Introduction
Lung cancer is the leading cause of cancer-related
deaths both in China and worldwide (1), and metastasis is the major cause of
cancer-related deaths (2).
Mechanistically, metastasis is a complex process including tumor
cell invasion of local vessels, survival of circulating tumor
cells, and colonization of tumor cells in distant organs. The
success of metastasis depends on intrinsic properties of the tumor
cells as well as interaction between tumor cells and tumor
environmental factors (3).
Investigation of the underlying mechanisms of tumor invasion and
metastasis may help us predict tumor aggressiveness, tailor
treatment according to metastatic potential and explore more
effective therapeutic targets.
MicroRNAs (miRNAs) are small non-coding RNAs that
regulate gene expression post-transcriptionally. In mammals, miRNAs
are predicted to regulate the activity of ~50% of all
protein-coding genes (4).
Deregulation of miRNAs has been implicated in various human
diseases, particularly cancer (5).
In addition to the regulation of tumor cell proliferation,
differentiation and apoptosis, miRNAs also regulate tumor cell
migration and invasion (6).
Numerous miRNAs have been demonstrated to play an essential role in
tumor metastasis (7–10).
miRNA-134 (miR-134) was initially identified as a
brain-specific miRNA that is involved in synapse development
(11). Subsequent investigations
revealed its essential role in stem cell differentiation (12–14).
Recently, numerous studies investigated the important functions of
miR-134 in cancer, presenting inconsistent results (15–24).
In non-small cell lung cancer (NSCLC), our previous data
demonstrated that miR-134 inhibited tumor cell growth both in
vitro and in vivo (25),
but its role as a suppressor or promoter of metastasis is still
controversial and warrants more investigation (16,17,23).
In the present study, we aimed to explore the role of miR-134 in
NSCLC cell migration and invasion, and identify potential targets
of miR-134.
Materials and methods
Cell culture
Human NSCLC cell lines (A549 and NCI-H1299) were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). Cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin and incubated in 5%
CO2 at 37°C.
miRNA and siRNA transfection
Cells were cultured to 40–50% confluency and
transiently transfected with 50 nM miRNA negative control (miR-NC),
or miRNA mimics using HiPerFect Transfection Reagent (Qiagen,
Germantown, MD, USA) according to the manufacturer's instructions.
For RNAi experiments, siRNAs were also transfected using HiperFect
Transfection Reagent at a final concentration of 50 nM. miRNA
mimics and NC were purchased from GenePharma (Shanghai, China), and
siRNAs were purchased from RiboBio Co., Ltd. (Guangzhou,
China).
Transwell migration and invasion
assays
For the migration assay, 2×104
transfected A549 or H1299 cells were seeded in the top chamber of
the Transwell insert (BD Biosciences, Sparks, MD, USA). For the
invasion assay, 3×105 transfected A549 or
1×105 transfected H1299 cells were seeded in the top
chamber of the insert (BD Biosciences), which had previously been
coated with 100 µl diluted Matrigel (Corning, Corning, NY, USA).
After 20–24 h of incubation at 37°C, the cells that had migrated or
invaded through the insert were fixed with 100% methanol, stained
with 0.1% crystal violet and counted.
RNA extraction and quantitative
real-time PCR
Total RNA containing miRNAs was isolated from cells
using miRNeasy Mini kit (Qiagen). For single-stranded complementary
DNA synthesis, 500 ng total RNA was reverse-transcribed using
PrimeScript™ RT reagent kit with gDNA Eraser (Takara, Dalian,
China). Real-time PCR was performed using UltraSYBR Mixture (CWBio,
Beijing, China) and LC 480 PCR System (Roche). Primers for integrin
β1 (ITGB1), KRAS, FOXM1 and GAPDH are shown in Table I. The expression levels of mRNA were
normalized to the endogenous control GAPDH, using the
2−ΔΔCt method.
 | Table I.Primers for real-time PCR. |
Table I.
Primers for real-time PCR.
| Gene | Forward | Reverse |
|---|
| ITGB1 |
5′-ATCCCAGAGGCTCCAAAGAT-3′ |
5′-CCCCTGATCTTAATCGCAAA-3′ |
| KRAS |
5′-TGGTGAGGGAGATCCGACAA-3′ |
5′-AGGCATCATCAACACCCAGA-3′ |
| FOXM1 |
5′-ATAGCAAGCGAGTCCGCATT-3′ |
5′-AGCAGCACTGATAAACAAAGAAAGA-3′ |
| GAPDH |
5′-CATGAGAAGTATGACAACAGCCT-3′ |
5′-AGTCCTTCCACGATACCAAAGT-3′ |
Western blotting
Cells were lysed using cell lysis buffer for western
blot analysis and IP with protease inhibitor phenylmethylsulfonyl
fluoride (PMSF) (Beyotime, Shanghai, China). Equal amounts of
protein were separated by SDS-PAGE, and then transferred onto
polyvinylidene difluoride membranes (Millipore, Billerica, MA,
USA). Membranes were blocked with 5% non-fat milk in TBS containing
0.1% Tween-20, and then incubated with corresponding primary
antibody. Primary antibodies against GAPDH (Abcam, Cambridge, UK)
were used at a dilution of 1:10,000, against E-cadherin and
vimentin (VIM) at a dilution of 1:1,000, and against ITGB1 (Abcam,
Cambridge, UK) at a dilution of 1:2,000. Secondary antibodies
conjugated with horseradish peroxidase (anti-mouse IgG and
anti-rabbit IgG) were used to detect primary antibodies. For HRP
detection, an ECL chemiluminescence kit (CWBio) was used.
Luciferase reporter assays
The wild-type 3 untranslated region (3′UTR) of ITGB1
and its target-site mutant 3′UTR were amplified by PCR. The PCR
products were then cloned into the XhoI/NotI site of
the psiCHECK-2 dual-luciferase reporter plasmid (Promega, Madison,
WI, USA). These vectors were named ITGB1-3′UTR, ITGB1-3′UTRm1,
ITGB1-3′UTRm2 and ITGB1-3′UTRm1+2, respectively. To perform
luciferase reporter assays, A549 and H1299 cells were plated into
96-well plates and co-transfected with the reporter vector and 50
nM miR-NC or miR-134 mimics using Attractene Transfection Reagent
(Qiagen). Forty-eight hours after transfection, Firefly and
Renilla luciferase activities were measured using a
Dual-Luciferase Reporter System (Promega).
Generation of A549 and H1299 cells
stably expressing ITGB1
Lentiviral vector expressing ITGB1 or an empty
lentiviral vector was purchased from GeneChem (Shanghai, China).
Cell infection was performed following the manufacturer's protocol.
Cells stably overexpressing ITGB1 or the empty vector were selected
by puromycin (2 µg/ml). A549 and H1299 cells that stably expressed
the empty vector or ITGB1 were designated A549-control and
H1299-control, or A549-ITGB1 and H1299-ITGB1.
Functional rescue experiments
Rescue experiments were carried out to determine
whether ITGB1 mediates the suppression effects on migration and
invasion mediated by miR-134. A549-control, A549-ITGB1,
H1299-control and H1299-ITGB1 cells were transfected with miR-NC or
miR-134 mimics, and then the Transwell assays were performed.
Animal experiments
To establish a lung cancer xenograft model,
2×106 A549 cells in 100 µl phosphate-buffered saline
were subcutaneously injected intox the right hind limb of BALB/c
nude mice [female, 4–5 weeks old, purchased from Beijing HFK
Bioscience Co., Ltd. (Beijing China)]. After 10 days, when the
diameter of the tumor reached ~5-6 mm, the nude mice were randomly
divided into 2 groups (n=4 each). miR-134 agomir or negative
control (NC) agomir (RiboBio Co., Ltd.) was then directly injected
into the implanted tumor at the dose of 5 nmol/mouse every 3 days
for 5 times. Tumor volume (V) was monitored every 3 days since the
first day of injection of agomir. Forty-eight hours after the last
injection, animals were sacrificed and tumor tissues were resected.
Lung tissues were also collected for inspection. Mice were
manipulated and housed according to protocols approved by the
Shandong Hospital Experimental Animal Care Commission.
Immunohistochemistry (IHC)
Tumor tissues were fixed in formalin and embedded in
paraffin. Sections (5-µm thick) were cut from the embedded tissues
and mounted on polylysine-coated slides. Tumor sections were
subjected to IHC staining for detection of E-cadherin, VIM and
ITGB1. Briefly, sections were deparaffinized in xylene, rehydrated
in gradient alcohol and treated with 0.3%
H2O2 for 15 min to quench endogenous
peroxidase activity. Following antigen retrieval, sections were
blocked in 10% normal serum with 1% BSA in TBS for 2 h at room
temperature, and then incubated at 4°C overnight with the
corresponding primary antibodies (E-cadherin, VIM and ITGB1;
Abcam). Negative controls were incubated with the negative control
antibody under the same condition. Next, the sections were
incubated with biotinylated secondary antibody for 1 h, followed by
incubation with conjugated horseradish peroxidase streptavidin for
1 h. Finally, the sections were incubated with diaminobenzidine and
counterstained with hematoxylin.
Statistical analysis
Experiments were performed at least three times.
Data were analyzed by Student's t-test when comparing two groups
and by one-way ANOVA followed Bonferronis post-test, when comparing
more than two groups. A P-value <0.05 was considered to indicate
a statistically significant result.
Results
miR-134 inhibits the migration and
invasion of NSCLC cell lines
We chose NSCLC cell lines A549 and H1299 to
investigate migration as well as invasion due to their innate
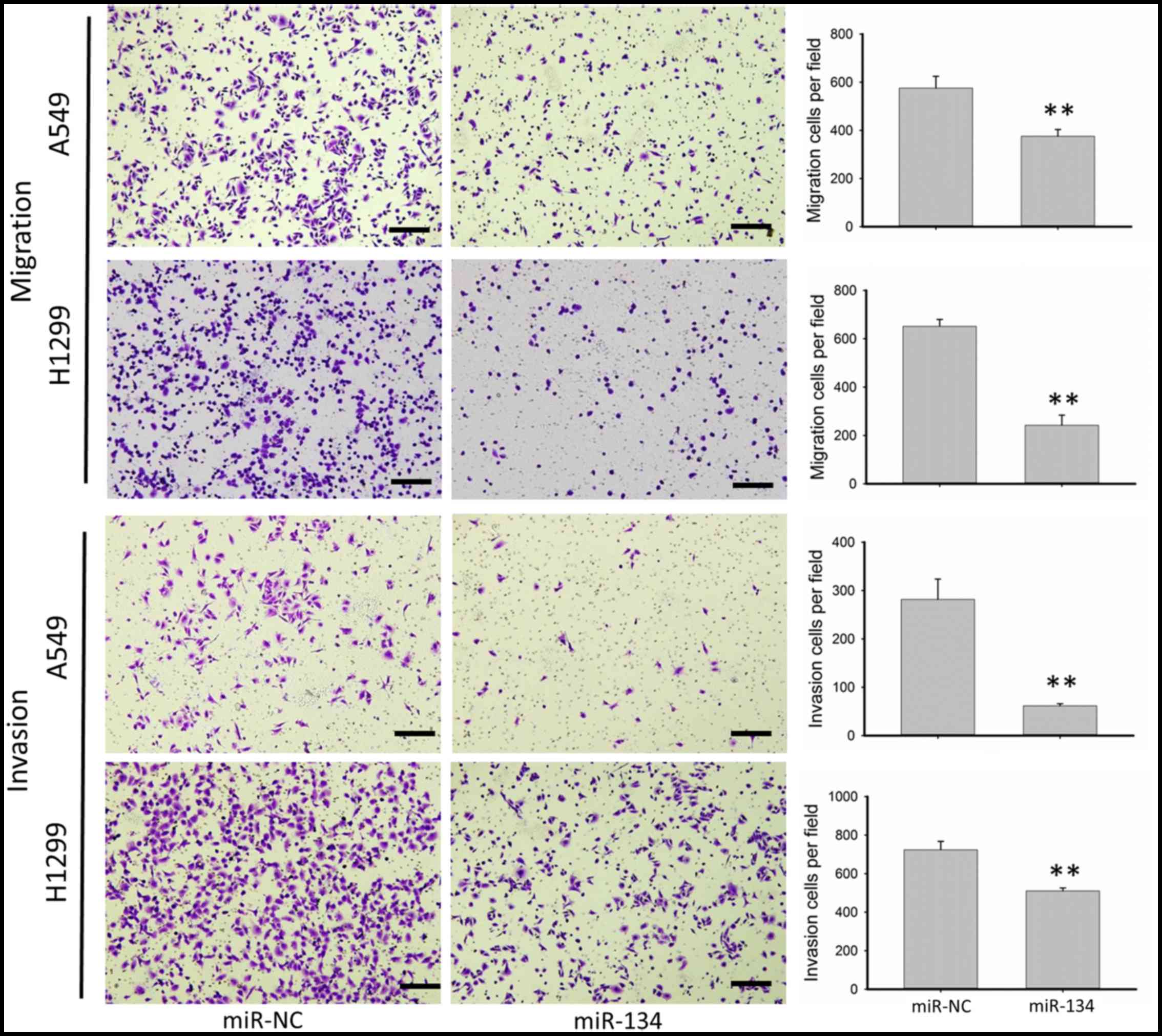
aggressiveness. As shown in Fig. 1,
miR-134 significantly inhibited the migration as well as the
invasion of the A549 and H1299 cells. Overexpression of miR-134 in
the A549 cells resulted in ~45 and 75% reduction in the number of
migratory and invasive cells, respectively, while in the H1299
cells, migratory and invasive cells were decreased 60 and 30%,
respectively, in the cells transfected with miR-134 compared with
cells transfected with the miR-NC.
miR-134 inhibits
epithelial-to-mesenchymal transition (EMT), and downregulates ITGB1
expression in NSCLC cell lines
EMT is an important process that increases the
aggressiveness and metastatic potential of cancer cells. After
revealing the suppressive roles of migration as well as invasion by
miR-134, we aimed to ascertain whether or not this suppression was
related to EMT inhibition. Thus, we detected the alteration in
expression of two essential molecules participating in EMT:
E-cadherin (epithelial marker) and VIM (mesenchymal marker). As
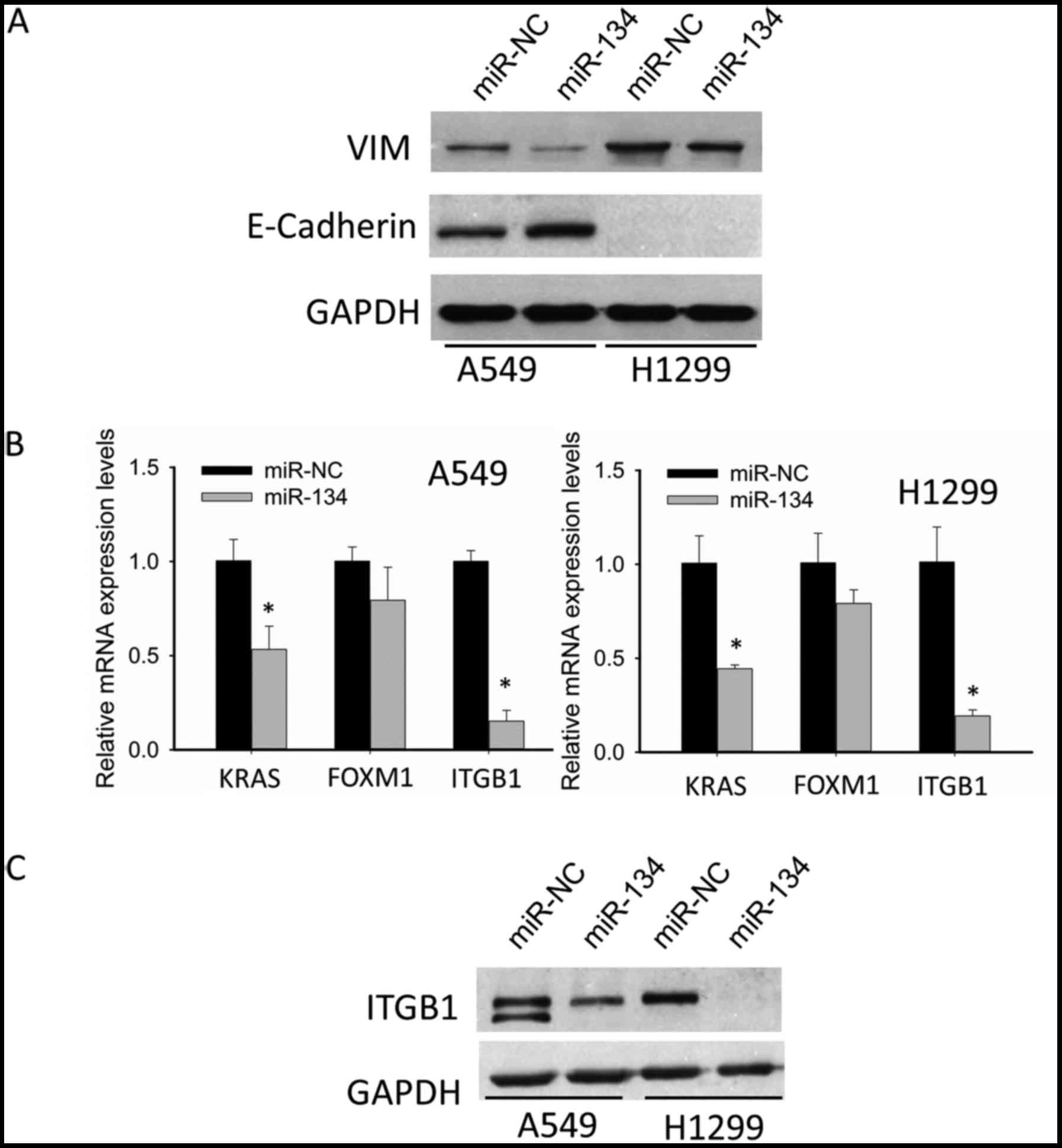
shown in Fig. 2A, in the A549
cells, miR-134 increased the expression of epithelial marker
E-cadherin, and decreased the expression of mesenchymal marker VIM;
in H1299 cells, there was no E-cadherin detected, while the
expression of VIM was significantly suppressed. These data
indicated that miR-134 inhibited EMT in NSCLC cells, which may be
partly responsible for suppression of migration and invasion by
miR-134.
Although we demonstrated that miR-134 inhibited EMT
of NSCLC cells, neither E-cadherin nor VIM was found to be the
predicted target of miR-134. Thus, we were motivated to identify
potential targets. Through integrating both target prediction as
well as literature review, we focused our attention on the
following 3 pertinent oncogenes, KRAS, FOXM1 and ITGB1, which have
been confirmed as authentic targets of miR-134. qRT-PCR suggested
that ITGB1 showed the most significant decrease after miR-134
transfection (Fig. 2B). Therefore,
we chose ITGB1 for further western blotting validation. As shown in
Fig. 2C, western blotting confirmed
the downregulation of ITGB1.
ITGB1 is a direct target of miR-134 in
NSCLC cell lines
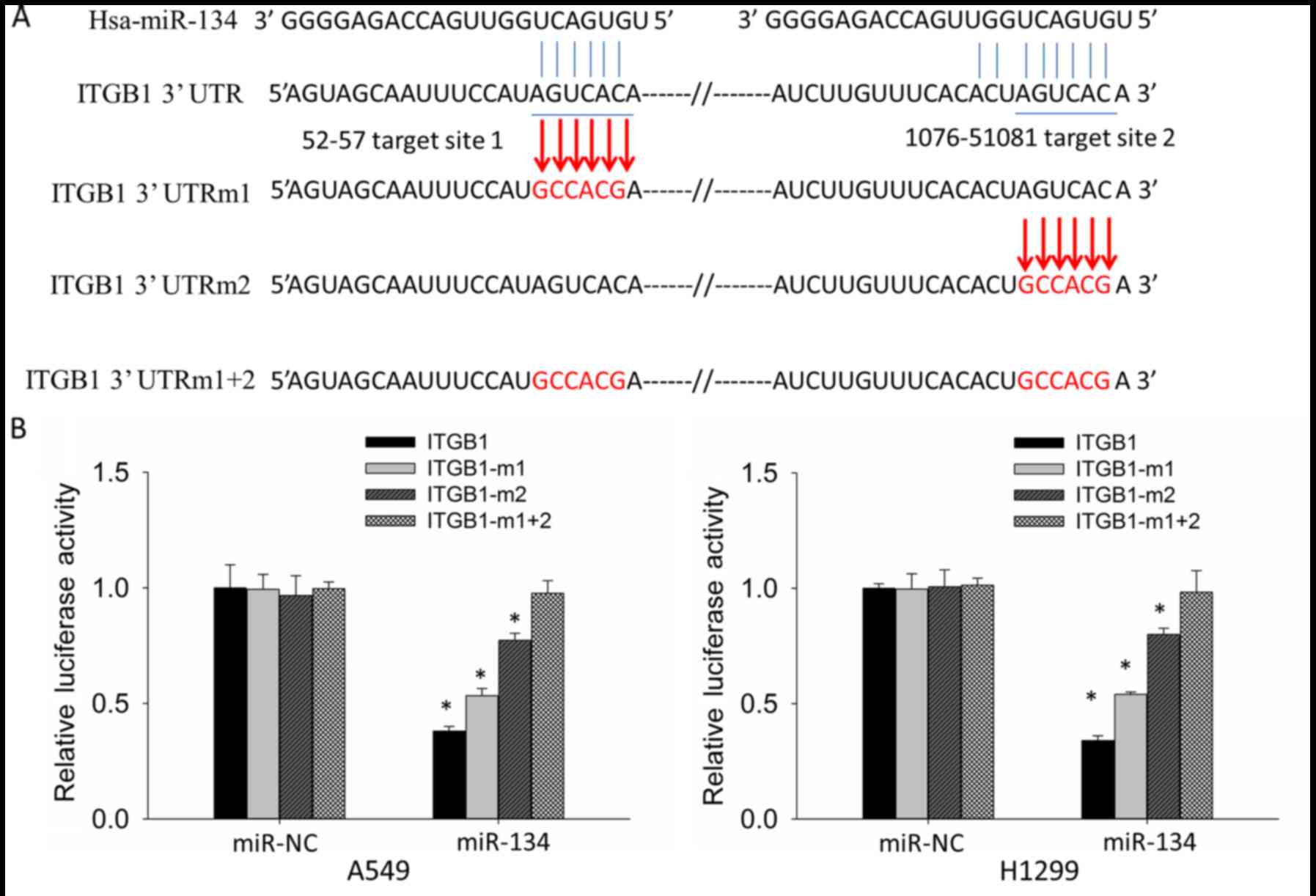
To determine whether downregulation of the ITGB1
expression levels are due to direct targeting of miR-134 to the
ITGB1-3′UTR, we constructed reporter vectors containing wild-type
(ITGB1-3′UTR) and target-site mutant (ITGB1-3′UTRm1, ITGB1-3′UTRm2
and ITGB1-3′UTRm1+2) 3′UTR of ITGB1 respectively to perform
luciferase reporter assay in the A549 and H1299 cells (Fig. 3A). Compared with miR-NC,
co-transfection of miR-134 and ITGB1-3′UTR showed significantly
decreased luciferase activity, whereas co-transfection of miR-134
and ITGB1-3′UTRm1+2 did not result in significant reduction in
luciferase activity. Co-transfection of miR-134 and ITGB1-3′UTRm1
or ITGB1-3′UTRm2 also resulted in reduced luciferase activity;
however, the degree of reduction was less than the co-transfection
of miR-134 and ITGB1-3′UTR (Fig.
3B). These results indicated that both target sites in
ITGB1-3′UTR are targeted by miR-134, and the second target site is
more potent than the first one.
ITGB1 mediates the suppressive effect
on migration and invasion by miR-134 in NSCLC cell lines
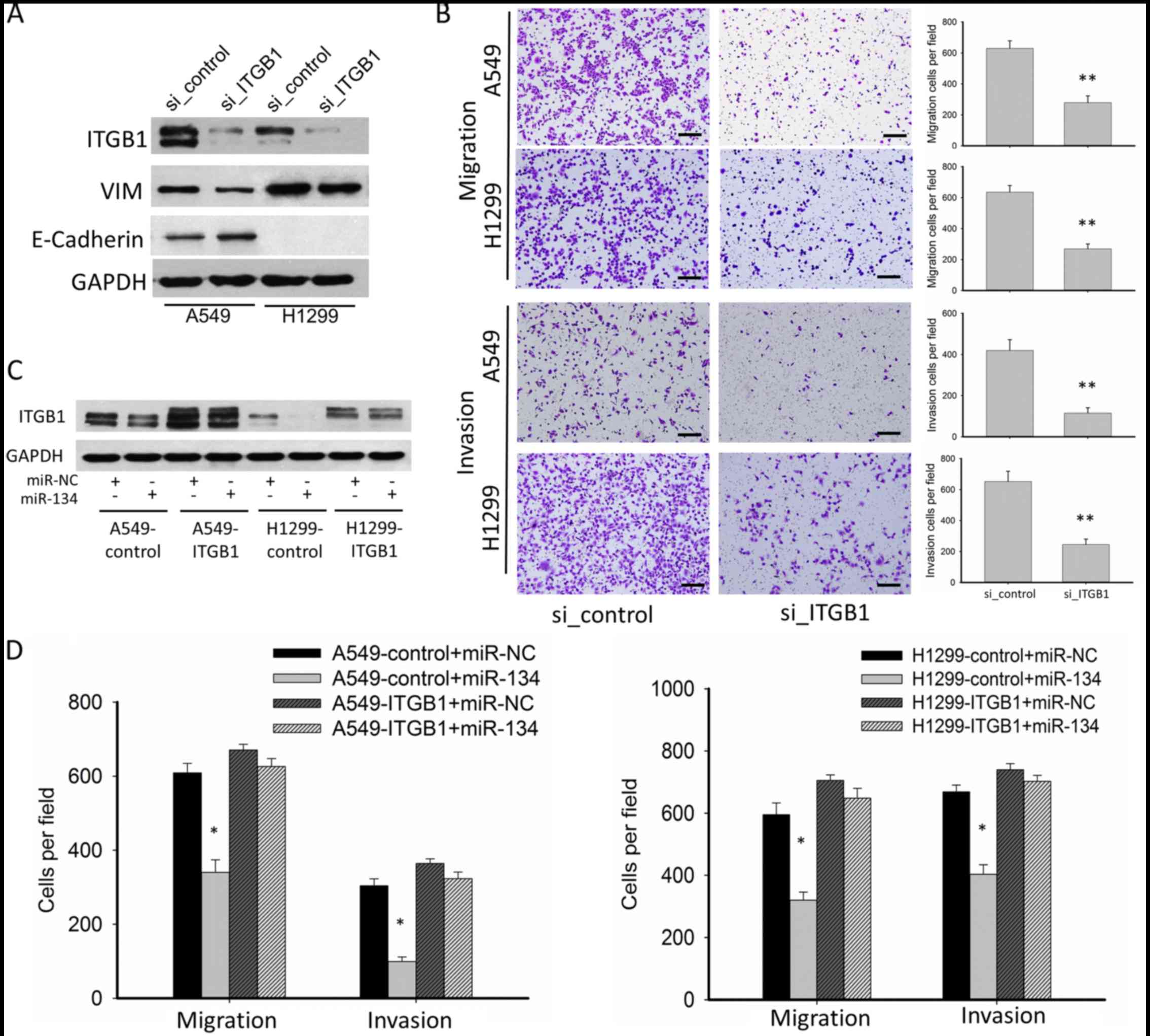
To determine whether miR-134 exerts its suppressive
functions on migration and invasion through downregulation of
ITGB1, we performed RNAi and rescue experiments. RNAi experiment
confirmed that knockdown of ITGBI significantly inhibited EMT, cell
migration and invasion, mimicking the phenotype of miR-134
overexpression (Fig. 4A and B). In
the functional rescue experiments, we constructed
ITGB1-overexpressing A549 and H1299 cells using a lentiviral vector
(A549-ITGB1 and H1299-ITGB1), in which the 3′UTR of ITGB1 was
missing. ITGB1 overexpression was confirmed by western blotting.
While miR-134 inhibited ITGB1 expression in the A549 and H1299
cells transfected with the lentiviral-vector control (A549-control
and H1299-control), no significant downregulation of ITGB1 was
detected in the A549-ITGB1 and H1299-ITGB1 cells (Fig. 4C). In the A549-ITGB1 and H1299-ITGB1
cells, the suppressive effects on tumor migration and invasion of
miR-134 were abolished (Fig. 4D),
suggesting that ITGB1 is a functional target of miR-134.
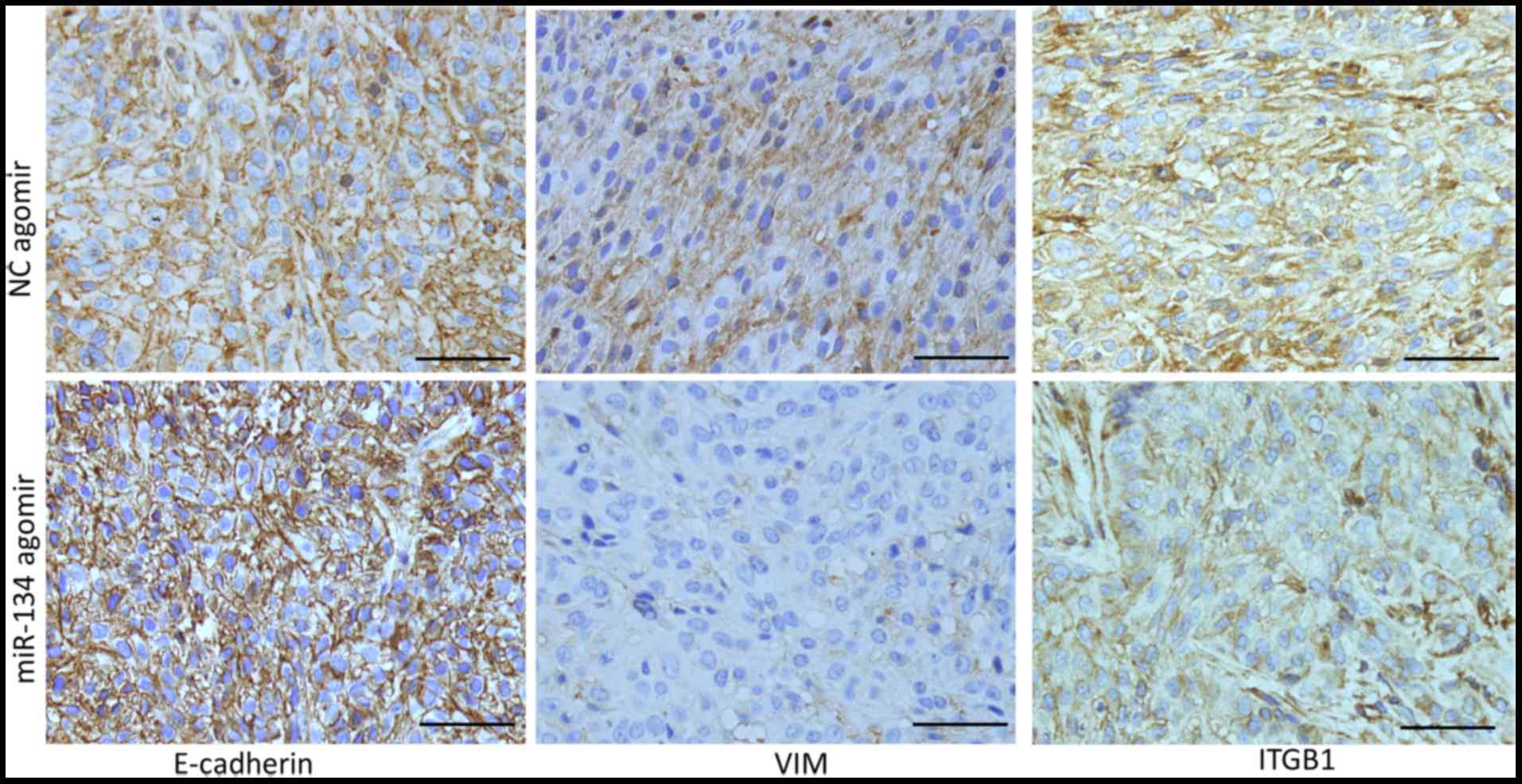
miR-134 inhibits EMT and downregulates
ITGB1 expression in an A549 lung cancer xenograft model
To investigate whether miR-134 inhibits NSCLC cell
metastasis in vivo, we constructed an A549 lung cancer
xenograft model, and treated the mice with an intro-tumor injection
of miR-134 agomir or NC agomir. miR-134 agomir treatment
significantly inhibited A549 xenograft growth (data not shown).
However, at the time of sacrifice, no metastasis was detected in
lung tissues, thus we performed IHC detection of E-cadherin, VIM
and ITGB1 as a surrogate to assess the metastatic potential of the
tumor cells. As shown in Fig. 5,
the expression level of E-cadherin was increased, while the
expression levels of VIM and ITGB1 were decreased in the tumors
injected with the miR-134 agomir compared to these levels in the
tumors injected with NC agomir. These data indicated that miR-134
increased E-cadherin and decreased VIM expression in vivo,
which was correlated with decreased EMT and aggressiveness, and
downregulated ITGB1 expression, which also suggested decreased
invasion in vivo.
Discussion
Most cancer-related deaths are caused by the
development of metastatic disease, since patients with systemically
disseminated disease hold more tumor burden and tumor cells possess
more comprehensive heterogeneity that render them more refractory
to currently available treatments (2). Considering the enormous discrepancies
of metastatic potential even among patients with the same tumor
type, it is worthwhile to explore any possible approach that may
reduce tumor metastasis.
miRNAs are key regulators in the initiation and
development of cancer. They function as tumor suppressors or
oncogenes and are potential targets for cancer treatment (6,26). In
the present study, we identified that miR-134 is a suppressor of
metastasis in NSCLC. Our results indicated that miR-134 inhibited
A549 and H1299 cell migration and invasion, which is consistent
with two previous studies (17,23),
and inconsistent with another study (16). As these studies and ours all
investigated the roles of miR-134 in A549 cells, the reasons for
such an inconsistence are difficult to explain.
EMT is a pivotal process that enables tumor cells to
acquire a more migratory and invasive mesenchymal phenotype, during
which tumor cells exhibit downregulation of the expression of
epithelial proteins such as E-cadherin, and upregulation of the
expression of mesenchymal proteins such as N-cadherin and VIM
(27). We found in the present
study that miR-134 increased the expression of E-cadherin and
decreased the expression of VIM, suggesting that miR-134 inhibits
or reverses the process of EMT. These results were similar to one
study (17), yet different to
another (16). Still, the
underlying reasons for such an apparent discordance are
unknown.
To identify potential targets of miR-134 that may be
responsible for its suppression of migration and invasion, we
selected 3 molecules: KRAS, FOXM1 and ITGB1, which were confirmed
to be authentic targets in other cancers [KRAS (19,21,24)
and ITGB1 (22)] or NSCLC [FOXM1
(17)], for validation. We found
that the expression level of ITGB1 showed the most significant
decline after transfection of miR-134. Considering such a
downregulation of ITGB1 and the importance of ITGB1 in lung cancer
metastasis (28), we chose ITGB1
for further testing.
ITGB1 is an essential subunit of the integrin family
which mediates cell-extracellular matrix (ECM) adhesion and
signaling that affect diverse cellular processes, including
proliferation, apoptosis, migration, invasion and survival
(29). Elevated expression or
activation of ITGB1 signaling not only correlates with poor
prognosis in some solid tumors including lung cancer (28,30,31),
but also confers resistance to chemotherapy (32), radiotherapy (33,34)
and targeted therapy (35–37), which renders ITGB1 as a potential
target. Strategies that target ITGB1 have been extensively
investigated in preclinical and clinical models, showing promising
efficacy (38–41). In the present study, we showed that
ITGB1 could be suppressed by miR-134 in NSCLC. Downregulation of
ITGB1 by miR-134 or siRNA both suppressed NSCLC cell migration and
invasion. However, miR-134 may be more advantageous than siRNA as
miRNAs may target a broad set of oncogenes simultaneously (26).
During the development of metastasis, regulation of
cell to cell adhesion and cell to extracellular matrix (EMC)
adhesion is essential for cancer cell migration and invasion
(2). In the present study, we
demonstrated that miR-134 increased E-cadherin expression, which is
a key mediator of cell to cell adhesion (27), thereby enhancing cell to cell
adhesion and reducing cancer cell mobility. Meanwhile, miR-134
decreased ITGB1 expression, which mediates cell to EMC adhesion
(29), thereby impairing cell to
ECM adhesion and suppressing the invasion of cancer cells.
In conclusion, we demonstrated that miR-134
downregulated ITGB1 expression and inhibited EMT, resulting in
suppressed migration and invasion of NSCLC both in vitro and
in vivo. ITGB1 is a direct functional target of miR-134.
Therefore, miR-134 replacement may be an approach to target ITGB1
and ITGB1-associated metastasis in NSCLC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81272501 and
81530060), and the Taishan Scholars Program of Shandong Province,
China (grant no. ts20120505).
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valastyan S and Weinberg RA: Tumor
metastasis: Molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010.PubMed/NCBI
|
|
5
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang H, Li Y and Lai M: The microRNA
network and tumor metastasis. Oncogene. 29:937–948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cui R, Meng W, Sun HL, Kim T, Ye Z, Fassan
M, Jeon YJ, Li B, Vicentini C, Peng Y, et al: MicroRNA-224 promotes
tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci
USA. 112:E4288–E4297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang RM, Xu JF, Fang F, Yang H and Yang
LY: MicroRNA-130b promotes proliferation and EMT-induced metastasis
via PTEN/p-AKT/HIF-1α signaling. Tumour Biol. 37:10609–10619. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schratt GM, Tuebing F, Nigh EA, Kane CG,
Sabatini ME, Kiebler M and Greenberg ME: A brain-specific microRNA
regulates dendritic spine development. Nature. 439:283–289. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poitz DM, Stölzel F, Arabanian L,
Friedrichs J, Docheva D, Schieker M, Fierro FA, Platzbecker U,
Ordemann R, Werner C, et al: MiR-134-mediated β1 integrin
expression and function in mesenchymal stem cells. Biochim Biophys
Acta. 1833:3396–3404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tay Y, Zhang J, Thomson AM, Lim B and
Rigoutsos I: MicroRNAs to Nanog, Oct4 and Sox2 coding regions
modulate embryonic stem cell differentiation. Nature.
455:1124–1128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tay YM, Tam WL, Ang YS, Gaughwin PM, Yang
H, Wang W, Liu R, George J, Ng HH, Perera RJ, et al: MicroRNA-134
modulates the differentiation of mouse embryonic stem cells, where
it causes post-transcriptional attenuation of Nanog and LRH1. Stem
Cells. 26:17–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen T, Gao F, Feng S, Yang T and Chen M:
MicroRNA-134 regulates lung cancer cell H69 growth and apoptosis by
targeting WWOX gene and suppressing the ERK1/2 signaling pathway.
Biochem Biophys Res Commun. 464:748–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kitamura K, Seike M, Okano T, Matsuda K,
Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K and Gemma A:
MiR-134/487b/655 cluster regulates TGF-β-induced
epithelial-mesenchymal transition and drug resistance to gefitinib
by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther.
13:444–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Wang Y, Luo J, Fu Z, Ying J, Yu Y
and Yu W: miR-134 inhibits epithelial to mesenchymal transition by
targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett.
586:3761–3765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu CJ, Shen WG, Peng SY, Cheng HW, Kao
SY, Lin SC and Chang KW: miR-134 induces oncogenicity and
metastasis in head and neck carcinoma through targeting WWOX gene.
Int J Cancer. 134:811–821. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Zhang M, Qian J, Bao M, Meng X,
Zhang S, Zhang L, Zhao R, Li S, Cao Q, et al: miR-134 functions as
a tumor suppressor in cell proliferation and
epithelial-to-mesenchymal Transition by targeting KRAS in renal
cell carcinoma cells. DNA Cell Biol. 34:429–436. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niu CS, Yang Y and Cheng CD: MiR-134
regulates the proliferation and invasion of glioblastoma cells by
reducing Nanog expression. Int J Oncol. 42:1533–1540.
2013.PubMed/NCBI
|
|
21
|
Yin C, Wang PQ, Xu WP, Yang Y, Zhang Q,
Ning BF, Zhang PP, Zhou WP, Xie WF, Chen WS, et al: Hepatocyte
nuclear factor-4α reverses malignancy of hepatocellular carcinoma
through regulating miR-134 in the DLK1-DIO3 region. Hepatology.
58:1964–1976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zha R, Guo W, Zhang Z, Qiu Z, Wang Q, Ding
J, Huang S, Chen T, Gu J, Yao M, et al: Genome-wide screening
identified that miR-134 acts as a metastasis suppressor by
targeting integrin β1 in hepatocellular carcinoma. PLoS One.
9:e876652014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Wang H, Zhang S, Song J, Zhang Y,
Wei X and Feng Z: MiR-134 functions as a regulator of cell
proliferation, apoptosis, and migration involving lung septation.
In Vitro Cell Dev Biol Anim. 48:131–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Kim J, Mueller AC, Dey B, Yang Y,
Lee DH, Hachmann J, Finderle S, Park DM, Christensen J, et al:
Multiple receptor tyrosine kinases converge on microRNA-134 to
control KRAS, STAT5B, and glioblastoma. Cell Death Differ.
21:720–734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qin Q, Wei F, Zhang J, Wang X and Li B:
miR-134 inhibits non-small cell lung cancer growth by targeting the
epidermal growth factor receptor. J Cell Mol Med. 20:1974–1983.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen Y, Gao DY and Huang L: In vivo
delivery of miRNAs for cancer therapy: Challenges and strategies.
Adv Drug Deliv Rev. 81:128–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oshita F, Kameda Y, Hamanaka N, Saito H,
Yamada K, Noda K and Mitsuda A: High expression of integrin beta1
and p53 is a greater poor prognostic factor than clinical stage in
small-cell lung cancer. Am J Clin Oncol. 27:215–219. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nikkola J, Vihinen P, Vlaykova T,
Hahka-Kemppinen M, Heino J and Pyrhönen S: Integrin chains beta1
and alphav as prognostic factors in human metastatic melanoma.
Melanoma Res. 14:29–37. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yao ES, Zhang H, Chen YY, Lee B, Chew K,
Moore D and Park C: Increased beta1 integrin is associated with
decreased survival in invasive breast cancer. Cancer Res.
67:659–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Damiano JS: Integrins as novel drug
targets for overcoming innate drug resistance. Curr Cancer Drug
Targets. 2:37–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cordes N, Seidler J, Durzok R, Geinitz H
and Brakebusch C: Beta1-integrin-mediated signaling essentially
contributes to cell survival after radiation-induced genotoxic
injury. Oncogene. 25:1378–1390. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park CC, Zhang HJ, Yao ES, Park CJ and
Bissell MJ: Beta1 integrin inhibition dramatically enhances
radiotherapy efficacy in human breast cancer xenografts. Cancer
Res. 68:4398–4405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang C, Park CC, Hilsenbeck SG, Ward R,
Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK and Schiff R: β1
integrin mediates an alternative survival pathway in breast cancer
cells resistant to lapatinib. Breast Cancer Res. 13:R842011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kanda R, Kawahara A, Watari K, Murakami Y,
Sonoda K, Maeda M, Fujita H, Kage M, Uramoto H, Costa C, et al:
Erlotinib resistance in lung cancer cells mediated by integrin
β1/Src/Akt-driven bypass signaling. Cancer Res. 73:6243–6253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jahangiri A, Aghi MK and Carbonell WS: β1
integrin: Critical path to antiangiogenic therapy resistance and
beyond. Cancer Res. 74:3–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park CC, Zhang H, Pallavicini M, Gray JW,
Baehner F, Park CJ and Bissell MJ: Beta1 integrin inhibitory
antibody induces apoptosis of breast cancer cells, inhibits growth,
and distinguishes malignant from normal phenotype in three
dimensional cultures and in vivo. Cancer Res. 66:1526–1535. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhaskar V, Zhang D, Fox M, Seto P, Wong
MH, Wales PE, Powers D, Chao DT, Dubridge RB and Ramakrishnan V: A
function blocking anti-mouse integrin alpha5beta1 antibody inhibits
angiogenesis and impedes tumor growth in vivo. J Transl Med.
5:612007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eke I, Zscheppang K, Dickreuter E,
Hickmann L, Mazzeo E, Unger K, Krause M and Cordes N: Simultaneous
β1 integrin-EGFR targeting and radiosensitization of human head and
neck cancer. J Natl Cancer Inst. 107:dju4192015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ricart AD, Tolcher AW, Liu G, Holen K,
Schwartz G, Albertini M, Weiss G, Yazji S, Ng C and Wilding G:
Volociximab, a chimeric monoclonal antibody that specifically binds
α5β1 integrin: Α phase I, pharmacokinetic,
and biological correlative study. Clin Cancer Res. 14:7924–7929.
2008. View Article : Google Scholar : PubMed/NCBI
|