Introduction
Diffuse intrinsic pontine glioma (DIPG) is an
incurable brain tumor that mostly affects young children (1). Treatment with radiation therapy (RT)
is usually transiently effective, but median progression-free
survival is only 7 months, and median overall survival is 11 months
(2). Hundreds of clinical trials
studying systemically delivered cytotoxic and targeted chemotherapy
agents have been conducted without showing any clinical benefit,
potentially due to inadequate tumor penetration (3). Investigators are therefore studying
novel drug delivery methods for these patients (4,5), and
some of these have reached clinical trials (6,7). As
these studies advance, better targeted therapeutic options will be
needed, especially as part of combination treatments.
The mechanistic target of rapamycin (MTOR) is a
serine-threonine kinase that has crucial roles in many cellular
pathways dysregulated in cancer, including metabolism, growth,
survival, and response to stress. It functions as part of two
distinct signaling complexes, MTOR complex 1 (MTORC1), which also
contains the regulatory associated protein of MTORC1 (RPTOR), and
MTOR complex 2 (MTORC2), containing the RPTOR independent companion
of MTORC2 (RICTOR). MTORC2 activates the V-Akt murine thymoma viral
oncogene (AKT) through phosphorylation at S473, which mediates many
of the cellular effects of MTORC2 (8–10).
Preclinical and clinical data suggest a role for MTOR in
gliomagenesis (11). First
generation MTOR inhibitors (rapamycin analogues or rapalogs), such
as everolimus, inhibit only MTORC1, and their clinical use in
high-grade glioma has been disappointing (12). Contrastingly, Kahn et al
reported preclinical findings showing that the MTORC1/2 inhibitor
AZD2014 enhanced the efficacy of RT in adult glioblastoma stem
cells in vitro and in vivo (13), generating hope that combined MTOR
inhibition may hold more clinical promise. DIPG has significant
biological differences from adult glioblastoma, however (14,15),
and prior to undertaking our project, MTOR inhibition of any kind
in DIPG had not been addressed in the literature.
In this study, we examined the effects of MTOR
inhibition preclinically in DIPG. We hypothesized that the addition
of MTORC2 inhibition would increase the antitumor efficacy over the
targeting of MTORC1 alone. Our overall goal was to determine how
MTOR inhibition could contribute to future clinical treatment
approaches to this devastating tumor.
Materials and methods
Aim and design
The aim of this study was to determine the efficacy
of MTORC1/2 compared to MTORC1 inhibitors in DIPG. We set out to
determine the phenotypic and molecular basis for the difference in
efficacy noted between inhibitors. We then studied how MTORC1/2
inhibition could be used in combination with other existing
treatments. All assays took place using three patient-derived
short-term culture cell lines in cell culture models.
Gene expression in tumor bank
We performed gene expression profiling on
patient-derived DIPG (n=16) and normal pons (n=2) samples. All
tumor samples were collected from consented pediatric patients
undergoing tumor biopsy using an IRB-approved study protocol
(COMIRB 95–500). Samples were snap frozen and stored in liquid
nitrogen. RNA was extracted from each sample using an RNeasy or
DNA/RNA AllPrep kit (Qiagen, Valencia, CA, USA) according to the
manufacturer's instructions. Four hundred nanograms of RNA was
processed using the Ambion MessageAmp™ Premier RNA Amplification
kit (Applied Biosystems, Foster City, CA, USA), according to the
manufacturer's instructions. RNA quality was verified using the
Nano Assay Protocol for the 2100 Bioanalyzer (Agilent, Santa Clara,
CA, USA) at two time-points: i) after initial extraction of the RNA
from the tumor sample, and ii) after preparation of the RNA for
chip hybridization. The prepared RNA was hybridized to HG-U133 Plus
2 GeneChips (Affymetrix, Santa Clara, CA, USA), according to the
manufacturer's instructions to measure gene expression. Mean and
standard error of fold changes for each sample set were calculated
for each measure indicated. We also conducted hallmark gene set
enrichment analysis (GSEA) on the sample sets.
Gene expression from public data
We performed gene expression analysis using the R2
genomics analysis and visualization platform (http://r2.amc.nl) using the default settings. We
compared expression of MTOR, RPTOR, and RICTOR in a set of normal
brain samples (n=172, Berchtold set) versus DIPG samples (n=37,
Paugh set) using one way analysis of variance.
Tumor lines and culture
conditions
Three primary human pediatric (DIPG 4 and DIPG 6,
derived from previously irradiated DIPGs at autopsy, provided by Dr
Michelle Monje, Stanford University, and SF7761, derived from a
biopsy sample, provided by Dr Nalin Gupta, University of
California, San Francisco) were grown in neurosphere (suspension)
culture conditions in ultra-low attachment flasks (Corning,
Corning, NY, USA, DIPG 4 and DIPG 6) or in tissue culture-treated
flasks (SF7761; Falcon/Corning) (16). Characteristics of the cell lines are
listed in Table I (17). For certain experiments, DIPG 4 cells
were also grown adherently in tissue culture-treated plates. The
identity of all lines was validated by molecular profiling and
compared to known results prior to and during this project. The
cell lines were maintained in Neurobasal-A medium mixed 1:1 with
Dulbecco's modified Eagle's medium/F-12 supplemented 1:100 by
volume with HEPES [4-(2-hydroxyethyl)-1-piperazine ethanesulfonic
acid] 1 M, sodium pyruvate 100 mM, MEM non-essential amino acids 10
mM, GlutaMAX-I, and antibiotic-antimycotic (all Gibco/Life
Technologies, Waltham, MA, USA); B27-A supplement 50X (1:50;
Invitrogen), heparin (2 µg/ml; Stemcell Technologies, Inc.,
Vancouver, BC, Canada) and human EGF, FGFb, and platelet derived
growth factor-AB (all 20 ng/ml; Shenandoah Biotech, Warwick, PA,
USA). For SF7761 cells, N2 supplement was also added (1:100; Life
Technologies). For all experiments, except as noted, cells were
plated at a concentration of 200,000 cells/ml of media in ultra-low
attachment plates (Corning). Prior to all end-point measurements,
neurospheres were dispersed by trituration using a
micropipette.
 | Table I.Cell line characteristics. |
Table I.
Cell line characteristics.
| Cell line | H3 mutation | Other genetic
features | When collected |
|---|
| DIPG 4 | H3. 1K27M | TP53 WT,
ACVR1 G328V | Autopsy |
| DIPG 6 | H3. 3K27M | TP53
mutant | Autopsy |
| SF7761 | H3. 3K27M | Exogenous
hTERT | Biopsy |
Dose-response curves
Cell proliferation was determined by the MTS [3-(4,
5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]
assay using CellTiter 96 AQueous One Solution (Promega, Madison,
WI, USA). Cells were seeded at 5,000 cells per well into a 96-well
plate (Costar) and allowed to attach overnight (DIPG 4) or in a
96-well plate (Corning) in neurosphere culture (DIPG 6 and SF7761),
in a total volume of 100 µl of media. Twenty-four hours later, the
cells were treated with a range of doses of everolimus and AZD2014
(Selleck, Houston, TX, USA) in triplicate. At the end of the drug
treatment period (72 h for DIPG 4 and DIPG 6, 120 h for SF7761), 20
µl of MTS reagent was added to each well to make a final volume of
120 µl and allowed to develop. Absorbance values for plate wells
were acquired using a BioTek Synergy 2 plate reader at a wavelength
of 490 nm after 3 h of incubation, and background absorbance was
subtracted. IC50 values were determined experimentally
through Prism, and IC90 values were calculated using the
IC50 and hill slope value using the Graphpad EC Anything
online calculator.
Neurosphere dilution assay
DIPG 4 and SF7761 cells were plated in 96-well
format in suspension, with 10 cells per well and 20 wells per
condition. Conditions for each cell line included DMSO control, the
calculated IC75 for AZD2014, or the same concentration
of everolimus. The presence and size (area using two perpendicular
axes) of a neurosphere in each well was measured after three
weeks.
Apoptosis measurement
SF7761 cells in neurosphere culture were treated in
a 96-well plate format with the indicated range of AZD2014 and
everolimus concentrations compared to the control, in triplicate.
Apoptosis was measured according to the manufacturer's instructions
using the Caspase Glo 3/7 assay (Promega) at 48 h of treatment
after 2 h of incubation with the Caspase-Glo reagent at room
temperature.
Cell cycle analysis
SF7761 cells in neurosphere culture were treated in
a 6-well format with the indicated range of AZD2014 and everolimus
concentrations compared to the control for 24 h. Cells were then
removed from compound solutions and fixed with 1 ml ice cold 70%
ethanol dropwise with gentle vortexing. Cells were incubated
overnight at 4°C. After 24 h, the cells were removed from 70%
ethanol and placed into 200 µl of propridium iodide (PI) and
incubated for 30 min. The stained cells then underwent cell cycle
analysis by flow cytometry with a Guava EasyCyte (Millipore,
Billerica, MA, USA). Data analysis was carried out using FlowJo
(Ashland, OR, USA).
Differentiation analysis
DIPG 4 cells were plated into 8-well chamber slides
treated with Poly-D-lysine to improve cell adhesion (354632;
Corning BioCoat) in 0.5 ml media per well. The cells were allowed
to gain adhesion overnight and were then treated for 72 h with the
indicated range of everolimus and AZD2014 concentrations compared
to the control. Following treatment, the cells were fixed,
permeabilized, and co-stained for TUBB3 (MAB1195; R&D Systems)
and GFAP (ag7260; Abcam), followed by conjugation respectively to
Alexa Fluor 555 and Alexa Fluor 488 fluorescent secondary
antibodies. Final staining with DAPI was performed to delineate
nuclei. Confocal images were acquired at a magnification of 400×
using a Zeiss Axio Observer Z1 microscope with Yokogawa CSU-X1
camera and lasers of 405 (DAPI), 488 and 561 nm wavelength for
fluorophore excitation. Mean values of fluorescence intensity were
computed on a cell by cell basis using ImageJ and then
averaged.
Senescence measurement by p21
immunofluorescence
Adherently growing DIPG 4 cells were plated at a
density of 20,000 cells per well in BioCoat 8-well chamber slides
coated with Poly-D-lysine and laminin (Corning) and allowed ~24 h
in which to develop adhesion before subjecting them to experimental
conditions. SF7761 cells were plated at a density of 1,000,000
cells per well of a 6-well plate (Corning). Cells were treated for
two days with 50 nM, 500 nM or 5 µM of AZD2014 or everolimus, along
with DMSO control. After two days of incubation, the cells were
placed into normal growth media for five days. Cells were fixed for
20 min in 37% formaldehyde diluted in 10X PBS (Sigma, St. Louis,
MO, USA), permeabilized in 0.1% Triton X (Sigma) in PBS for 10 min,
and blocked for 45 min in 4% bovine serum albumin (BSA) in PBS
supplemented with 0.05% Triton X. Cells were then incubated in
primary antibody to p21 (2947S; 1:200; Cell Signaling Technology,
Inc.), with 4% BSA (in PBS) and 0.05% Triton X, for 1 h at room
temperature or overnight at 4°C. SF7761 cells to be stained were
fixed and permeabilized in Foxp3/Transcription Factor
Fixation/Permeabilization Concentrate and Diluent (Affymetrix
EBioscience, Santa Clara, CA, USA) according to the manufacturer's
instructions. Cells were incubated in primary antibody to p21,
along with Permeabilization Buffer (Affymetrix EBioscience). After
rinsing to remove the primary antibody, adherent and suspension
cells were incubated in secondary antibody (1:500; Alexa Fluor 488;
Life Technologies) for 1 h at room temperature. Suspension cells
were affixed to slides using a Cytospin 4 Centrifuge (Thermo Fisher
Scientific) at 2,000 rpm for 10 min. Slides were fixed using
ProLong antifade reagent with DAPI (Life Technologies). Confocal
imaging was performed at a magnification of ×400 using 405 (DAPI)
and 488 nm (Alexa Fluor 488) lasers on a 3I Marianas imaging system
(Intelligent Imaging Innovations). Images were obtained using an
Evolve 16-bit EMCCD camera (Photometrics, Tucson, AZ, USA).
Senescence measurement by
β-galactosidase staining
Adherently growing DIPG 4 cells were plated at a
density of 100,000 cells per well in a 6-well plate and allowed ~24
h in which to develop adhesion before subjecting them to
experimental conditions. Cells were treated for two days with 50
nM, 500 nM or 5 µM of AZD2014 or everolimus, along with DMSO
control. After two days of incubation, the cells were placed into
normal growth media for five days. Staining for β-galactosidase was
performed using a Senescence β-galactosidase Staining kit (Cell
Signaling Technology, Inc.) according to the manufacturer's
instructions. Cells were washed with 2 ml of PBS, followed by a
15-min incubation using 1X fixative. Cells were again washed twice
with PBS and then incubated with 1 ml of β-galactosidase staining
solution. The cells were incubated overnight in a dry 37°C
incubator without CO2. Senescencent cells were imaged
via brightfield microscopy (Nikon DS-L2).
Autophagy measurement
DIPG 4 and SF7761 cells were plated in neurosphere
culture in 6-well plates and exposed to one of the following
conditions for 4 h: 0.2% DMSO, 10 µM chloroquine (MP Biomedicals),
1 µM AZD2014, 1 µM everolimus, 1 µM AZD2014 combined with 10 µM
chloroquine, or 1 µM everolimus combined with 10 µM chloroquine.
Immediately following treatment, the cells were lysed, and western
blotting was conducted as described below.
AKT/phospho-AKT measurement
DIPG 4 and SF7761 cells in neurosphere culture were
treated in a 6-well format with the indicated range of AZD2014 and
everolimus versus the control for 2 h. Western blotting was then
conducted as below using antibodies specific to phosphorylated and
unphosphorylated AKT.
Western blotting
Cells were lysed in 200 µl radioimmunoprecipitation
assay buffer with 100X protease and phosphatase inhibitor (Pierce;
Thermo Fisher Scientific) by gentle vortexing for 5 sec. Cells were
incubated on ice for 5 min before centrifugation at 4°C at 14,000 ×
g for 10 min. Protein concentration was measured using the Pierce
BCA assay. Pre-cast 4–20% Mini-PROTEAN TGX gels (Bio-Rad, Hercules,
CA, USA) were loaded with ~30 µg of protein per lane and then run
for 90 min at 125 V. Transfer was then performed to a
polyvinylidine fluoride membrane via a Bio-Rad transfer set at 4°C
at 50 V for 90 min. Blocking was carried out in 5% non-fat dry
milk, as was all staining. Primary staining was performed as
indicated for α-tubulin (#2125 rabbit anti-human mAb at 1:1,000
overnight at 4°C; Cell Signaling Technology, Inc.); pAKT (#4060
rabbit anti-human mAb at 1:1,000 overnight at 4°C; Cell Signaling
Technology, Inc.); and total AKT (#9272 rabbit anti-human pAb at
1:1,000 overnight at 4°C; Cell Signaling Technology, Inc.).
Secondary staining was carried out with anti-rabbit HRP at 1:3,000
for 1 h at room temperature. Blots were developed using Western
Lightning Plus-ECL chemiluminescent (PerkinElmer, Waltham, MA,
USA).
AKT small molecule inhibition
DIPG 4 and SF7761 cells in neurosphere culture were
treated in a 96-well format in triplicate at the indicated
concentration range of MK-2206 (Selleck Chemicals, Houston, TX,
USA) compared to the control for 120 h. Cell viability was then
determined by MTS assay as above.
Constitutive AKT activation
Introduction of a mutation from serine to aspartate
at amino acid 473 of AKT (S473D) mimics serine phosphorylation at
this position and results in constitutive activation of the protein
(18). DIPG 4 cells were plated at
a density of 200,000 cells per well in 2 ml of medium in a 6-well
plate and allowed to adhere overnight. AKT cDNA containing the
S473D mutation (gift of Dr N. Rosen) was transfected into the cells
using jetPRIME® transfection reagent (2 µg cDNA per well
mixed into 200 µl jetPRIME buffer, followed by 4 µl of jetPRIME
reagent; 5-min incubation; Thermo Fisher). The medium was changed
at 18 h following transfection. At 48 h following transfection,
western blotting was conducted as above. Transfected and wild-type
cells were then plated in a 96-well format, allowed to adhere, and
then treated for 120 h as follows with the indicated range of
AZD2014 concentrations compared to the control, in sextuplicate.
Cell viability was then measured by MTS assay as above.
Radiation combination
SF7761 cells in neurosphere culture were plated in a
96-well format in triplicate. The next day, they were exposed to
irradiation at the indicated dose range using a cesium irradiator.
They were then treated starting the next day with the indicated
dose range of AZD2014 compared to control for 120 h. Cell viability
was then measured by MTS assay as above. The combination index at
each dose level combination was determined by the Chou Talalay
method (19).
Immunofluorescence staining
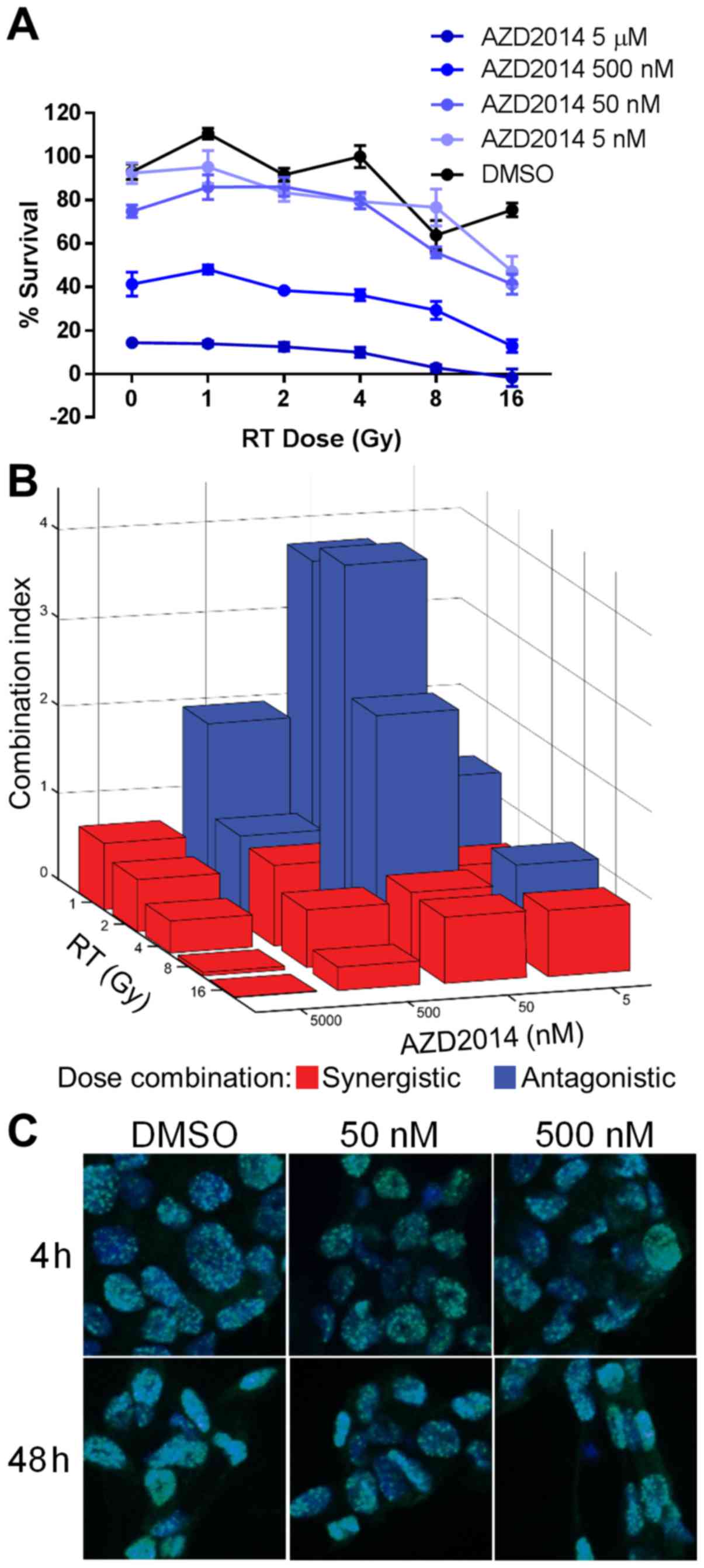
Immunofluorescence staining for γH2AX was performed
on cells exposed to AZD2014 followed by radiation to identify
double-stranded DNA (dsDNA) damage patterns. SF7761 cells were
plated at a density of 20,000 cells per well in BioCoat
Poly-D-lysine/laminin-coated chamber slides (#354688; Corning) and
allowed ~24 h in which to develop adhesion before subjecting them
to experimental conditions. Cells were treated with vehicle or
AZD2014 at 50 and 500 nM dose levels for 4 or 48 h and then
irradiated with 4 Gy from a Cs137 source in a single
dose. At 4 h post-radiation, the cells were fixed for 20 min in 37%
formaldehyde diluted in 10X PBS (Sigma), permeabilized in 0.1%
Triton X in PBS for 10 min, and blocked for 45 min in 4% BSA in PBS
supplemented with 0.05% Triton X. Cells were incubated in primary
γH2AX antibody (#2577; Cell Signaling Technology, Inc.) diluted
1:600 with 4% BSA, dissolved in PBS and 0.05% Triton X, for 1 h at
room temperature. After rinsing, cells were incubated in secondary
antibody (1:500; Alexa Fluor 488; Life Technologies) for 1 h at
room temperature. Slides were fixed using ProLong antifade reagent
with DAPI (Life Technologies). Confocal imaging was performed at a
magnification of ×400 using 405 (DAPI) and 488 nm (Alexa Fluor 488)
lasers on a 3I Marianas imaging system (Intelligent Imaging
Innovations). Images were obtained using an Evolve 16-bit EMCCD
camera (Photometrics).
Chemotherapy combination panel
SF7761 cells in neurosphere culture were plated in a
96-well format. One set of cells was treated with the
IC50 concentration of AZD2014, while another set of
cells was treated with DMSO. All drugs from the Approved Oncology
Drugs Set VII (National Cancer Institute, Bethesda, MD, USA) were
then added to one well of each set of cells at a concentration of 1
µM. In addition, 6 wells of cells were treated with the
IC50 concentration of AZD2014 alone, and 6 with DMSO
alone. Cells were exposed to these compounds for 120 h, and then
cell viability was determined by MTS assay as above. The relative
cell viability for each combination was then compared to DMSO
alone. The therapeutic relationship for each combination was then
calculated by dividing this value by the product of the relative
cell viability for each drug and the relative cell viability for
AZD2014 alone.
Ponatinib combination
DIPG 4, DIPG 6, and SF7761 cells were treated in a
96-well format in triplicate with the indicated ranges of AZD2014,
ponatinib, and the two compounds together, compared to the control,
for 120 h. Cell viability was then determined by MTS assay as
above. The combination index at each dose level combination was
determined by the Chou Talalay method (19).
Data analysis
All data analysis and chart creation was performed
using GraphPad Prism. All charts show mean ± standard error of the
mean (SEM) unless otherwise noted. Comparison of the percentage of
neurospheres in the neurosphere dilution assay was carried out via
Chi-square comparison of proportions test. Comparison of mean
neurosphere area was performed by the unpaired t-test. Statistical
significance is represented as follows: p>0.05 is indicated by
ns (not significant); p=0.05–0.01 is indicated by *; p=0.01–0.001
is indicated by **; p=0.001–0.0001 is indicated by ***; p<0.0001
is indicated by ****.
Results
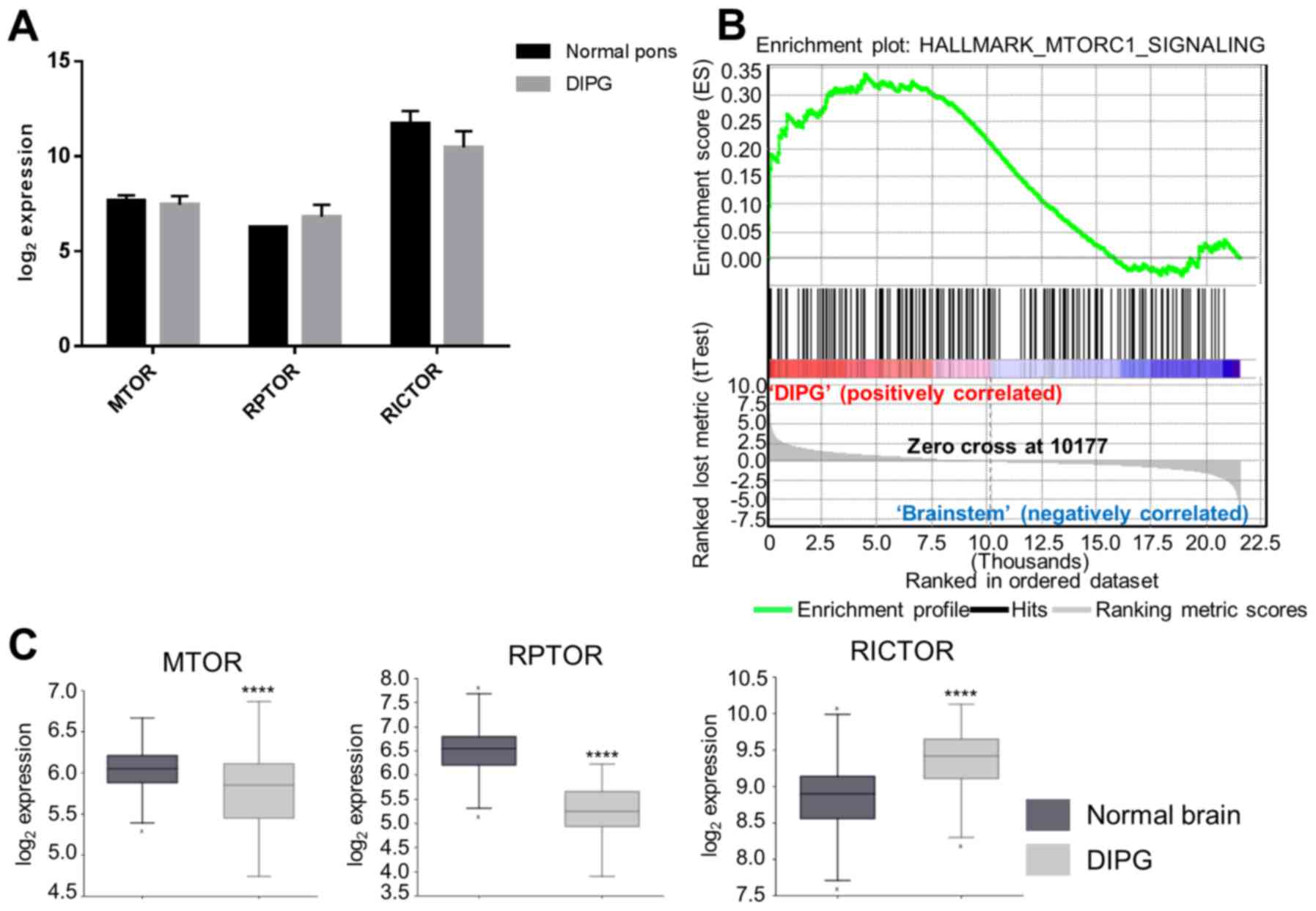
Variable MTOR expression in human DIPG
samples versus normal brain
We first performed measurements of gene expression
of MTOR and the MTOR components RPTOR (MTORC1) and RICTOR (MTORC2)
in a panel of human DIPG samples versus normal pons samples in our
tissue bank. Multiple measures showed no significant difference in
expression between the two sample sets (Fig. 1A). On hallmark GSEA, however, the
MTORC1 signaling pathway was enriched in the DIPG versus normal
samples (NES 1.3, p=0.021, Fig.
1B). We then compared expression of the same three genes using
publically-available sets of normal brain (n=172) and DIPG (n=37)
samples. MTOR (p=4.4×10−6) and RPTOR
(p=4.2×10−29) were both underexpressed in DIPG compared
to normal brain, while RICTOR (p=3.3×10−9) was
overexpressed (Fig. 1C).
MTORC1/2 inhibition decreases cell
survival in vitro versus MTORC1 inhibition alone
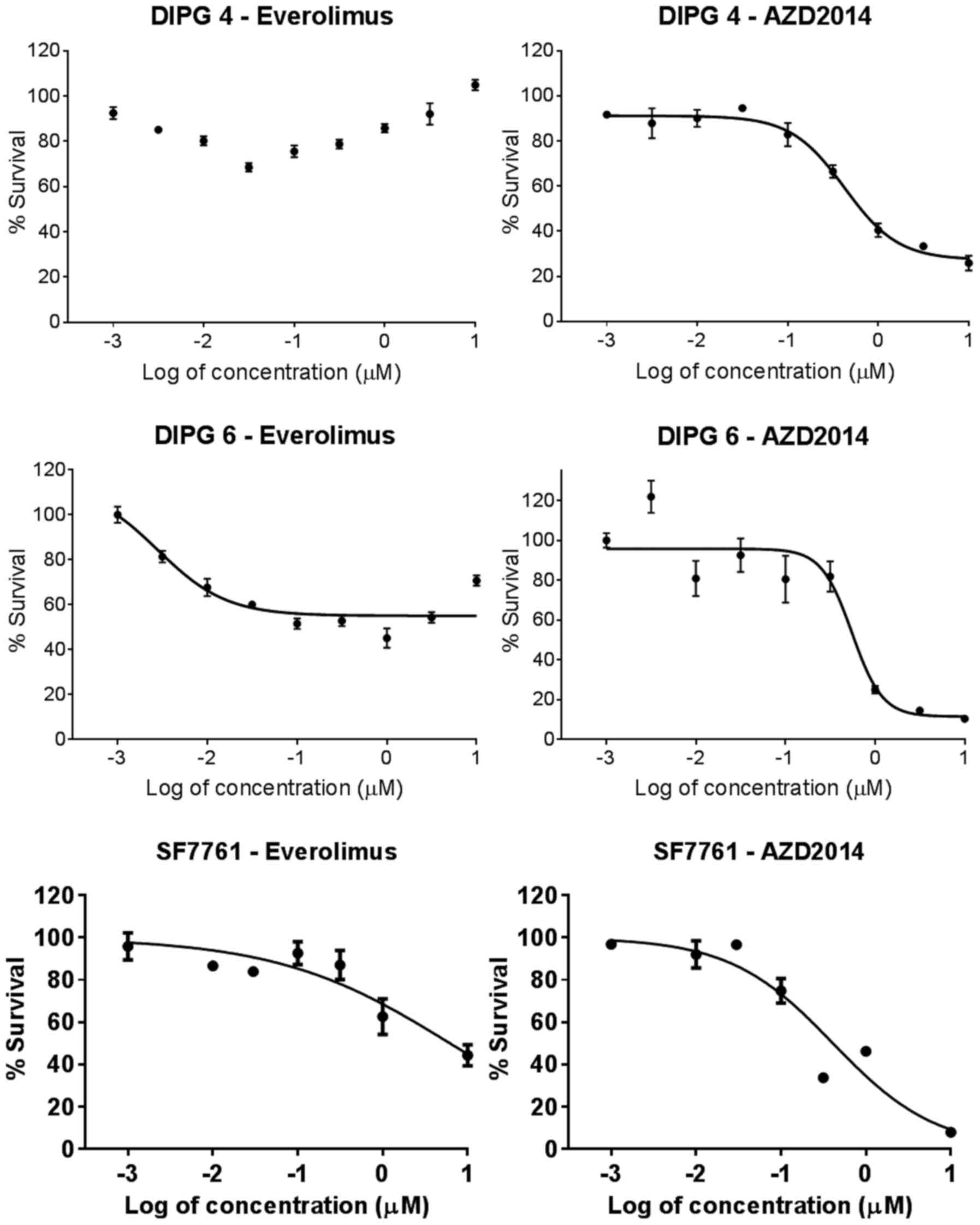
We measured cell survival after five days of
continuous drug exposure by MTS assay for three primary human DIPG
cell lines, DIPG 4, DIPG 6, and SF7761 in neurosphere culture.
Antitumor efficacy, especially in terms of the IC90, was
far greater for AZD2014 compared to everolimus (Fig. 2). For DIPG 4, the IC50
and IC90 were not reached for everolimus and were 0.425
and 1.90 µM for AZD2014, respectively (Table II). For DIPG 6, the IC50
and IC90 were also not reached for everolimus and were
0.552 and 1.30 µM for AZD2014. For SF7761, the IC50 and
IC90 were 6.22 and 1,070 µM for everolimus and 0.410 and
8.86 µM for AZD2014.
| Table II.IC50 and IC90
values. |
Table II.
IC50 and IC90
values.
|
| Everolimus | AZD2014 |
|---|
|
|
|
|
|---|
| Cell line | IC50
(µM) | IC90
(µM) | IC50
(µM) | IC90
(µM) |
|---|
| DIPG 4 | Not reached | Not reached | 0.425 | 1.90 |
| DIPG 6 | Not reached | Not reached | 0.552 | 1.30 |
| SF7761 | 6.22 | 1,070 | 0.410 | 8.86 |
Cell self-renewal difference but no
clear difference in other phenotypic measures
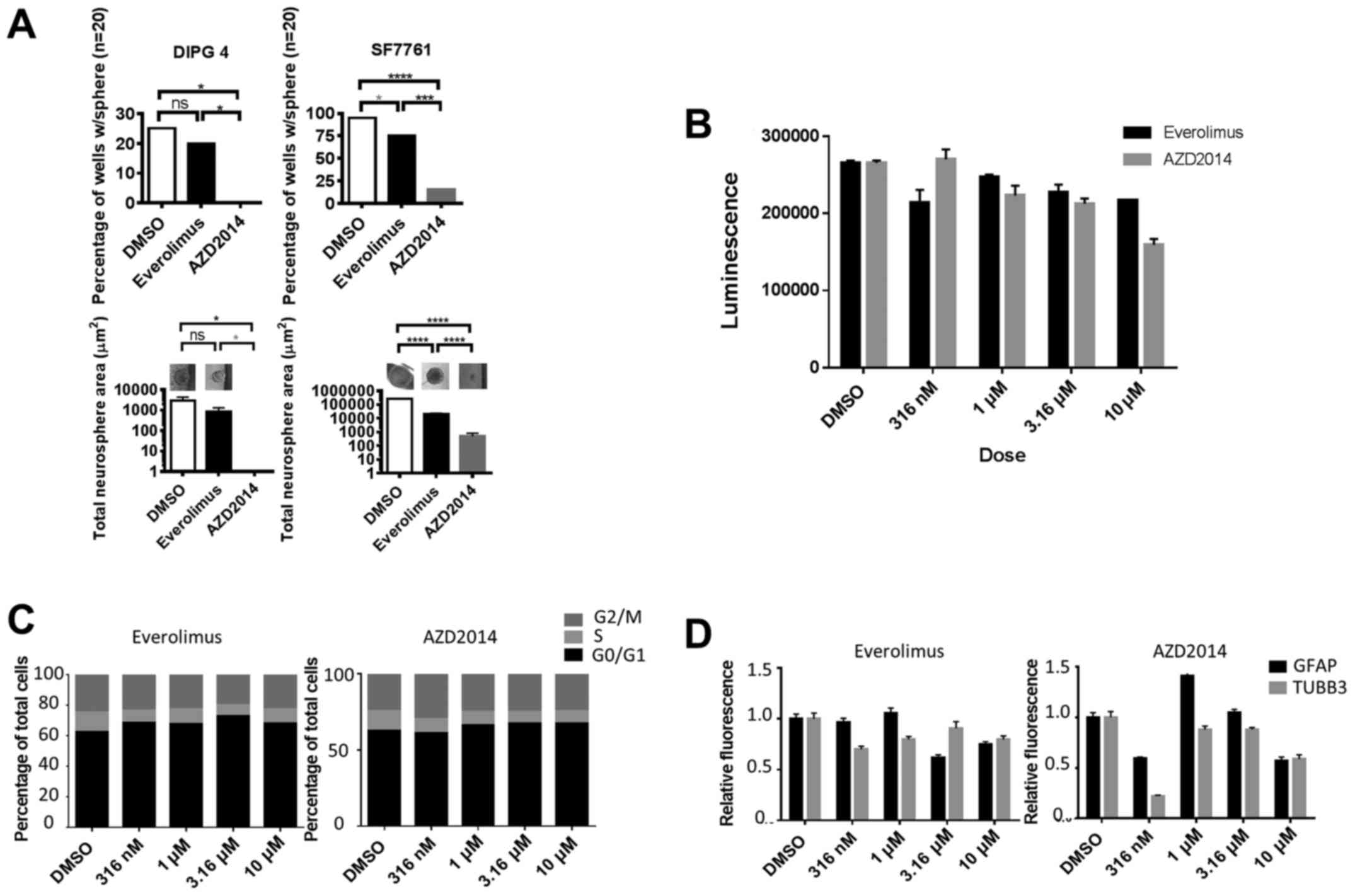
We next examined the general mechanism of action for
differences in the effects between the two drugs. When DIPG 4 and
SF7761 cells were plated in 96-well format at 10 cells per well and
20 wells per condition to test cell self-renewal capability, those
exposed continuously to the IC75 dose of AZD2014 were
less likely to form spheres and formed smaller spheres than those
exposed to the same concentration of everolimus, or control
(Fig. 3A). After 48 h of treatment,
everolimus and AZD2014 both caused a decrease in apoptosis compared
to the control, as measured by caspase 3/7 luminescence assay
(Fig. 3B); there was no significant
difference between the two drugs. We noted no significant
difference in cell cycle distribution with increasing doses of
either drug as measured by propidium iodide flow cytometry
(Fig. 3C). We also saw no clear
trend to demonstrate an increase or decrease in glial or neuronal
differentiation as measured by GFAP or TUBB3 immunofluorescence,
respectively (Fig. 3D).
When we measured induction of senescence in DIPG 4
and SF7761 cells by immunofluorescence for the senescence marker
p21, neither compound showed a consistent increase in senescence
compared to the control (Fig. 4A).
We also measured by staining for the senescence marker
β-galactosidase in DIPG 4 cells; here, both compounds appeared to
increase the percentage of senescent cells in a dose-dependent
manner, but there was no clear difference between the two (Fig. 4B). AZD2014 and everolimus both
induced greater levels of autophagic flux compared to the control,
as determined by western blotting for LC3-II, an autophagy
biomarker (Fig. 4C). Neither
compound induced consistently more autophagy than the other,
however.
Inhibition of AKT phosphorylation is
necessary and sufficient for the effect of AZD2014 on DIPG
We next set out to determine the specific mechanism
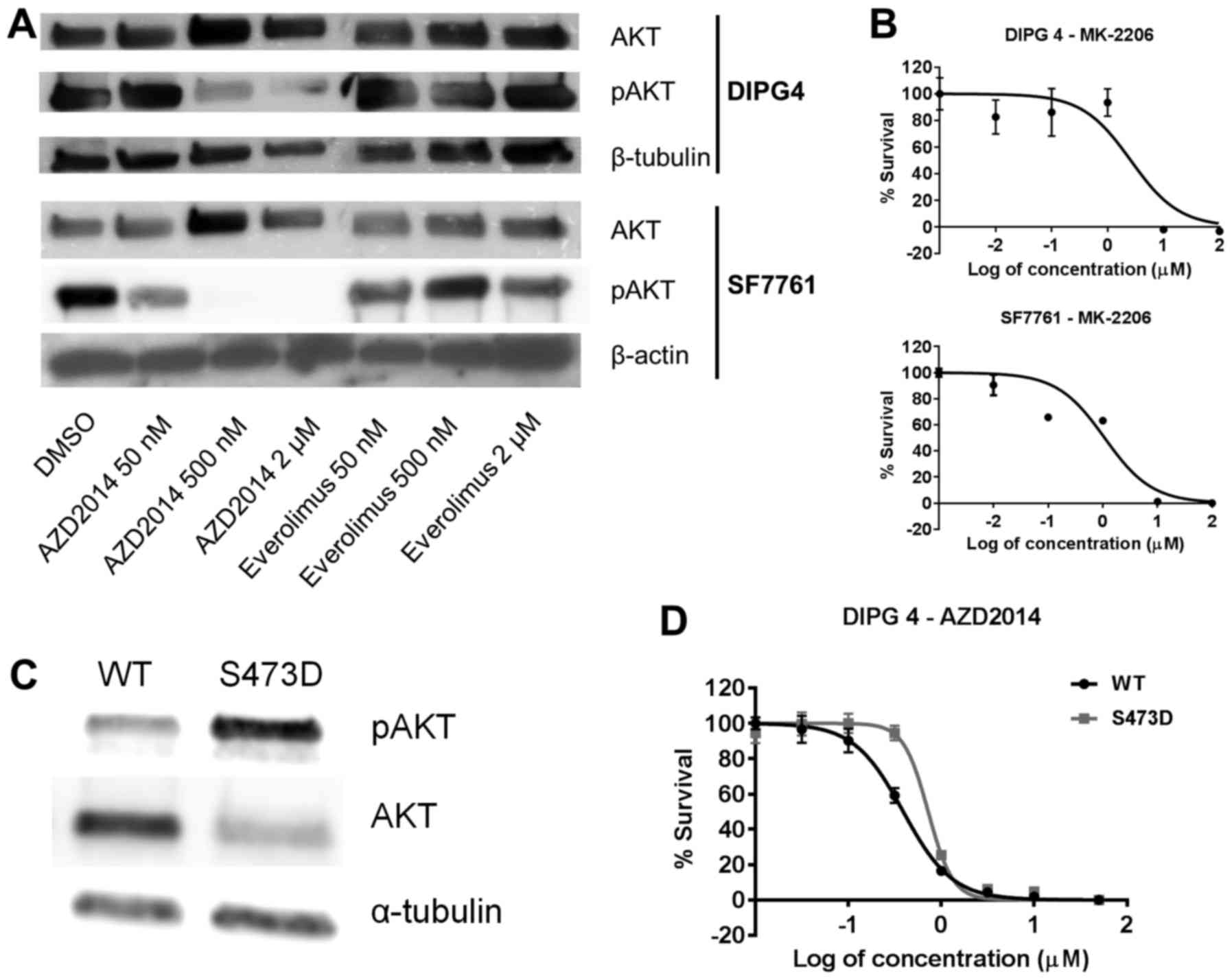
of action differentiating AZD2014 from everolimus, focusing on AKT,
which is activated when phosphorylated by MTORC2 but is upstream of
MTORC1. We found that increasing doses of AZD2014 caused a
dose-dependent decrease in pAKT relative to total AKT, as
determined by western blotting (Fig.
5A). We saw no such decrease with everolimus. We then used a
direct AKT inhibitor, MK-2206, and demonstrated that exposing DIPG
4 and SF7761 cells in neurosphere culture to this drug for five
days resulted in IC50 levels of 2.60 µM in DIPG 4 and
1.08 µM in SF7761 cells (Fig. 5B).
These values were higher but within the range of those obtained
with AZD2014. We then transfected DIPG 4 cells with a cDNA for a
mutant form of AKT (S473D) intending to confer constitutive
activation. Compared to the wild-type, the transfected cells
exhibited increased apparent pAKT (due to the S473D mutation, which
binds the phospho-AKT antibody), accompanied by a decrease in
unphosphorylated AKT, suggesting that total AKT (measured by the
sum of the pAKT and AKT bands) remained relatively constant between
the wild-type and S473D cells in the experiment. Transfection with
S473D thus accurately models the phosphorylation of AKT by MTORC2
(Fig. 5C). Cells transfected with
S473D mutant AKT demonstrated resistance to AZD2014 as compared to
wild-type cells, with an increase in IC50 from 0.390 to
0.724 µM (p<0.01, Fig. 5D),
strongly suggesting that AZD2014 operates at least in part by
inhibiting AKT activation through MTORC2 phosphorylation.
AZD2014 has a variable therapeutic
relationship with RT in DIPG depending on dosing
We then tested the therapeutic relationship between
AZD2014 and RT, since RT is the current standard of care treatment
in DIPG. Because DIPG 4 and DIPG 6 are derived from previously
irradiated tumor samples and resistant to RT, we treated
radiation-naïve SF7761 cells with increasing doses of AZD2014 and
exposed them to various doses of cesium RT in one fraction. We
demonstrated a dose-dependent decrease in cell survival with both
treatments as measured by MTS assay (Fig. 6A). Maximal cell killing with RT
alone was only in the range of 40%, even though SF7761 is our most
radiosensitive cell line, because the single fraction delivered is
well below the total dose delivered clinically over many fractions.
We then determined the therapeutic relationship at each dose
combination by the Chou Talalay method (19) to calculate combination indices. The
relationship was variable but was generally synergistic
(combination index <1) at higher doses of each treatment and
antagonistic (combination index >1) at lower doses (Fig. 6B). We found that, while AZD2014 and
RT were synergistic in killing cells at high doses, the level of
dsDNA damage caused by this combination did not exceed the damage
caused by RT alone as assessed by γH2AX staining (Fig. 6C).
AZD2014 has variable therapeutic
relationships with FDA-approved chemotherapy agents, including
synergy with ponatinib
Finally, we conducted a drug screen of all
FDA-approved chemotherapy agents in combination with AZD2014, using
SF7761 in a 96-well format (data not shown; goo.gl/3XCCqk). Aside
from antagonistic relationships with microtubule inhibitors, there
was no clear trend to AZD2014's therapeutic relationship with any
category of drug. AZD2014 did show synergistic relationships with
drugs from multiple classes, however, including microtubule
stabilizers, topoisomerase inhibitors, and tyrosine kinase
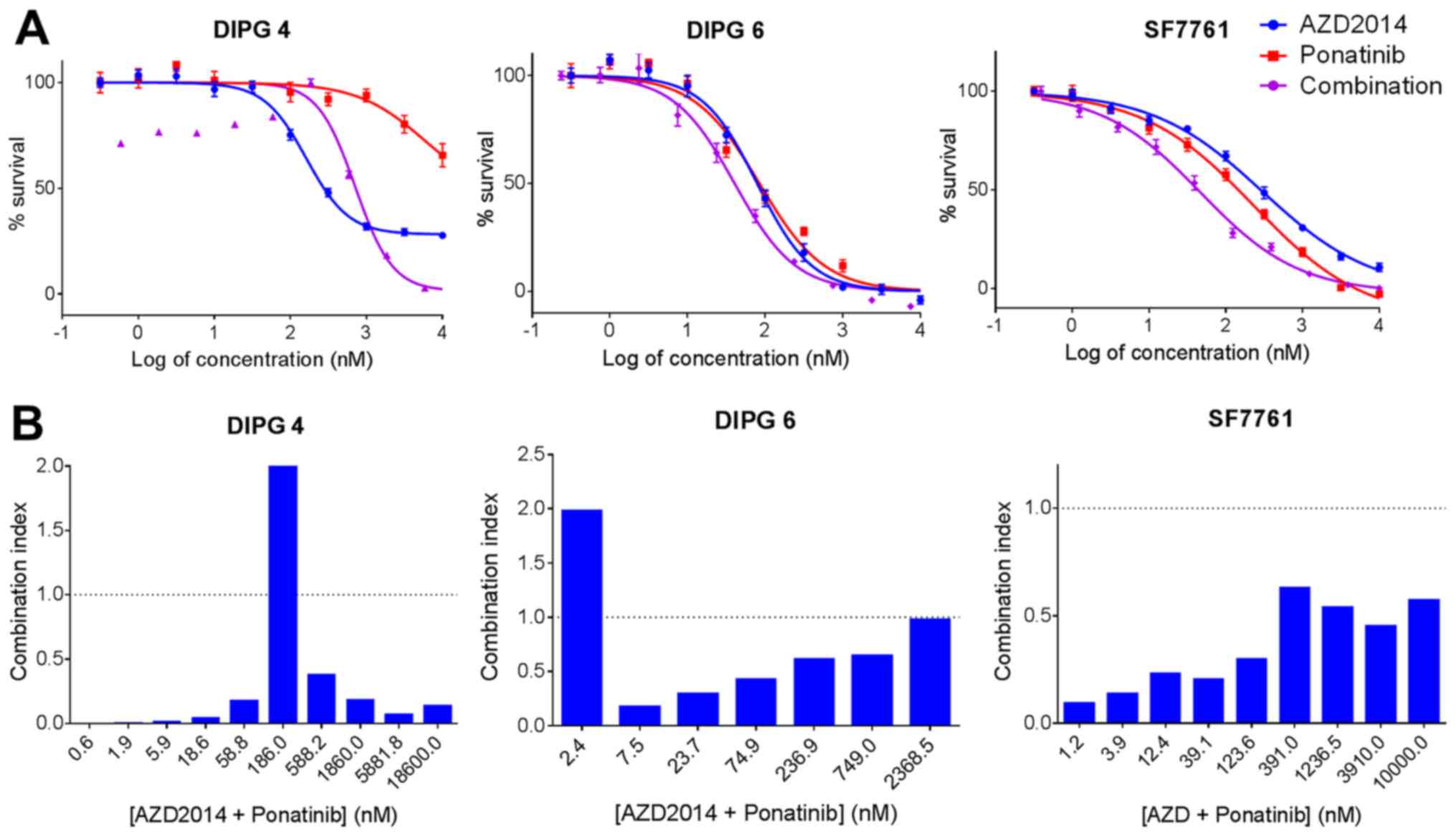
inhibitors. We then validated our screening finding of a
potentially synergistic relationship between AZD2014 and the
multikinase inhibitor ponatinib using multiple dose levels of the
AZD2014-ponatinib combination in DIPG 4, DIPG 6, and SF7761
(Fig. 7A). We demonstrated synergy
between AZD2014 and ponatinib at most dose levels in all three cell
lines, especially dose levels exceeding the combined
IC50 of the two agents (Fig.
7B).
Discussion
In this study, we demonstrated that MTORC1/2
inhibition shows greatly increased antitumor efficacy in a panel of
patient-derived DIPG cell lines compared to MTORC1 inhibition
alone, which showed little to no effect. The phenotype behind this
advantage appears to be a decrease in cell self-renewal; assays
testing other potential general mechanisms of action showed no
significant or consistent differences between the two drugs. On a
molecular level, the decrease in AKT phosphorylation caused by
MTORC2 inhibition appears to be sufficient and at least partially
necessary to the augmented tumor inhibition when MTORC2 is
targeted. This phenotype and molecular mechanism are consistent
with the known role of AKT in cancer cell self-renewal (20–22).
Finally, we showed that AZD2014 has the potential to act
synergistically with RT, as well as with cytotoxic and targeted
chemotherapeutic agents of various classes.
It is unclear whether the efficacy of AZD2014 in
this study depends on overexpression of MTORC2 components.
Expression levels of MTOR, RPTOR, and RICTOR were similar in the
DIPG samples in our tumor bank compared to levels in normal pons,
although the MTOR pathway was enriched in DIPG on GSEA. MTOR and
RPTOR were underexpressed in a large public dataset of DIPG
samples, while RICTOR was overexpressed, which could contribute to
an explanation for the difference seen between compounds, since
RICTOR is unique to MTORC2. It should be noted in this analysis,
however, that the available comparison group was tissue samples
from throughout the brain, not the pons alone. In terms of previous
studies, a large study of 43 DIPG samples that examined copy number
abnormalities and expression profiles did not identify MTOR in any
of the abnormalities (23). This
study did find AKT as a potential gene of interest in focal
recurrent gains, however. Another group working with a genetically
engineered mouse model of DIPG found that AKT was overexpressed in
cell lines derived from this model, and when they conducted a
high-throughput drug screen against these cells, they identified
antitumor activity in a multikinase inhibitor that decreases levels
of pAKT (24). These findings
support our results showing that AKT inhibition through MTORC1 and
MTORC2 is necessary and sufficient to the antitumor effect of
AZD2014, and that this is the reason for everolimus' lack of
activity.
RT is currently the only effective primary therapy
in DIPG, and clinical trial data are also emerging to suggest its
utility at recurrence as well (25,26).
Therefore, the therapeutic relationship with RT will be relevant to
any new targeted drug proposed. Our findings suggest that AZD2014
and RT have a synergistic therapeutic relationship at higher doses
of each. These higher RT doses are used in DIPG, and AZD2014 levels
at and above the IC50 will also be necessary to achieve
therapeutic effect. We did not observe greater levels of dsDNA
damage from the combination versus RT alone and continue to
investigate the underlying mechanism of synergy. The prior study
examining this combination in adult glioblastoma found that the
reason for synergy is most likely inhibition of DNA repair
(13). On our chemotherapy
combination screen, AZD2014 also appeared to show synergy with
several other DNA damaging agents. Another candidate drug from our
chemotherapy screen, ponatinib, primarily targets BCR-ABL but also
inhibits VEGFR, PDGFR, and EGFR (27), all of which have relevance to DIPG
(28). Ponatinib may also inhibit
AKT (29), raising another possible
mechanism of synergy with AZD2014. Given that development of
resistance to targeted therapies is a major issue with their
efficacy, using combinations such as this to target multiple key
oncogenic pathways may be crucial to successful DIPG treatment. Our
consistent results with ponatinib in each of the lines validates
our screening method. The availability of these preclinical models
in DIPG is of great translational value. Besides their lack of
efficacy, the drugs used in previous DIPG clinical trials have also
caused harm through adverse effects, mandating that future
treatments undergo rational preclinical testing before they are
deemed worthy of the risk they carry for patients.
Our study has several limitations. We did not show
that AZD2014 reverts to an everolimus-like dose-response curve with
constitutive AKT activation, although the difference in
IC50 was statistically significant. This may be because
AZD2014 was still able to achieve some level of AKT inhibition due
to imperfect transfection efficiency in this experiment. Also,
AZD2014's blood-brain barrier (BBB) penetration is unknown, but
given the concerns with drug penetration in DIPG in general, it is
likely that local delivery methods that bypass the BBB, such as
convection-enhanced delivery, will at least partially obviate this
consideration. Finally, we do not yet have in vivo
validation of our findings. However, while our study was under
review, Miyahara et al published their findings on the
effect of another MTORC1/2 inhibitor, TAK228, in DIPG, including
its efficacy in a patient-derived xenograft model (30). Our study delves further into the
molecular mechanism of action and potential combination
chemotherapy approaches, and together, these two studies on
different MTORC1/2 inhibitors provide strong preclinical rationale
for this strategy in DIPG.
In conclusion, the present study demonstrates that
DIPG does not respond preclinically to MTORC1 inhibition alone but
does respond well to combined MTORC1/2 inhibition, due to the
inhibitory effect of MTORC2 on AKT. AZD2014, an MTORC1/2 inhibitor,
shows synergy with RT and with selected chemotherapy agents in
DIPG. This strategy should be studied further as a potential
component of combinatorial approaches to treatment of this
currently incurable tumor.
Acknowledgements
The authors wish to thank Radu Moldovan and Alireza
Hemmati of the Anschutz Medical Campus Advanced Light Microscopy
Core Facility for their support. This study was supported by a
grant from the Morgan Adams Foundation. A.L.G. is the Luke's Army
Pediatric Cancer Research Fund St. Baldrick's Fellow and a Hyundai
Hope on Wheels Young Investigator.
Glossary
Abbreviations
Abbreviations:
|
AKT
|
V-Akt murine thymoma viral
oncogene
|
|
BBB
|
blood-brain barrier
|
|
DIPG
|
diffuse intrinsic pontine glioma
|
|
GSEA
|
gene set enrichment analysis
|
|
MTOR
|
mechanistic target of rapamycin
|
|
MTORC1
|
MTOR complex 1
|
|
MTORC2
|
MTOR complex 2
|
|
RICTOR
|
RPTOR independent companion of
MTORC2
|
|
RPTOR
|
regulatory associated protein of
MTORC1
|
|
RT
|
radiation therapy
|
|
SEM
|
standard error of the mean
|
References
|
1
|
Fangusaro J: Pediatric high-grade gliomas
and diffuse intrinsic pontine gliomas. J Child Neurol.
24:1409–1417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Massimino M, Spreafico F, Biassoni V,
Simonetti F, Riva D, Trecate G, Giombini S, Poggi G, Pecori E,
Pignoli E, et al: Diffuse pontine gliomas in children: Changing
strategies, changing results? A mono-institutional 20-year
experience. J Neurooncol. 87:355–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Green AL and Kieran MW: Pediatric
brainstem gliomas: New understanding leads to potential new
treatments for two very different tumors. Curr Oncol Rep.
17:4362015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hashizume R, Ozawa T, Gryaznov SM, Bollen
AW, Lamborn KR, Frey WH II and Deen DF: New therapeutic approach
for brain tumors: Intranasal delivery of telomerase inhibitor
GRN163. Neuro-oncol. 10:112–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goodwin CR, Xu R, Iyer R, Sankey EW, Liu
A, Abu-Bonsrah N, Sarabia-Estrada R, Frazier JL, Sciubba DM and
Jallo GI: Local delivery methods of therapeutic agents in the
treatment of diffuse intrinsic brainstem gliomas. Clin Neurol
Neurosurg. 142:120–127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barua NU, Lowis SP, Woolley M, O'Sullivan
S, Harrison R and Gill SS: Robot-guided convection-enhanced
delivery of carboplatin for advanced brainstem glioma. Acta
Neurochir (Wien). 155:1459–1465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cohen K, Jones A, Raabe E and Pearl M:
Highly selective intra-arterial chemotherapy for the treatment of
progressive diffuse intrinsic pontine gliomas (DIPG). In: 20th
International Conference on Brain Tumor Research and Therapy
Neuro-Oncology, Lake Tahoe, CA. Neurooncology. 16:iii292014.
|
|
8
|
Zeng Z, Sarbassov D, Samudio IJ, Yee KW,
Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M and
Konopleva M: Rapamycin derivatives reduce mTORC2 signaling and
inhibit AKT activation in AML. Blood. 109:3509–3512. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guertin DA, Stevens DM, Thoreen CC, Burds
AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ and Sabatini DM:
Ablation in mice of the mTORC components raptor, rictor, or mlST8
reveals that mTORC2 is required for signaling to Akt-FOXO and
PKCalpha, but not S6K1. Dev Cell. 11:859–871. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akhavan D, Cloughesy TF and Mischel PS:
mTOR signaling in glioblastoma: Lessons learned from bench to
bedside. Neuro-oncol. 12:882–889. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galanis E, Buckner JC, Maurer MJ,
Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR,
Morton RF, et al North Central Cancer Treatment Group, : Phase II
trial of temsirolimus (CCI-779) in recurrent glioblastoma
multiforme: A North Central Cancer Treatment Group Study. J Clin
Oncol. 23:5294–5304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp
T, Camphausen K and Tofilon PJ: The mTORC1/mTORC2 inhibitor AZD2014
enhances the radiosensitivity of glioblastoma stem-like cells.
Neuro-oncol. 16:29–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwartzentruber J, Korshunov A, Liu XY,
Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA,
Tönjes M, et al: Driver mutations in histone H3.3 and chromatin
remodelling genes in paediatric glioblastoma. Nature. 482:226–231.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Buczkowicz P, Hoeman C, Rakopoulos P,
Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E,
Bartels U, et al: Genomic analysis of diffuse intrinsic pontine
gliomas identifies three molecular subgroups and recurrent
activating ACVR1 mutations. Nat Genet. 46:451–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehta S, Huillard E, Kesari S, Maire CL,
Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon
KL, et al: The central nervous system-restricted transcription
factor Olig2 opposes p53 responses to genotoxic damage in neural
progenitors and malignant glioma. Cancer Cell. 19:359–371. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagaraja S, Vitanza NA, Woo PJ, Taylor KR,
Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al:
Transcriptional dependencies in diffuse intrinsic pontine glioma.
Cancer Cell. 31:635–652 e636. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rodrik-Outmezguine VS, Chandarlapaty S,
Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J,
Guichard S and Rosen N: mTOR kinase inhibition causes
feedback-dependent biphasic regulation of AKT signaling. Cancer
Discov. 1:248–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bahena-Ocampo I, Espinosa M,
Ceballos-Cancino G, Lizarraga F, Campos-Arroyo D, Schwarz A,
Garcia-Lopez P, Maldonado V and Melendez-Zajgla J: miR-10b
expression in breast cancer stem cells supports self-renewal
through negative PTEN regulation and sustained AKT activation. EMBO
Rep. 17:10812016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin Y, Yang Y, Li W, Chen Q, Li J, Pan X,
Zhou L, Liu C, Chen C, He J, et al: Reciprocal regulation of Akt
and Oct4 promotes the self-renewal and survival of embryonal
carcinoma cells. Mol Cell. 48:627–640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh S, Trevino J, Bora-Singhal N,
Coppola D, Haura E, Altiok S and Chellappan SP: EGFR/Src/Akt
signaling modulates Sox2 expression and self-renewal of stem-like
side-population cells in non-small cell lung cancer. Mol Cancer.
11:732012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Paugh BS, Broniscer A, Qu C, Miller CP,
Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS,
et al: Genome-wide analyses identify recurrent amplifications of
receptor tyrosine kinases and cell-cycle regulatory genes in
diffuse intrinsic pontine glioma. J Clin Oncol. 29:3999–4006. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Halvorson KG, Barton KL, Schroeder K,
Misuraca KL, Hoeman C, Chung A, Crabtree DM, Cordero FJ, Singh R,
Spasojevic I, et al: A high-throughput in vitro drug screen in a
genetically engineered mouse model of diffuse intrinsic pontine
glioma identifies BMS-754807 as a promising therapeutic agent. PLoS
One. 10:e01189262015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fontanilla HP, Pinnix CC, Ketonen LM, Woo
SY, Vats TS, Rytting ME, Wolff JE and Mahajan A: Palliative
reirradiation for progressive diffuse intrinsic pontine glioma. Am
J Clin Oncol. 35:51–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wolff JE, Rytting ME, Vats TS, Zage PE,
Ater JL, Woo S, Kuttesch J, Ketonen L and Mahajan A: Treatment of
recurrent diffuse intrinsic pontine glioma: The MD Anderson Cancer
Center experience. J Neurooncol. 106:391–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frankfurt O and Licht JD: Ponatinib - a
step forward in overcoming resistance in chronic myeloid leukemia.
Clin Cancer Res. 19:5828–5834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jansen MH, van Vuurden DG, Vandertop WP
and Kaspers GJ: Diffuse intrinsic pontine gliomas: A systematic
update on clinical trials and biology. Cancer Treat Rev. 38:27–35.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim DH, Kwak Y, Kim ND and Sim T:
Antitumor effects and molecular mechanisms of ponatinib on
endometrial cancer cells harboring activating FGFR2 mutations.
Cancer Biol Ther. 17:65–78. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyahara H, Yadavilli S, Natsumeda M,
Rubens JA, Rodgers L, Kambhampati M, Taylor IC, Kaur H, Asnaghi L,
Eberhart CG, et al: The dual mTOR kinase inhibitor TAK228 inhibits
tumorigenicity and enhances radiosensitization in diffuse intrinsic
pontine glioma. Cancer Lett. 400:110–116. 2017. View Article : Google Scholar : PubMed/NCBI
|