayee A: Targeting the STAT3
signaling pathway in cancer: Role of synthetic and natural
inhibitors. Biochim Biophys Acta. 1845:136–154. 2014.PubMed/NCBI
|
|
18
|
Zhao C, Li H, Lin HJ, Yang S, Lin J and
Liang G: Feedback activation of STAT3 as a cancer drug-resistance
mechanism. Trends Pharmacol Sci. 37:47–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mukherjee N, Lu Y, Almeida A, Lambert K,
Shiau CW, Su JC, Luo Y, Fujita M, Robinson WA, Robinson SE, et al:
Use of a MCL-1 inhibitor alone to de-bulk melanoma and in
combination to kill melanoma initiating cells. Oncotarget. Apr
12–2016.(Epub ahead of print).
|
|
20
|
Jangi SM, Asumendi A, Arlucea J, Nieto N,
Perez-Yarza G, Morales MC, de la Fuente-Pinedo M and Boyano MD:
Apoptosis of human T-cell acute lymphoblastic leukemia cells by
diphenhydramine, an H1 histamine receptor antagonist. Oncol Res.
14:363–372. 2004.PubMed/NCBI
|
|
21
|
Jangi SM, Ruiz-Larrea MB, Nicolau-Galmés
F, Andollo Y, Arroyo-Berdugo N, Ortega-Martínez I, Díaz-Pérez JL
and Boyano MD: Terfenadine-induced apoptosis in human melanoma
cells is mediated through Ca+2 homeostasis modulation
and tyrosine kinase activity, independently of H1 histamine
receptors. Carcinogenesis. 29:500–509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
García-Quiroz J and Camacho J: Astemizole:
An old anti- histamine as a new promising anti-cancer drug.
Anticancer Agents Med Chem. 11:307–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roger S, Gillet L, Le Guennec JY and
Besson P: Voltage-gated sodium channels and cancer: Is excitability
their primary role? Front Pharmacol. 6:1522015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fofaria NM, Frederick DT, Sullivan RJ,
Flaherty KT and Srivastava SK: Overexpression of Mcl-1 confers
resistance to BRAFV600E inhibitors alone and in
combination with MEK1/2 inhibitors in melanoma. Oncotarget.
6:40535–40556. 2015.PubMed/NCBI
|
|
25
|
Liu F, Cao J, Wu J, Sullivan K, Shen J,
Ryu B, Xu Z, Wei W and Cui R: Stat3-targeted therapies overcome the
acquired resistance to vemurafenib in melanomas. J Invest Dermatol.
133:2041–2049. 2013. View Article : Google Scholar : PubMed/NCBI
|