Introduction
HCC is the fifth leading cause of cancer-related
deaths in the world (1,2). The high recurrence rate and poor
prognosis of HCC are responsible for the high mortality resulted
from this cancer. Surgery, loco-regional therapy, transcatheter
arterial chemoembolization (3), and
chemotherapy are available for HCC treatment, however, they provide
limited success in reducing cancer-related mortality. Targeted
therapy with multiple tyrosine kinase inhibitor sorafenib has
improved HCC treatment (4), but it
is of major concern to identify critical targets and underlying
mechanisms involved in HCC development to further optimize
therapeutic efficiency.
Among hallmarks in cancer formation, dependence on
glycolysis is one of the important features in various kinds of
cancer including HCC (5–9). In addition to glucose, cancer cells
remodel the metabolism of other macromolecules, including amino
acids (5,10) and fatty acids (11,12),
to support neoplasia growth. Our laboratory has demonstrated that
dysregulated lipid metabolism by long-chain acyl-CoA synthetase
(ACSL) expression (13) and
dysregulated amino acid metabolism by argininosuccinate lyase (ASL)
expression (14) play important
roles in cancer formation.
Among dysregulated amino acid metabolism, glutamine,
serine, and glycine are reported to regulate cancer formation and
are potential therapeutic targets (10,15).
In addition, decreased arginine production is frequently observed
in HCC and melanoma. Therefore, these tumors are susceptible to
arginine deprivation therapy (15,16).
Arginase or arginine deiminase has been reported to display
effective anticancer potential in HCC and melanoma in vitro
(17–24), in vivo (17–19,21,24),
and in clinical trials (25–28).
However, arginine deprivation therapy may also, like
other cancer chemotherapies and targeted therapies (29,30),
confront the problem of drug resistance (15,16).
To this end, molecular mechanisms in arginine metabolic
enzyme-mediated cancer formation and arginine deprivation therapy
deserve further elucidation.
Our laboratory previously identified that knockdown
of the arginine metabolic enzyme ASL inhibits HCC formation in part
through reduction of cyclin A2 (14). In this study, we further studied the
mechanism by which ASL regulates cyclin A2 protein level. We found
that ASL directly interacted with cyclin A2 in the cytoplasm of HCC
cells. Mutant ASL which is devoid of arginine metabolic activity
retained the ability to interact with cyclin A2 and promoted
anchorage-independent growth, suggesting ASL/cyclin A2 interaction
may be important for tumor growth. Furthermore, ASL overexpression
modulated liver cancer progression regarding drug resistance
especially to arginine deprivation therapy, with potential
therapeutics counteracting above phenomenon being identified with
bioinformatics analysis, which may provide an opportunity for
improvement of treatment efficiency.
Materials and methods
Cell lines
Human HCC cell line Huh7 was kindly provided by I.J.
Su at National Health Research Institute. Huh7 and another human
HCC cell line HepG2 were cultured in DMEM media containing 10% FBS
(Biological Industries, Beit Haemek, Israel) and 1%
penicillin-streptomycin. Cells were kept in incubator at 37°C and
5% CO2. shASL-Huh7 and shASL-HepG2 were established as
previously described (14).
Chemicals, reagents, plasmids, and
antibodies
Gelatin, bovine serum albumin (BSA), 5-FU,
cisplatin, and sorafenib were purchased from Sigma (St. Louis, MO,
USA). Micro- BCA™ protein assay reagent kit was purchased from
Pierce (Woburn, MA, USA). DMEM and antibiotic mixture were
purchased from Invitrogen (Carlsbad, CA, USA). Turbofect
transfection reagent was purchased from Fermentas (Glen Burnie, MD,
USA). Plasmids of ASL-Myc and cyclin A2-HA were as described
previously (14). Antibodies
against ASL (GTX113629; polyclonal; rabbit anti-human; GeneTex,
Hsinchu, Taiwan), Myc (51-1485GR; monoclonal; mouse anti-Myc; BD
Biosciences, Franklin Lakes, NJ, USA), Cyclin A2 (sc-596;
polyclonal; rabbit anti-human; Santa Cruz Biotechnology, Dallas,
TX, USA), HA (11867431001; monoclonal; rat anti-HA; Roche, Basel,
Switzerland), HA (MMS-101P; monoclonal; mouse anti-HA; Covance,
Princeton, NJ, USA), GAPDH (GTX100118; polyclonal; rabbit
anti-human; GeneTex), Lamin A/C (ab108595; monoclonal; rabbit
anti-human; Abcam, Cambridge, UK), GFP (GTX628528; monoclonal;
mouse anti-GFP; GeneTex), and HALO (G9211; monoclonal; mouse
anti-HALO; Promega, Sunnyvale, CA, USA) were used in western
blotting (all 1:2000 except 1:1000 for ASL and 1:5000 for GAPDH),
immunofluorescence (all 1:100), or co-immunoprecipitation. ADI-PEG
was kindly provided by Polaris Pharmaceuticals (San Diego, CA,
USA).
Immunofluorescence and confocal
microscopy
Cancer cells (5×104) were seeded onto
6-well plates containing cover glasses pre-coated with 0.1% gelatin
and cultured overnight. Cells were then washed with PBS and fixed
with 3.7% paraformaldehyde. Following wash with 0.1 M glycine,
cells were treated with permeabilization/blocking buffer (2% FBS,
0.4% Triton X-100 in PBS). After washing with wash/staining buffer
(0.2% BSA, 0.2% Triton X-100 in PBS), cells were probed with
primary antibodies overnight at 4°C. Following washing, cells were
probed with secondary antibodies at room temperature for 1 h in the
dark. Cells were then stained with DAPI after washing. Subcellular
localization and co-localization of ASL and/or cyclin A2 were then
analyzed by confocal microscope Olympus FV1000MPE (Olympus, Tokyo,
Japan) with 63X oil immersion objective. Fluorescent images were
taken in sequential scanning mode (instead of simultaneous one) to
avoid non-specific fluorescent signal during image acquisition. IgG
was used as a negative control.
Nuclear-cytosolic fractionation
Cells were subjected to nuclear-cytosolic
fractionation according to the manufacturer's instructions (Thermo
Fisher Scientific, Waltham, MA, USA).
Western blotting
Cells were lysed in modified RIPA buffer with
protease inhibitors and let stand on ice for 20 min. After
centrifugation at 15,000 × g at 4°C for 10 min, supernatant was
harvested and protein concentration was assayed by micro-BCA
protein assay reagent kit (Thermo Fisher Scientific). Samples with
same amount of protein and 4X sample buffer were mixed, heated at
95°C for 5 min, and subjected to electrophoresis. The protein was
then transferred onto PVDF membrane (Millipore, Bedford, MA, USA)
by Hoefer Semiphor Semi-Dry transfer unit (Amersham Pharmacia
Biotech, Inc., San Francisco, CA, USA), and blocked in 5% non-fat
milk at room temperature for 1 h. Membrane was then probed with
primary antibody overnight at 4°C. Following washing with 0.1%
TBS-T, membrane was probed with secondary antibody at room
temperature for 1 h. After washing, the quantity of targets were
identified by adding chemiluminescence reagent ECL (Millipore) onto
membrane and luminescent intensity was recorded by BioSpectrum AC
imaging system (UVP Inc., Upland, CA, USA).
Co-immunoprecipitation
Cells were lysed in immunoprecipitation buffer
containing NP-40 or Triton X-100 with protease inhibitors and
harvested as in western blotting or in nuclear-cytosolic
fractionation, and protein was quantified. Samples (5%) were loaded
in input lane. Remaining samples were added with 2 µg primary
antibody for immunoprecipitation at 4°C overnight with agitation.
30 µl protein G magnetic beads (Millipore) or 100 µl protein G
agarose beads (Millipore) were then washed, mixed gently with
samples, and incubated at 4°C for 2 h with agitation. Following
washing, samples were eluted from beads by 4X sample buffer with
heating at 95°C for 5 min, and subjected to electrophoresis.
Site-directed mutagenesis and
polymerase chain reaction (PCR)
Plasmids of ASL-Myc and cyclin A2-HA were subjected
to site-directed mutagenesis using QuikChange Site-Directed
Mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA, USA),
HiFi HotStart PCR kit (Kapa Biosystems, Wilmington, MA, USA), and
Applied Biosystems 2720 Thermal Cycler (Applied Biosystems, Foster
City, CA, USA) according to the manufacturer's instructions to
establish loss-of-enzymatic activity mutant ASL-Myc and cyclin
A2-HA. Primers for site-directed mutagenesis were purchase from
MDBio, Inc. (Taipei, Taiwan) and listed below: ASL G532A: (forward)
5′-ATTCTGAGCCACGCCATGGCACTGACCCGAG-3′, (reverse)
5′-CTCGGGTCAGTGCCATGGCGTGGCTCAGAAT-3′; ASL A857G: (forward)
5′-GCAGCCTGATGCCCCGGAAGAAAAACCCCGA-3′, (reverse)
5′-TCGGGGTTTTTCTTCCGGGGCATCAGGCTGC-3′; CCNA2 M210A: (forward)
5′-AGCCAGACATCACTAACAGTGCGAGAGCTATCCTCGTGGACT-3′, (reverse)
5′-AGTCCACGAGGATAGCTCTCGCACTGTTAGTGATGTCTGGCT-3′; CCNA2 L214A:
(forward) 5′-ACTAACAGTGCGAGAGCTATCGCCGTGGACTGGTTAGTTGAAGTA-3′,
(reverse) 5′-TACTTCAACTAACCAGTCCACGGCGATAGCTCTCGCACTGTAGCTCTCAT-3′.
Final concentration for reagents for site-directed mutagenesis were
described as below: 1X KAPA HiFi buffer (GC), 0.3 mM KAPA dNTP Mix,
0.3 µM forward/reverse primer, 50 ng cDNA, 1 U/µl KAPA HiFi
HotStart DNA polymerase in 50 µl reaction. PCR for site-directed
mutagenesis is described below: at 95°C for 30 sec, 1 cycle; 95°C
for 30 sec, 55°C for 1 min, 68°C for 1 min 30 sec, 18 cycles.
RNA interference
The shASL plasmid-carrying bacterial clones and
corresponding pseudo lentivirus were obtained from RNAi core
facility (Academia Sinica, Taipei, Taiwan) and the shRNA targets 3′
UTR sequences: 5′-AGGAGGCTGCTGTGTGTTT-3′. Cells were infected with
pseudo lentivirus against ASL or luciferase vector control,
selected by 2 µg/ml puromycin for 2 days, and subjected to further
experiments.
Anchorage-dependent growth by colony
formation assay
Cancer cells (1×103) were seeded onto
6-well plates and cultured for 9 days to assay the
anchorage-dependent growth ability of cancer cells. The number of
colonies was identified by 2% methyl blue staining, counted and
analyzed.
Anchorage-independent growth by soft
agar growth assay
Cells (5×103) were seeded into 1 ml 0.3%
agar-containing medium and then onto the 0.6% agar-covered 6-well
plates and cultured for 14 days to assay the anchorage-independent
growth ability of cancer cells. The number of colonies was
identified by 0.05% crystal violet staining, counted and
analyzed.
Microarray analysis
Total RNA of Huh7 and shASL-Huh7 stable
transfectants were extracted using TRIzol reagent (MDBio, Inc.,
Taiwan). The microarray analysis was performed using Whole Human
Genome Oligo Microarray kit (4×44 K) (Agilent Technologies, Inc.)
by Welgene Biotech, Co., Ltd. (Taipei, Taiwan).
Metacore bioinformatics analysis
The pathway analysis database Metacore (https://portal.genego.com/cgi/data_manager.cgi#)
(31) was applied with default
setting. The analyses with false discovery rate (FDR) <0.05 were
displayed as pathway maps (pathways from literature consensus).
Connectivity Map bioinformatics
analysis
The gene-drug interaction database Connectivity Map
(https://www.broad institute.org/cmap/) (32,33)
was applied with default setting. The differentially expressed
genes in shASL-Huh7 were divided into top 500 upregulated and
downregulated groups and uploaded to Connectivity Map database for
analysis of potential therapeutics with gene expression signature
mimicking ASL knockdown in Huh7.
Statistical analysis
All statistical analyses were performed with the
GraphPad Prism version 5 (GraphPad Software, San Diego, CA, USA).
All error bars of the figures represent SEM. Student's t-test and
two-way ANOVA followed by Bonferroni post-test were used for
analysis of difference between each experimental group. P-value of
<0.05 was considered to be significant.
Results
ASL colocalizes and interacts with
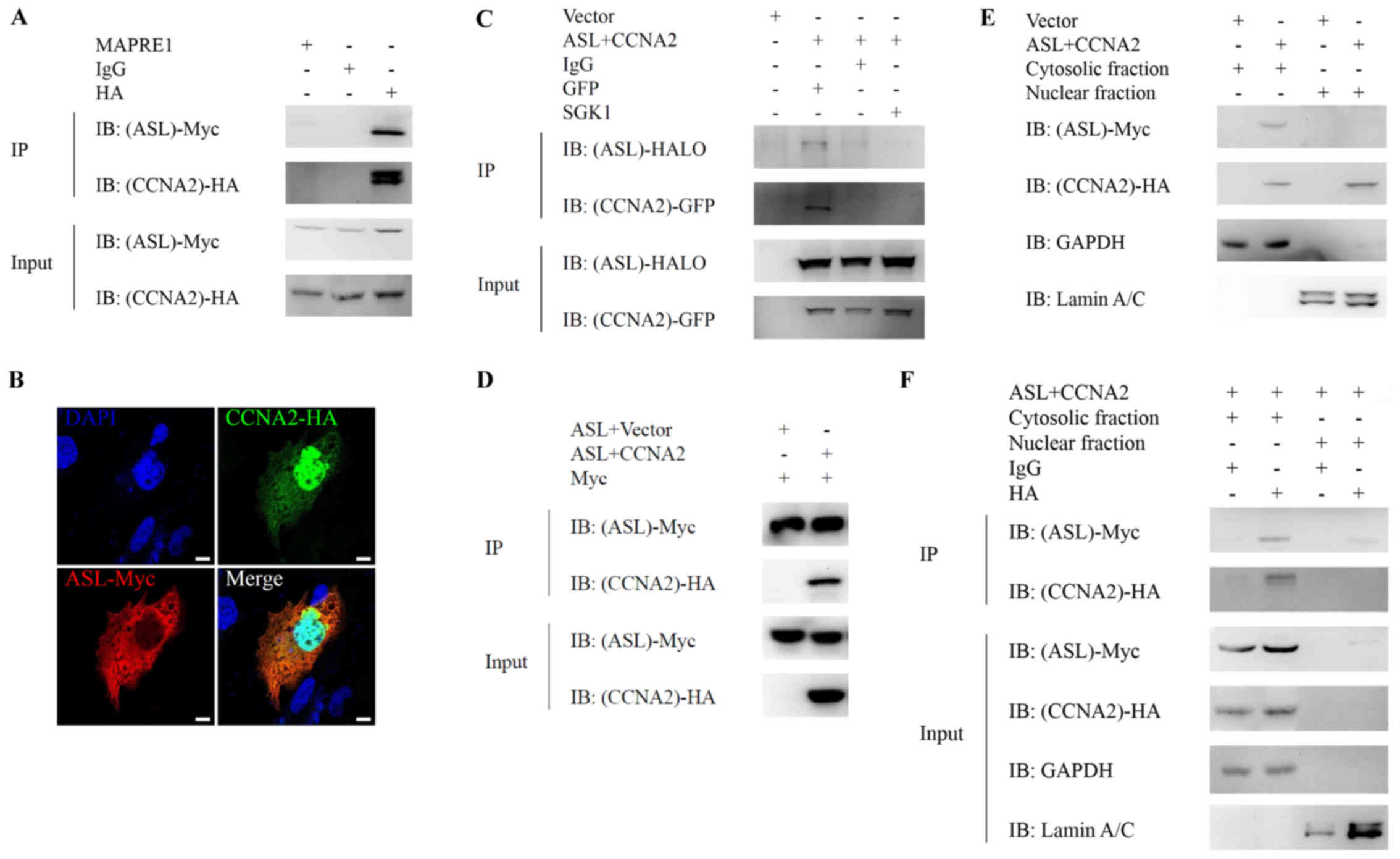
cyclin A2 in the cytoplasm of HCC cells
We have demonstrated that downregulation of ASL by
shRNA inhibits tumor growth which is in part mediated through
downregulating cyclin A2 expression. The reduction of cyclin A2 is
probably regulated at the protein level because cyclin A2 mRNA was
not altered (14). Based on this
information, we hypothesized that ASL might directly interact with
cyclin A2 and regulate its protein expression. We cotransfected
ASL-Myc and cyclin A2-HA into Huh7 and investigated their
colocalization and interaction by immunofluorescence and
immunoprecipitation, respectively. ASL interacted (Fig. 1A) and colocalized (Fig. 1B) with cyclin A2 in Huh7 liver
cancer cells. Double-band was observed in immunoprecipitation of
HA-cyclin A2, but not in the input of HA-cyclin A2. As cyclin A2 is
easily degraded, we speculated that the double bands may be
resulted from the degradation of cyclin A2 during
immunoprecipitation. To avoid IgG heavy chain interruption during
co-immunoprecipitation, we applied EasyBlot IgG HRP secondary
antibody and observed no obvious signal resulting from IgG heavy
chain. This interaction is also observed by coexpression of
exogenous ASL-HALO and cyclin A2-GFP in 293 cells (Fig. 1C). In addition, we have performed
the experiments with ASL-Myc and cyclin A2-HA or HA-vector control
and we observed no interaction between ASL-Myc and vector control,
supporting that the specific interaction between ASL and cyclin A2
indeed occurred under co-overexpression (Fig. 1D). Altogether, the arginine
metabolic enzyme ASL co-localized and interacted with cell cycle
regulator cyclin A2.
To further identify the subcellular localization of
ASL/cyclin A2 interaction, ASL-Myc and cyclin A2-HA were
cotransfected into Huh7 and nuclear-cytosolic fractionation without
or with subsequent immunoprecipitation. ASL is mainly localized in
the cytosol of Huh7, and cyclin A2 is located in both cytosol and
the nucleus (Fig. 1E), suggesting
the subcellular localization for ASL/cyclin A2 interaction was in
the cytosol. Furthermore, the interaction between ASL and cyclin A2
was detected in the cytosolic fraction of Huh7 with
immunoprecipitation (Fig. 1F). This
result indicated that the interaction between ASL and cyclin A2
mainly occurs in the cytosol of liver cancer cells.
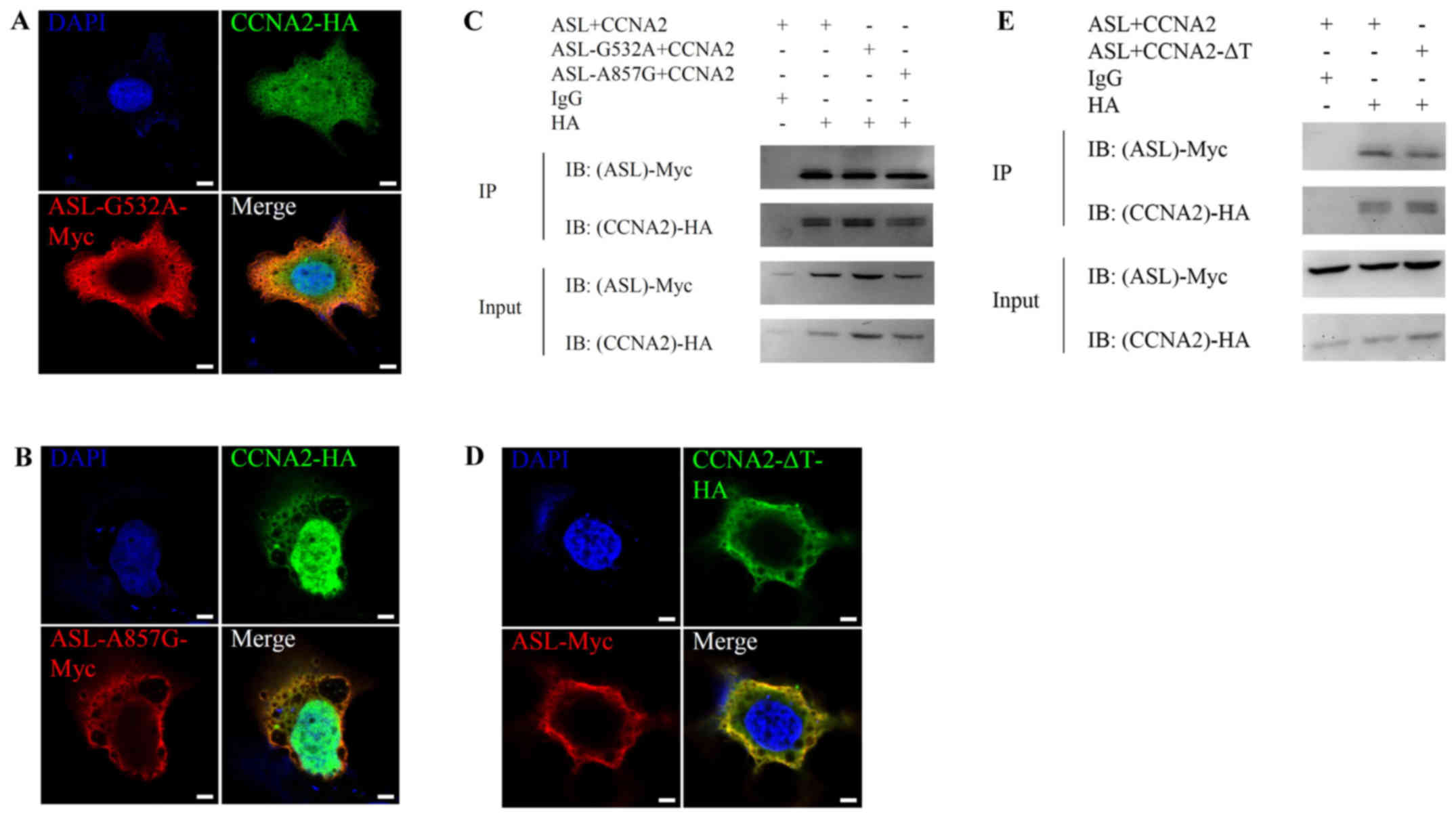
Loss-of-enzymatic activity mutant ASL retains the
ability to interact with cyclin A2 and promotes cell growth. In
order to determine whether the metabolic enzymatic activity of ASL
is required for the interaction, we constructed mutant ASL-Myc
without enzymatic activity. Previous results indicated that genomic
mutations in ASL leading to G532A or A857G amino acid substitution
were observed in patients with ASL function deficiency (34). On the other hand, cyclin A2
containing M210A, L214A, W217A triple mutation (∆Triple) is unable
to influence cyclin-dependent kinase activity in human (35,36).
We then investigated the interaction between wild-type ASL and
mutant cyclin A2 or mutant ASL and wild-type cyclin A2.
Immunofluorescence and immunoprecipitation analysis revealed that
mutant ASL or cyclin A2 still retained the ability to colocalize
(Fig. 2A, B and D) and interact
(Fig. 2C and E) with wild-type
interaction partner. This result revealed that ASL enzymatic
activity is not essential for the interaction with cyclin A2.
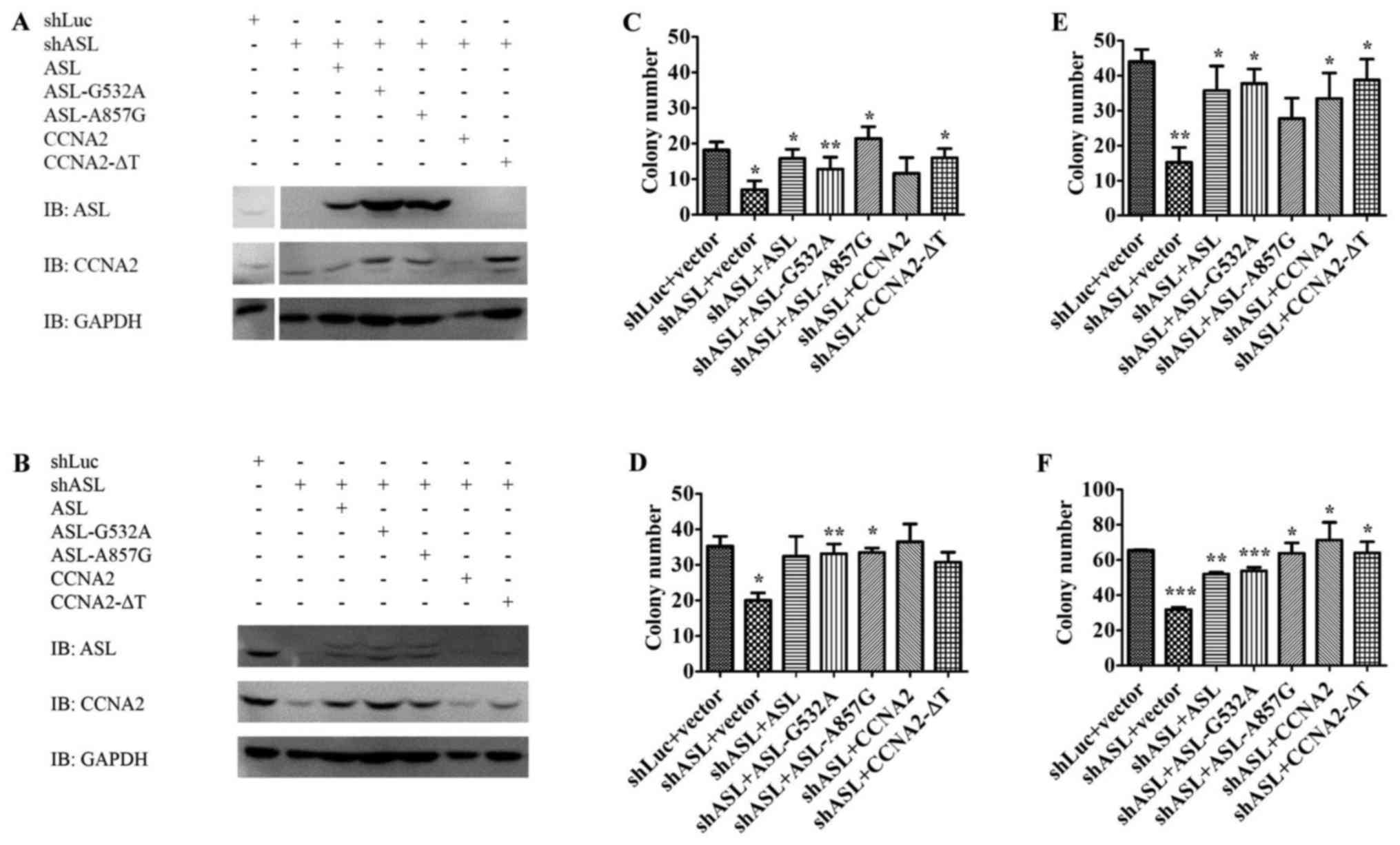
To elucidate the importance of the interaction
between ASL and cyclin A2 in liver cancer promotion, we delivered
mutant ASL or mutant cyclin A2 into Huh7 ASL shRNA stable
transfectants, and observed whether they could restore the growth
inhibition by ASL shRNA. Both wild-type and loss-of-enzymatic
activity ASL or cyclin A2 were able to restore the growth of
shASL-Huh7 stable transfectants in colony formation assay and soft
agar growth assay in vitro (Fig.
3). Therefore, loss-of-enzymatic activity ASL or cyclin A2
still maintained the ability to promote anchorage-dependent growth
and anchorage-independent growth in liver cancer cells. Since these
mutants retained the ability to form the ASL/cyclin A2 complex,
these results imply that the novel ASL/cyclin A2 interaction may
play a role in mediating cell growth (Figs. 2 and 3).
ASL overexpression promotes drug
resistance to arginine deprivation therapy
To identify the impact of ASL on tumor progression
in terms of drug resistance, we transfected ASL-Myc or vector
control into Huh7 and HepG2 and investigated their resistance
toward chemotherapies with 5-FU, cisplatin, multiple tyrosine
kinase sorafenib (4), and arginine
deprivation therapy ADI-PEG (Fig. 4A
and B). Overall, ectopic expression of ASL resulted in an
increased tendency of resistance toward these anticancer drugs in
colony formation assay although the difference did not reach
statistical significance (Fig. 4C, D,
E, G, H and I). Importantly, ASL overexpression conferred drug
resistance to arginine deprivation therapy ADI-PEG with
anchorage-dependent growth (Fig. 4F and
J) and anchorage-independent growth (Fig. 4K and L).
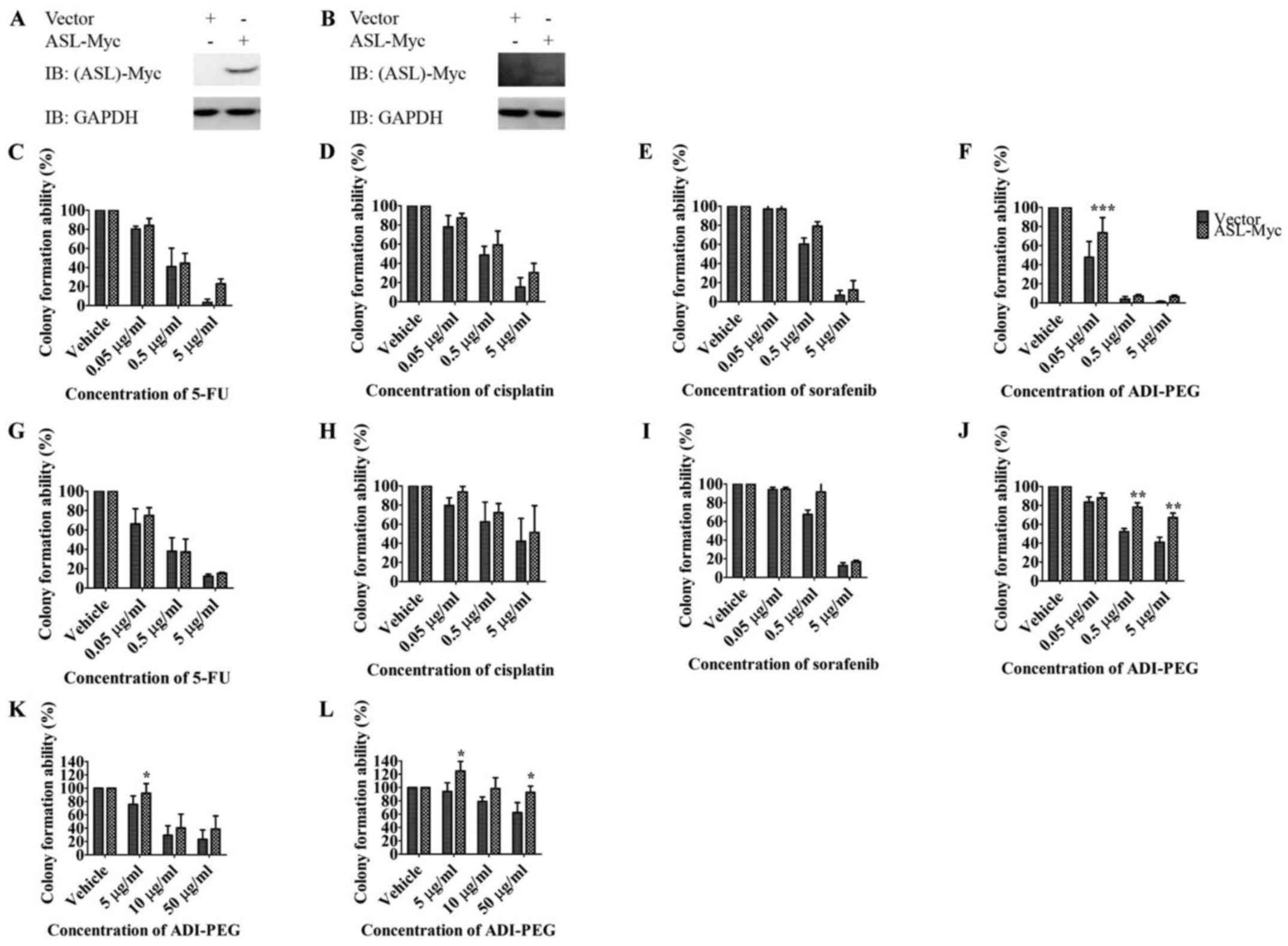 | Figure 4.ASL overexpression promotes drug
resistance against arginine deprivation therapy. Huh7 (A, C, D, E,
F and K) or HepG2 (B, G, H, I, J and L) was transfected with
ASL-Myc or vector control, and the expression was determined by
western blotting (A and B). The growth of cells in the presence of
anticancer drugs including the chemotherapies with 5-FU (C and G)
and cisplatin (D and H), targeted therapy sorafenib (E and I), and
arginine deprivation therapy ADI-PEG (F, J, K and L) was determined
by colony formation assay (C-J) and soft agar growth assay (K and
L). Results are from three independent experiments, and error bars
represent SEM, the statistical difference between the
anchorage-dependent and -independent growth ability of above
mentioned overexpression clones and control clone was examined with
two-way ANOVA followed by Bonferroni post-test (*P<0.05;
**P<0.01, ****P<0.001). |
Bioinformatics analyses reveal
potential therapeutics in ASL-regulated HCC formation
Since ASL played an important role in liver tumor
formation, we performed microarray gene expression analysis on the
comparison of Huh7 cells and ASL shRNA stable transfectants. The
downregulated genes by ASL shRNA in Huh7 cells were selected, and
uploaded to Metacore bioinformatics database for identification of
important pathways with its default setting. The analyses were
displayed as pathway maps. The gene expression signature resulted
from ASL knockdown in Huh7 was associated with cell cycle
progression, cytoskeleton remodeling, apoptosis, and immune
responses (Table I).
 | Table I.Bioinformatics analysis with Metacore
database revealed important pathways in ASL-regulated HCC
formation. |
Table I.
Bioinformatics analysis with Metacore
database revealed important pathways in ASL-regulated HCC
formation.
| Rank | Maps | FDR |
|---|
| 1 |
Transport_Clathrin-coated vesicle
cycle | 4.287E-15 |
| 2 | Cytoskeleton
remodeling_TGF, WNT and cytoskeletal remodeling | 4.287E-15 |
| 3 | Cell cycle_Start of
DNA replication in early S phase | 3.031E-11 |
| 4 | DNA damage_ATM/ATR
regulation of G1/S checkpoint | 5.234E-10 |
| 5 | Cell
cycle_Influence of Ras and Rho proteins on G1/S Transition | 6.190E-10 |
| 6 | Cytoskeleton
remodeling_Cytoskeleton remodeling | 8.999E-10 |
| 7 | DNA damage_Role of
Brca1 and Brca2 in DNA repair | 2.617E-09 |
| 8 | Apoptosis and
survival_Endoplasmic reticulum stress response pathway | 3.117E-09 |
| 9 |
Development_Differentiation of white
adipocytes | 1.489E-08 |
| 10 | Immune
response_Role of PKR in stress-induced antiviral cell response | 1.489E-08 |
| 11 |
Transcription_Epigenetic regulation of
gene expression | 1.489E-08 |
| 12 | Immune
response_IL-1 signaling pathway | 1.843E-08 |
| 13 |
Development_Regulation of telomere length
and cellular immortalization | 1.843E-08 |
| 14 |
Transcription_Sirtuin6 regulation and
functions | 2.722E-08 |
| 15 | Apoptosis and
survival_TNFR1 signaling pathway | 4.026E-08 |
| 16 | G-protein
signaling_RhoA regulation pathway | 4.062E-08 |
| 17 | Apoptosis and
survival_Role of PKR in stress-induced apoptosis | 4.952E-08 |
| 18 |
Translation_Regulation of EIF4F
activity | 4.952E-08 |
| 19 | Apoptosis and
survival_Role of IAP-proteins in apoptosis | 6.565E-08 |
| 20 | Oxidative
phosphorylation | 7.756E-08 |
| 21 | Apoptosis and
survival_FAS signaling cascades | 8.099E-08 |
| 22 | IGF family
signaling in colorectal cancer | 9.132E-08 |
| 23 | Cell
cycle_Transition and termination of DNA replication | 9.906E-08 |
| 24 | Regulation of
degradation of deltaF508-CFTR in CF | 1.317E-07 |
| 25 | DNA damage_Brca1 as
a transcription regulator | 1.317E-07 |
| 26 | Signal
transduction_Additional pathways of NF-κB activation (in the
nucleus) | 1.317E-07 |
| 27 |
Transcription_Transcription regulation of
aminoacid metabolism | 1.433E-07 |
| 28 | Signal
transduction_AKT signaling | 1.495E-07 |
| 29 | Development_WNT
signaling pathway. Part 2 | 1.687E-07 |
| 30 |
Development_NOTCH1-mediated pathway for
NF-κB activity modulation | 2.002E-07 |
| 31 | Cell cycle_The
metaphase checkpoint | 2.346E-07 |
| 32 | Role of Tissue
factor-induced Thrombin signaling in cancerogenesis | 2.416E-07 |
| 33 | Cell cycle_Role of
SCF complex in cell cycle regulation | 2.746E-07 |
| 34 |
Proteolysis_Putative SUMO-1 pathway | 2.746E-07 |
| 35 | Apoptosis and
survival_Apoptotic TNF-family pathways | 2.920E-07 |
| 36 | Development_IGF-1
receptor signaling | 3.075E-07 |
| 37 | Development_TGF-β
receptor signaling | 3.075E-07 |
| 38 | Cell cycle_ESR1
regulation of G1/S transition | 4.112E-07 |
| 39 | Cell cycle_Cell
cycle (generic schema) | 4.236E-07 |
| 40 | Cell
cycle_Chromosome condensation in prometaphase | 4.236E-07 |
| 41 | DNA damage_ATM/ATR
regulation of G2/M checkpoint | 4.236E-07 |
| 42 | Cell
cycle_Regulation of G1/S transition (part 2) | 4.236E-07 |
| 43 | Apoptosis and
survival_DNA-damage-induced apoptosis | 4.314E-07 |
| 44 | Transport_RAN
regulation pathway | 5.083E-07 |
| 45 | Signal
transduction_Additional pathways of NF-κB activation (in the
cytoplasm) | 5.433E-07 |
| 46 | IL-6 signaling in
multiple myeloma | 5.433E-07 |
| 47 | Signal
transduction_NF-κB activation pathways | 5.433E-07 |
| 48 | Oxidative
stress_Role of Sirtuin1 and PGC1-α in activation of antioxidant
defense system | 8.745E-07 |
| 49 | Apoptosis and
survival_Caspase cascade | 9.568E-07 |
| 50 | Cell
cycle_Initiation of mitosis | 9.751E-07 |
To further identify therapeutic options for
ASL-regulated HCC formation, we applied the gene expression
signature of ASL-knockdown in Huh7 cells in Connectivity Map
bioinformatics database (32,33)
with default setting. The gene expression signature was associated
with the treatment on cancer cells of 755 drugs in Connectivity Map
analysis, in which 60 drugs overlapped with the FDA drug library
available for testing the drug repurposing. Since drugs used in
neural and cardiovascular diseases may confer stronger side effects
to patients, we further excluded drugs with indications in these
two fields. The potential novel therapeutics were narrowed down to
i) bisoprolol, a β1-adrenergic receptor blocker, ii) celecoxib, a
COX-2 selective non-steroidal anti-inflammatory drug (NSAID), and
iii) ipratropium bromide, an anticholinergic function regulator
(Fig. 5). This result pointed out
the potential therapeutics for ASL-regulated HCC formation. These
FDA-approved drugs deserve further elucidation on the combination
with current cancer therapy for HCC.
Discussion
In the present study, we demonstrated that ASL
interacted with cyclin A2 in the cytosol of HCC cells and the
interaction might be important for tumor growth. We further found
that ASL overexpression conferred drug resistance especially
against arginine deprivation therapy. Bioinformatics analysis
revealed that several drugs might be used to target
ASL-overexpressing liver tumors.
The interaction was detected by the exogenous
expression of ASL and cyclin A2. We have tried to determine the
interaction between endogenous proteins, but was unable to detect
the interaction between endogenous ASL and cyclin A2. This could be
due to limited expression of ASL and cyclin A2 or the antibody
binding site may interfere with the binding between ASL and cyclin
A2. Therefore, we have used two different types of tagged exogenous
ASL and cyclin A2 for IP in two different cell lines to sustain our
conclusion (Fig. 1). It is
interesting to note that the interaction between ASL and cyclin A2
is mainly localized in the cytosolic fraction. We further examined
whether cytosolic cyclin A2 is attenuated by ASL shRNA.
Nuclear/cytosolic fraction clearly demonstrated that cytosolic
cyclin A2 is downregulated specifically. In contrast, nuclear
cyclin A2 remained constant (data not shown). This result supports
the notion that cytosolic ASL/cyclin A2 interaction influences its
protein level. It should be noted that cyclin A2 may shuttle
between nucleus and cytosol (37).
The distribution of cyclin A2 is dependent on the cell cycle, and
may be variable between different experiments. Jackman and
coworkers (37) applied nuclear
export assay to specifically address whether cyclin A could export
from nucleus to cytosol spontaneously since it does not have
nuclear localization signal (NLS), and found out that cyclin A
indeed shuttled between nucleus and cytosol. This suggests that it
is the interaction with other proteins, such as other cell cycle
regulators containing NLS, that determines the subcellular
localization of cyclin A. This phenomenon may also occur in the
present study since upregulated ASL expression during HCC formation
may interact, protect, and retain cyclin A2 in cytosol to execute
biological functions in addition to the cell cycle regulator.
The interaction between wild-type and mutant forms
of ASL and cyclin A2 is similar in co-immunoprecipitation (Fig. 2). The results indicate that the
functional enzymatic mutation does not influence the interaction
between ASL and cyclin A2. This observation supports our hypothesis
that non-enzymatic function of ASL may influence cell cycle
progression and tumor formation. Non-enzymatic functions of
metabolic gene products have been reported to participate in cancer
promotion and progression. For example, non-enzymatic function of
heparanase promotes glioblastoma growth in vivo via
activation of Akt signaling (38,39).
Non-enzymatic function of metabolic enzyme
methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2)
promotes colon cancer growth in vitro via association with
DNA synthesis (40); non-enzymatic
function of ATP-citrate lyase also enhances colon cancer growth
in vitro by inhibiting AMPK signaling (41); non-enzymatic function of pyruvate
kinase M2 (PKM2) promotes glioblastoma growth in vivo via
activation of β-catenin signaling (42); non-enzymatic function of
fructose-1,6-bisphosphatase 1 (FBP1) inhibits clear cell renal cell
carcinoma progression in patients by reducing HIF function
(43). The present study on ASL
adds the importance of non-enzymatic functions of enzymes in
regulating cancer formation (40,44).
Based on the critical roles of ASL in drug
resistance and cancer progression, the therapeutic potential
targeting ASL overexpression deserves further attention. From the
bioinformatics analysis with Connectivity Map database in the
present study, bisoprolol, celecoxib, and ipratropium bromide were
identified to be capable of counteracting ASL-regulated HCC
formation. Our bioinformatics predictions are in accordance with a
previous study on the targets of these drugs. These targets,
including β1-adrenergic receptor, COX-2, and cholinergic receptor,
are involved in various kinds of cancer (45–48).
In summary, we identified the cytosolic interaction
between ASL and cyclin A2. This interaction may be important for
liver cancer growth. Overexpression of ASL may be related with drug
resistance especially for arginine deprivation therapy.
Acknowledgements
Computational analyses were supported by the system
provided by the Bioinformatics Core at National Cheng Kung
University, Tainan, Taiwan. This study was supported by the grant
to M.-D.L. 101-2320-B-006-028-MY3 from Ministry of Science and
Technology, Taiwan and NHRI-EX100-9927B1 from National Health
Research Institute, Taiwan.
Glossary
Abbreviations
Abbreviations:
|
ASL
|
argininosuccinate lyase
|
|
CCNA2
|
cyclin A2
|
|
5-FU
|
5-fluorouracil
|
|
ADI-PEG
|
arginine deiminase formulated with
polyethylene glycol
|
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruix J, Sala M and Llovet JM:
Chemoembolization for hepatocellular carcinoma. Gastroenterology.
127:(Suppl 1). S179–S188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu AX: Development of sorafenib and other
molecularly targeted agents in hepatocellular carcinoma. Cancer.
112:250–259. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pelicano H, Martin DS, Xu RH and Huang P:
Glycolysis inhibition for anticancer treatment. Oncogene.
25:4633–4646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heiden MG Vander: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Locasale JW: Serine, glycine and
one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer.
13:572–583. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tirado-Vélez JM, Joumady I, Sáez-Benito A,
Cózar-Castellano I and Perdomo G: Inhibition of fatty acid
metabolism reduces human myeloma cells proliferation. PLoS One.
7:e464842012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Currie E, Schulze A, Zechner R, Walther TC
and Farese RV Jr: Cellular fatty acid metabolism and cancer. Cell
Metab. 18:153–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang YS, Tsai CT, Huangfu CA, Huang WY,
Lei HY, Lin CF, Su IJ, Chang WT, Wu PH, Chen YT, et al: ACSL3 and
GSK-3β are essential for lipid upregulation induced by endoplasmic
reticulum stress in liver cells. J Cell Biochem. 112:881–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang HL, Hsu HP, Shieh SC, Chang YS, Chen
WC, Cho CY, Teng CF, Su IJ, Hung WC and Lai MD: Attenuation of
argininosuccinate lyase inhibits cancer growth via cyclin A2 and
nitric oxide. Mol Cancer Ther. 12:2505–2516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Delage B, Fennell DA, Nicholson L, McNeish
I, Lemoine NR, Crook T and Szlosarek PW: Arginine deprivation and
argininosuccinate synthetase expression in the treatment of cancer.
Int J Cancer. 126:2762–2772. 2010.PubMed/NCBI
|
|
16
|
Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr
M, Spector S and Savaraj N: Arginine deprivation as a targeted
therapy for cancer. Curr Pharm Des. 14:1049–1057. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ensor CM, Holtsberg FW, Bomalaski JS and
Clark MA: Pegylated arginine deiminase (ADI-SS PEG20,000 mw)
inhibits human melanomas and hepatocellular carcinomas in vitro and
in vivo. Cancer Res. 62:5443–5450. 2002.PubMed/NCBI
|
|
18
|
Cheng PNM, Lam TL, Lam WM, Tsui SM, Cheng
AW, Lo WH and Leung YC: Pegylated recombinant human arginase
(rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation
of human hepatocellular carcinoma through arginine depletion.
Cancer Res. 67:309–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li YY, Wu C, Chen SM, Shah SS,
Wangpaichitr M, Feun LG, Kuo MT, Suarez M, Prince J and Savaraj N:
BRAF inhibitor resistance enhances vulnerability to arginine
deprivation in melanoma. Oncotarget. 7:17665–17680. 2016.PubMed/NCBI
|
|
20
|
Bobak Y, Kurlishchuk Y,
Vynnytska-Myronovska B, Grydzuk O, Shuvayeva G, Redowicz MJ,
Kunz-Schughart LA and Stasyk O: Arginine deprivation induces
endoplasmic reticulum stress in human solid cancer cells. Int J
Biochem Cell Biol. 70:29–38. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miraki-Moud F, Ghazaly E, Ariza-McNaughton
L, Hodby KA, Clear A, Anjos-Afonso F, Liapis K, Grantham M, Sohrabi
F, Cavenagh J, et al: Arginine deprivation using pegylated arginine
deiminase has activity against primary acute myeloid leukemia cells
in vivo. Blood. 125:4060–4068. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang H-L, Chen W-C, Hsu H-P, Cho CY, Hung
YH, Wang CY and Lai MD: Argininosuccinate lyase is a potential
therapeutic target in breast cancer. Oncol Rep. 34:3131–3139.
2015.PubMed/NCBI
|
|
23
|
Khoury O, Ghazale N, Stone E, El-Sibai M,
Frankel AE and Abi-Habib RJ: Human recombinant arginase I
(Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is
selectively cytotoxic to human glioblastoma cells. J Neurooncol.
122:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shan Y-S, Hsu H-P, Lai M-D, Yen MC, Chen
WC, Fang JH, Weng TY and Chen YL: Argininosuccinate synthetase 1
suppression and arginine restriction inhibit cell migration in
gastric cancer cell lines. Sci Rep. 5:97832015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ott PA, Carvajal RD, Pandit-Taskar N,
Jungbluth AA, Hoffman EW, Wu BW, Bomalaski JS, Venhaus R, Pan L,
Old LJ, et al: Phase I/II study of pegylated arginine deiminase
(ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs.
31:425–434. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang TS, Lu SN, Chao Y, Sheen IS, Lin CC,
Wang TE, Chen SC, Wang JH, Liao LY, Thomson JA, et al: A randomised
phase II study of pegylated arginine deiminase (ADI-PEG 20) in
Asian advanced hepatocellular carcinoma patients. Br J Cancer.
103:954–960. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glazer ES, Piccirillo M, Albino V, Di
Giacomo R, Palaia R, Mastro AA, Beneduce G, Castello G, De Rosa V,
Petrillo A, et al: Phase II study of pegylated arginine deiminase
for nonresectable and metastatic hepatocellular carcinoma. J Clin
Oncol. 28:2220–2226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tomlinson BK, Thomson JA, Bomalaski JS,
Diaz M, Akande T, Mahaffey N, Li T, Dutia MP, Kelly K, Gong IY, et
al: Phase I trial of arginine deprivation therapy with ADI-PEG 20
plus docetaxel in patients with advanced malignant solid tumors.
Clin Cancer Res. 21:2480–2486. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hegedüs C, Truta-Feles K, Antalffy G,
Várady G, Német K, Ozvegy-Laczka C, Kéri G, Orfi L, Szakács G,
Settleman J, et al: Interaction of the EGFR inhibitors gefitinib,
vandetanib, pelitinib and neratinib with the ABCG2 multidrug
transporter: Implications for the emergence and reversal of cancer
drug resistance. Biochem Pharmacol. 84:260–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ekins S, Bugrim A, Brovold L, Kirillov E,
Nikolsky Y, Rakhmatulin E, Sorokina S, Ryabov A, Serebryiskaya T,
Melnikov A, et al: Algorithms for network analysis in
systems-ADME/Tox using the MetaCore and MetaDrug platforms.
Xenobiotica. 36:877–901. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lamb J: The Connectivity Map: A new tool
for biomedical research. Nat Rev Cancer. 7:54–60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lamb J, Crawford ED, Peck D, Modell JW,
Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et
al: The Connectivity Map: Using gene-expression signatures to
connect small molecules, genes, and disease. Science.
313:1929–1935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Linnebank M, Tschiedel E, Häberle J,
Linnebank A, Willenbring H, Kleijer WJ and Koch HG:
Argininosuccinate lyase (ASL) deficiency: Mutation analysis in 27
patients and a completed structure of the human ASL gene. Hum
Genet. 111:350–359. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Arsic N, Bendris N, Peter M, Begon-Pescia
C, Rebouissou C, Gadéa G, Bouquier N, Bibeau F, Lemmers B and
Blanchard JM: A novel function for Cyclin A2: Control of cell
invasion via RhoA signaling. J Cell Biol. 196:147–162. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bendris N, Lemmers B, Blanchard J-M and
Arsic N: Cyclin A2 mutagenesis analysis: A new insight into CDK
activation and cellular localization requirements. PLoS One.
6:e228792011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jackman M, Kubota Y, den Elzen N, Hagting
A and Pines J: Cyclin A- and cyclin E-Cdk complexes shuttle between
the nucleus and the cytoplasm. Mol Biol Cell. 13:1030–1045. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fux L, Ilan N, Sanderson RD and Vlodavsky
I: Heparanase: Busy at the cell surface. Trends Biochem Sci.
34:511–519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ilan N, Elkin M and Vlodavsky I:
Regulation, function and clinical significance of heparanase in
cancer metastasis and angiogenesis. Int J Biochem Cell Biol.
38:2018–2039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sheppard N Gustafsson, Jarl L, Mahadessian
D, Strittmatter L, Schmidt A, Madhusudan N, Tegnér J, Lundberg EK,
Asplund A, Jain M, et al: The folate-coupled enzyme MTHFD2 is a
nuclear protein and promotes cell proliferation. Sci Rep.
5:150292015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee JH, Jang H, Lee SM, Lee JE, Choi J,
Kim TW, Cho EJ and Youn HD: ATP-citrate lyase regulates cellular
senescence via an AMPK- and p53-dependent pathway. FEBS J.
282:361–371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li B, Qiu B, Lee DS, Walton ZE, Ochocki
JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, et al:
Fructose-1,6-bisphosphatase opposes renal carcinoma progression.
Nature. 513:251–255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jeffery CJ: Why study moonlighting
proteins? Front Genet. 6:2112015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cole SW and Sood AK: Molecular pathways:
Beta-adrenergic signaling in cancer. Clin Cancer Res. 18:1201–1206.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Harris RE, Alshafie GA, Abou-Issa H and
Seibert K: Chemoprevention of breast cancer in rats by celecoxib, a
cyclooxygenase 2 inhibitor. Cancer Res. 60:2101–2103.
2000.PubMed/NCBI
|
|
47
|
Jendrossek V: Targeting apoptosis pathways
by Celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao C-M, Hayakawa Y, Kodama Y,
Muthupalani S, Westphalen CB, Andersen GT, Flatberg A, Johannessen
H, Friedman RA, Renz BW, et al: Denervation suppresses gastric
tumorigenesis. Sci Transl Med. 6:250ra115. 2014. View Article : Google Scholar
|