Introduction
Chemotherapy, surgery and radiotherapy are
considered to be the most important types of cancer treatments
available for patients with solid or hematological malignancies
(1,2). Cancer patients are frequently unable
to tolerate chemotherapy due to the cytotoxic nature of the drugs.
Chemotherapy and radiotherapy are thought to be important in the
treatment of cancerous tissues. However, non-cancerous tissues are
equally targeted by these therapies (3,4). These
treatments are also associated with poor prognosis and survival of
cancer patients. Currently, targeting the immune system to fight
cancer is thought to be more effective as it is associated with
fewer cytotoxic side-effects (5).
Immunotherapy is classified as being much safer than chemotherapy
and radiotherapy as it targets specific antigens associated with
cancer cells and tumors. Therefore immunotherapy, specifically
using T-cells, has potential in the field of cell therapy due to
the following: i) T-cells have the potential to differentiate
between cancerous and non-cancerous cells (6,7); ii)
upon activation, T-cells undergo rapid clonal expansions (8,9); iii)
T-cells have the ability to maintain therapeutic responses for a
longer period of time (10); and
iv) T-cells have the ability to target specific antigens at a
variety of different sites (8,11),
therefore, they are important for the treatment of local and
distant metastases.
Prostate cancer is caused by the malignant growth of
the cells of the prostate gland, a walnut shaped organ in males
located below the bladder and behind the rectum, which produces and
stores seminal fluid. The American Cancer Society projects that
164,690 novel cases of prostate cancer will be diagnosed with
29,430 men expected to succumb to this disease in 2018 (12). Prostate cancer has been classified
as one of the leading causes of mortality among men worldwide
(13,14). However, over the past few decades,
the mortality rate has stabilized due to early diagnosis, which
helps to maintain or cure the disease (12,15).
A broad range of treatment options are available for
men diagnosed with prostate cancer, including surgery
(prostatectomy), radiation, chemotherapy (drug therapy), high
intensity focused ultrasound or through an integrated cancer
program (16–18). Another method, which is the subject
of the present review, is the application of immunotherapy.
The aforementioned methods of treatment have been
widely used, however, they frequently leave the patient with
adverse side-effects; therefore, there is a need to develop a more
efficient method with fewer side-effects. Immunotherapeutic
techniques have now been employed for over a century in the
treatment of tumors and in the last few decades have greatly
improved and produced effective applications (19,20).
Methods including cytokine induced killers, tumor infiltrating
lymphocytes, monoclonal antibodies (mAbs), tumor vaccines, immune
checkpoint blockades, bispecific antibodies and chimeric antigen
receptor (CAR)T-cell are a few of the immunological techniques
employed in cancer treatments (21,22).
Among the techniques and approaches employed, CART-cell therapy is
an advanced method with promising curative potential.
Nonetheless, while the use of CART-cell therapy is
advanced in hematological malignancies, it is also presenting
potential in solid tumors, though at a limited rate. The delay over
the years has been as a result of the difficulty of immune cells
targeting and penetrating tumors and cancerous cells, as well as
overcoming the metabolically hostile tumor microenvironment
(23,24).
The CART-cell
CAR is an antigen targeting protein receptor
engineered to fuse with T-cells and designed to specifically bind
to antigens on tumors or cancerous cells that are mostly
unrecognized by normal T-cells. The CAR exhibits the following
features: i) An antigen binding part; ii) a hinge region; iii) a
transmembrane domain; and iv) an intracellular co-stimulatory
domain (Fig. 1) (25–27).
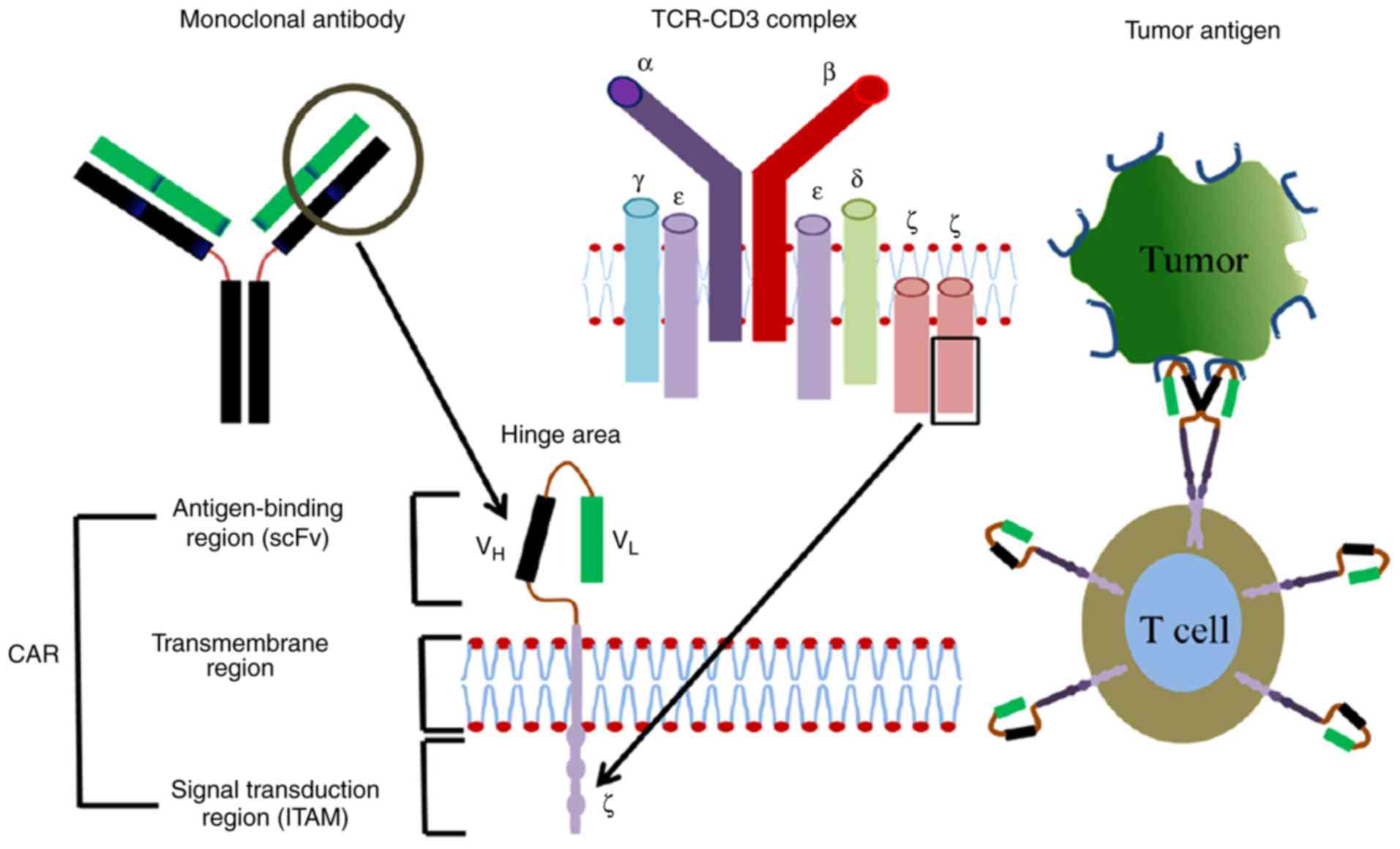 | Figure 1.CARs are comprised of a surface
antigen-binding region, transmembrane region and intracellular
signal transduction region. Derived from a mono-antibody, the
antigen-binding region consists of VH and VL
chains, which are joined by a flexible hinge area. Dimer membrane
proteins including CD3, CD8 and CD28 are demonstrated in the
transmembrane region. The intracellular signal transduction region
is comprised of immuno-tyrosine-based activation motifs, including
CD3ζ or FcƐRlƴ. CARs, chimeric antigen receptors; CD, cluster of
differentiation; Fc, fragment crystallizable domain; TCR, T-cell
receptor; VH, variable heavy chain; VL,
variable light chain; ITAM, immunoreceptor tyrosine-based
activation motif; scFv, single-chain variable fragment. |
The antigen-binding domain is an extracellular
single-chain variable fragment (scFv) antibody that specifically
recognizes an antigen on the surface of cancerous cells. scFvs are
designed from full length mAbs to reduce the undesirable effects
accompanied by the fragment crystallizable domain (Fc) and to
increase antigen binding affinity. An scFv fragment consists of
variable heavy and light chains joined together by a flexible
peptide linker (28). The
hydrophilic nature of the linker residue enhances flexibility
through its glycine and serine sequences, whereas the interspersed
glutamine and lysine sequences enhance solubility (29,30).
In normal T-cell receptors (TCRs), the antigen-binding domain is
the cluster of differentiation (CD)3-ζ chain, which recognizes
antigen in a major histocompatibility complex (MHC).
The hinge region is a flexible part that directs the
scFv to the target antigen. The transmembrane domain and its
associated proteins also serve a critical role in the signal
transduction and surface expression of the receptor (31). It has also been demonstrated to
provide stability to the CAR and could dimerize to form complexes
with endogenous TCRs resulting in enhanced T-cell activation
(32).
The intracellular signaling domain, which usually
originates from the signal transduction subunit of T-cells or
natural killer cells, includes 4-1BB and CD28, and its function is
to transduce extracellular binding signals to initiate the
activation of downstream signaling cascades. T-cell activation
relies on the phosphorylation of immunoreceptor tyrosine-based
activation motifs (ITAMs) present in the cytoplasmic CD3-ζ domain.
While the majority of current CAR endo-domains contain an
activation domain derived from CD3-ζ, other ITAM-containing domains
have been investigated including the Fc receptor of the
immunoglobulin (Ig)E-γ domain, however, this has been demonstrated
to be less effective (32).
Modes of T-leucocyte mediated tumor
action
The T-lymphocyte mediated immune response serves a
major role in adaptive immunity by attacking infected cells or
invading the microorganism and memorizing the targeted antigens to
protect against subsequent infections (8,33).
TCRs bind antigens presented by antigen presenting cells (APCs),
which in turn activate the T-cell. The binding of TCRs to the
antigen releases cytokines and other inflammatory molecules which
help to kill the infected cells or remove the foreign organism
(34,35). In the case of cancer, MHCs are
downregulated which limits its antigen presenting ability and
therefore, cancerous cells and tumors are able to escape the
surveillance of T-cells (36).
Production and autologous transfer of
CART-cells
The processes involved in the creation of
CART-cells, its autologous transfer and monitoring have been
described by Porter et al (37,38).
To produce CART-cells, CD4+ and CD8+ T
lymphocytes are separated from the patient or donor's blood through
leukapheresis. Isolated T-cells are then transfected with a viral
vector including lentivirus, containing genes for CAR. The virus
integrates CAR genes into the T-cell genome and expresses the cell
surface CAR protein. The genetically modified T-cells are then
cultured with a sufficient amount of interleukin (IL)-2 in
vitro. Subsequently, autologous CART-cells are injected back
into the patient and the patient is then monitored for treatment
responses and for the function of the modified T-cells (37–39).
CART-cell antitumor mechanism
Normal T-cell mediated immune responses require the
presence of an antigen in an MHC-dependent complex and a
co-stimulatory signal, which involves the recruitment of CD28 on
the T-cell surface with the co-stimulatory molecule CD80 or CD86 on
the APC (34,35). However, activation of CART is
independent of MHC. Upon recognition of the cancer antigen by
surface CAR scFv, the T-cell is activated by ITAMs, which leads to
the subsequent induction of cytotoxic cytokine secretion and T-cell
proliferation. The cytotoxic role of the activated CART-cells is
initiated through the secretion of perforin and granzyme granules
and the activation of death receptor signaling through Fas/FasL and
tumor necrosis factor (TNF)/TNF receptor. The enhanced secretion
and proliferation of proinflammatory cytokines [IL-2, interferon
(IFN)-γ, IL-12 and TNF] following the activation of CART-cells is
critical for targeted cell lysis (22).
CAR generations
A broader understanding of CAR signaling has led to
an improvement in the engineering of various signaling domains that
are potent in enhancing the complete activation of T-cells. A total
of four generations of CAR are currently under investigation in
preclinical and ongoing clinical studies (Fig. 2). These generations are
distinguished by their intracellular signaling domains attached to
the scFv receptor molecule. The first-generation CAR has only CD3ζ
as the intracellular signaling domain. However, the
second-generation CAR has an additional co-stimulatory domain,
which may be CD27, CD28, CD137 (4-1BB) or OX40. The
third-generation CAR contains CD3ζ and two other co-stimulatory
domains such as CD28, CD137 (4-1BB) or other co-stimulatory
molecules (40–42). To enhance the efficacy of CARs, the
T-cells are modulated through the introduction of additional genes,
including those encoding potent antitumor cytokines including IL-2
and IL-15 or co-stimulatory ligands including 4-1BBL, thereby
generating engineered T-cells that are more potent and cytotoxic to
tumors. This is the fourth-generation CAR that is still under
investigation to improve the expression of the required cytokines
and to control the excessive release of other inflammatory
cytokines (7,43–45).
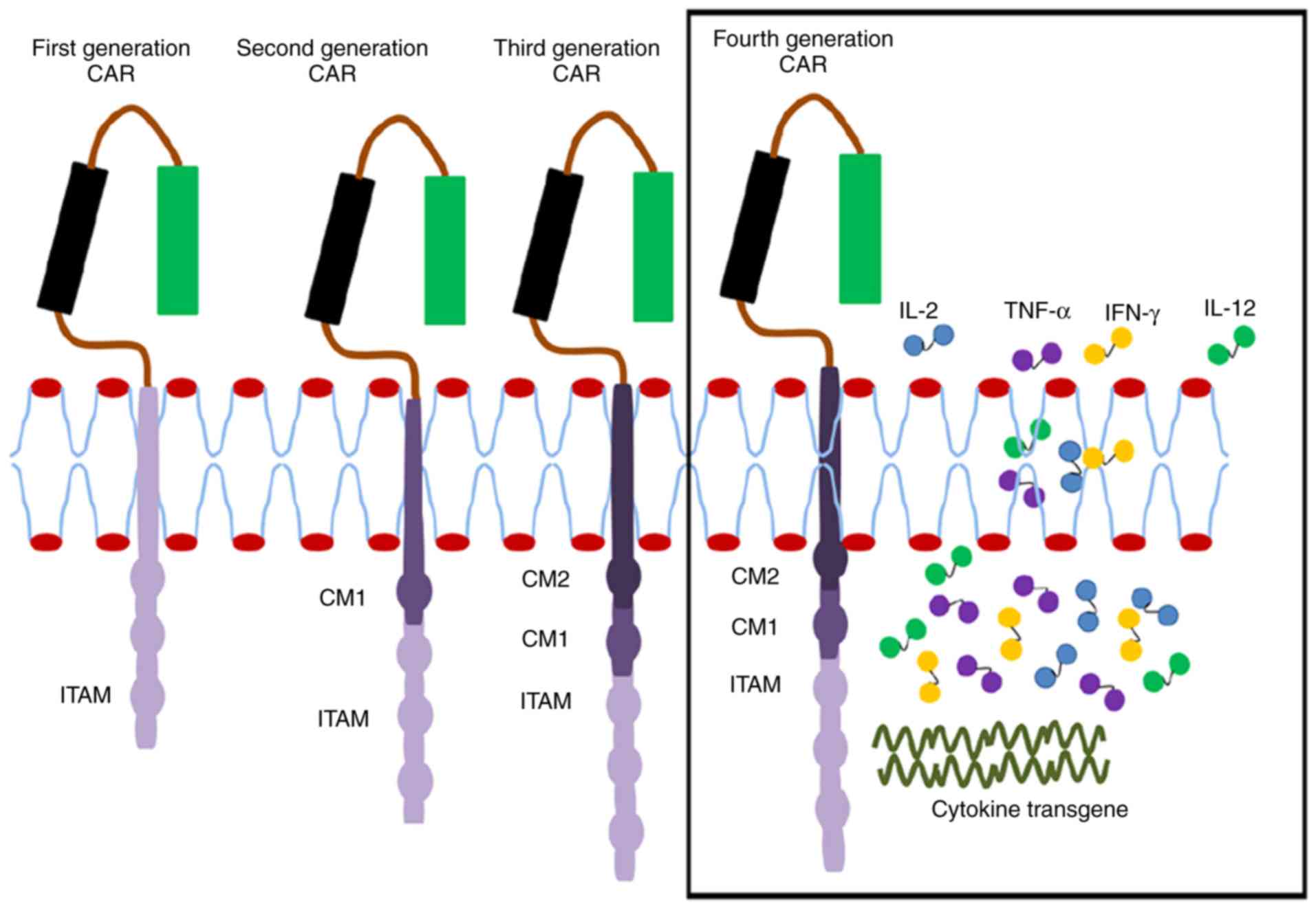 | Figure 2.Single chain antibody links with the
ITAM (CD24ζ or FcƐRlƴ) of the first-generation CAR forming a
transmembrane region. In the second generation, CM1 (including
CD28) has been engineered to the signal transduction region. The
third generation has another CM2 based on the second generation,
combining CD134 or CD137. The fourth generation is further enhanced
to produce more cytotoxic cytokines. CAR, chimeric antigen
receptor; CD, cluster of differentiation; ITAM, immunoreceptor
tyrosine-based activation motif; Fc, fragment crystallizable
domain; CM, co-stimulatory molecule; IL, interleukin; TNF-α, tumor
necrosis factor; IFN, interferon. |
Sipuleucel-T is a treatment option for asymptomatic
or minimally symptomatic metastatic castrate-resistant prostate
cancer approved by the Food and Drug Administration. This therapy
reintroduces activated T-cells specific for the prostatic acid
phosphate (PAP) antigen into the patient. Sipuleucel-T consists of
autologous peripheral blood mononuclear cells obtained via
leukapheresis, which are then cultured and activated with a
recombinant human protein consisting of PAP linked to
granulocyte-macrophage colony-stimulating factor (46–48).
Sipuleucel-T does not require the genetic engineering applied in
CART and the activation of T-cells is performed in vitro
using the PAP antigen which is highly expressed in prostate cancer
(49).
Classified prostate associated antigens
A few protein and glycoprotein molecules
preferentially expressed in malignant prostate tissues, which serve
as targets for CART have been described previously. These include
PAP, prostate-specific antigen, prostate-specific membrane antigen
(PSMA), prostate stem cell antigen (PSCA), T-cell receptor γ
alternate reading frame protein, transient receptor potential-p8
and six-transmembrane epithelial antigen of the prostate 1
(49).
The PSMA
PSMA is a cell surface glycoprotein expressed in the
prostate epithelial cell membrane of normal prostates but is
expressed to a substantially higher degree in prostate carcinomas.
It contains 750 amino acids, with a type II transmembrane domain
similar to that of the transferrin receptor (50). An ELISA performed by Sokollof et
al (51), revealed high
expression levels of PSMA in prostatic tissues but low expression
in the cell membranes of the breast, colon, liver and kidneys with
negligible levels of expression on other membranes. Semen content
is high in PSMA and therefore, PSMA is observed in high expression
levels in prostate epithelial cells. As the PSMA is a
membrane-bound antigen with high prostate tissue specificity, it
has become a major prostate cancer marker for metastatic
castrate-resistant prostate cancer and other types of cancer
associated with the prostate. PSMA is a promising target for the
treatment of metastatic prostate cancer (52), although it is not absolutely
specific to prostate tissue (53).
Even though PSMA is a promising immunotherapeutic target for
metastatic prostate cancer, using it as an exclusive target can
lead to different types of autoimmune disorders as other cells also
express the surface glycoprotein. The PSCA is also a cell membrane
glycoprotein that is highly expressed in the prostate. Its
expression is upregulated in cases of prostate tumors. In addition
to being highly expressed in the prostate, it is also expressed in
the kidneys and urinary bladder as illustrated by Silva et
al (53), as well as in the
placenta, colon, and stomach (52,54,55).
Likewise, exclusive targeting of only the PSCA in the treatment of
cancer would also lead to the induction of an autoimmune disorder.
Therefore, a careful combinatorial approach that targets PSCA and
PSMA is expected to increase prostate cancer targeting and reduce
the reactivity against healthy tissues expressing either antigen
alone (55,56). Feldmann et al (57) designed a Universal CAR that serves
as the effector module activated by a target module, which has
specificity for PSCA or PSMA.
CART-cells target PSMA
The application of engineered hybrid antigen
receptors on T-cells have been effective in the treatment of a
number of hematological types of cancer (37). While engineered CART-cells have seen
effective improvements in the treatment of hematological cancer,
its application in solid tumors has proven to be difficult. The
impediment surrounding the success of CART-cell therapy against
solid tumors may be due to the following factors: i) The lack of a
unique tumor-associated antigen in the majority of different types
of cancer; ii) inefficient trafficking of CART-cells to tumor
sites; iii) heterogeneous expression of the targeted antigen(s)
leading to outgrowth of antigen-negative tumor variants; iv)
inadequate supply of growth factors (including IL-2); v) the
presence of immunosuppressive agents; and vi) the metabolically
hostile tumor microenvironment (23).
Targeting prostate tumors using mAbs is a technique
that has been employed for years. mAbs have proven valuable in
cancer treatment by delivering cytotoxic agents to the cancer cells
and PSMA serves as an ideal antigen target in prostate cancer. J591
is noted to be the first IgG mAb developed to target the
extracellular domain of PSMA and has been humanized to allow for
repeated dosing in patients. Prostate cancer cytotoxicity has been
reported to occur when there is a coupling of the J591 mAb with
ricin A (19,49).
Buhler et al (58,59)
demonstrated that the use of the PSMA × CD3 bispecific diabody to
selectively activate PSMA-specific CD8+ and
CD4+T-cells and to recruit them to the tumor site
produced efficient inhibition of tumor growth in a xenograft model
which further revealed the effective elimination of human prostate
cancers in in vitro and in vivo trials. Another
approach for PSMA-specific targeting is based on engineered T-cells
expressing chimeric anti-PSMA immunoglobulin-TCR constructs which
has been demonstrated to specifically lyse PSMA-expressing prostate
cancer cells and retard tumor growth in a mouse xenograft model
(60).
CART-cell persistence and trafficking
The efficacy of T-cell function depends on its
ability to be directed to the target site and persist. The survival
rate or time period of infused modified T-cells (including CART)
determines its efficiency in fighting and killing cancerous tumors.
While first-generation CARs have a poor persistence rate, the
second and third generations have been demonstrated to exhibit
higher persistence rates (23,61).
Activation of CAR with two or more co-stimulatory domains has been
reported to enhance the persistence of infused CART-cells. The
persistence of CART-cells can also be improved through
lymphodepleting chemotherapy and conditioning chemotherapy, which
removes suppressive cells including myeloid cells and regulatory
T-cells. Administration of IL-2 can also help improve the
persistence of CART-cells (41).
Furthermore, for effective tumor eradication to occur, CART-cells
should be able to migrate to tumor sites in large numbers and
persist for a long period of time. The homing potential of
CART-cells to target sites is called trafficking, an essential
requirement for T-cell-tumor eradication. Homing of CART-cells has
been observed to be enhanced by transducing T-cells with the
chemokine receptor expressing gene IL-8Rβ which recognizes
chemokine ligand 1 expressed in melanomas (23).
Immunosuppressive agents
Immunosuppressive agents are cells or molecules that
partially or completely prevent immune response. Immune suppression
occurs to maintain tolerance to self-cells and prevent the
development of autoimmune diseases. Immunosuppressive molecules
have been demonstrated to be present in markedly high levels at
tumors sites, which shields the tumor from immune surveillance and
subsequent attack. Immunosuppressive agents that have been reported
to protect tumors include regulatory T-cells (Tregs) (62,63),
transforming growth factor-β (TGF-β) (64) and myeloid-derived suppressor cells
(65), together with other
immunosuppressive molecules. To escape immunosuppressive effects,
genetic modification to CARs can help improve T-cell resistance to
immunosuppression. The incorporation of co-stimulatory molecules
including CD28 and inhibitory cytokines into CARs can assist
T-cells to become more resistant to Tregs and TGF-β as well as
other associated immunosuppressive molecules (24,41,66,67).
Individuals with recurrent prostate cancer normally
undergo androgen deprivation therapy, a technique, which stimulates
and activates the overexpression of a serine/threonine kinase,
known as protein kinase B, to regulate cancer cell growth and
survival. A combination therapy of androgen deprivation and
CART-cell infusion can protect CARs against immunosuppression
(68,69). In addition, CART-cells can be
further improved to express cytokines such as IL-2, IL-12, TNF-α
and IFNγ in order to escape immunosuppression (70–72).
Advantages of the CART-cell
CART-cell MHC-independent recognition of tumor
antigens allows for universal application as it can recognize
surface antigens including proteins, carbohydrates and glycolipids
making it difficult for the target tumor to escape antibody
recognition (32,73,74).
The CART-cell has a broad range of antigen targeting potential
compared with normal T-cells due to its ability to produce large
numbers of tumor specific T-cells in a moderately short period of
time which gives it an advantage in clinical applications (37,38,75).
Furthermore, there is reduced autoimmune and cytotoxic responses
when the correct gene is inserted into the T-cell (7).
Challenges associated with CART-cell
therapy
Toxicity of CART-cells
The infusion of CART-cells into cancer patients is
associated risks regardless of the number of benefits. CART-cells
have been proven to be successful in a number of the clinical
trials. However, the majority of the time it also led to expected
and unexpected toxicities including cytokine release syndrome
(CRS), neurologic toxicity, ‘on target/off tumor’ recognition, and
anaphylaxis (76). CRS has been
noted as one of the prevalent adverse effects that arises from CART
infusion. The activation of infused autologous CART-cells produces
inflammatory cytokines that could produce mild to severe CRS and
can be life threatening (77).
Clinical features, as noted by Bonifant et al (76) and Lee et al (78), include high fever, malaise, fatigue,
myalgia, nausea, anorexia, tachycardia/hypotension, capillary leak,
cardiac dysfunction, renal impairment, hepatic failure and
disseminated intravascular coagulation.
In addition to CRS, another form of toxicity likely
to arise from CART-cell infusion is the ‘On-target/off-tumor
recognition’. This occurs as the targeted antigen on the tumor may
not be restricted only to the tumor cells, but may also be present
on certain normal cells (79).
Activated CART-cells attacking normal self-cells can lead to
autoimmune disease and organ/tissue damage.
Insertional oncogenesis
The primary concern associated with engineered
T-cells is the insertion or activation of oncogenes in the host's
DNA. CART-cells carry a foreign gene transferred through a viral
vector with high efficiency, including lentiviral vectors,
adenovirus carrier and adeno-associated virus vectors. Integration
of the target gene into T-cells may possibly trigger oncogenesis;
however, no insertional oncogenesis has been recorded with respect
to CART-cells due to the high efficiency vector delivery.
Nevertheless, when the gene is redirected to a different site and
does not integrate into the specific site, it could lead to
insertional mutagenesis (76,80).
Hostile tumor microenvironment
Another challenge impeding the improvement of
CART-cell therapy in tumors involves the hostile microenvironment
that surrounds tumors. The presence of immunosuppressive cells and
increased inflammatory activity around tumors hinders exposure to
T-cell activity. Tumor surroundings are known to harbor
substantially high concentrations of reactive oxygen species (ROS),
which impedes antitumor function. T-cells co-expressing catalase
are hypothesized to perform efficiently in such tumor sites. To
confirm this, Ligtenberg et al (81) investigated catalase-CART-cells and
reported that catalase reduced the oxidative state of the tumor
with less accumulation of ROS. The catalase-CART-cell was also
observed to maintain antitumor activity (23,41,81).
Conclusion
The present review has demonstrated that compared
with normal T-cells and first-generation CART-cells, the second and
third-generation CARs have superior antitumor functions, providing
immunotherapy against prostate tumors and other target tissue
tumors. Recently, CART-cell therapy has greatly improved in the
treatment of hematological malignancies; however, developments are
still ongoing in targeting tumor-associated antigens for future
treatments. Due to the absence of specific antigens associated with
tumors, it is essential to design T-cells with more than one CAR as
this method will increase the efficiency and reduce the risk of
autoimmune associated diseases. More importantly, the use of
CART-cell therapy for the treatment of prostate cancer is
advantageous compared with surgery, radiotherapy and chemotherapy,
as it is associated with fewer cytotoxic effects. Therefore,
targeting the immune system with CART-cells may be important in
treating prostate cancer.
Even though CART-cell therapy demonstrates immense
promise as a potential cure for cancer, this system also has
potentially life-threatening complications including CRS and the US
FDA has registered safety concerns for its application. Although
the FDA has approved a number of CART therapy drugs for certain
cases of B-cell lymphoma, a number of life-threatening complication
cases have been reported in clinical trials because different CARs
and delivery methods elicit different cytotoxic responses. Another
challenge researchers face is the Federal common rule that provides
guidance on human-subject research with CART-cell therapy, which
limits its wide application and development. A detailed discussion
on the regulatory policy for this technique is essential for its
application in the biomedical industry and scientific community.
However this is beyond the scope of this review paper.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31271272 and
31600952), the Jiangsu Natural Science Foundation (grant no.
BK20151347) and the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HS and FY conceived the study. KU and FAP researched
the literature, and draft the manuscript. HS and FY critically
revised the article. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arruebo M, Vilaboa N, Sáez-Gutierrez B,
Lambea J, Tres A, Valladares M and González-Fernández Á: Assessment
of the evolution of cancer treatment therapies. Cancers.
3:3279–3330. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ethun CG, Bilen MA, Jani AB, Maithel SK,
Ogan K and Master VA: Frailty and cancer: Implications for oncology
surgery, medical oncology, and radiation oncology. CA Cancer J
Clin. 67:362–377. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alfarouk KO, Stock C-M, Taylor S, Walsh M,
Muddathir AK, Verduzco D, Bashir AH, Mohammed OY, Elhassan GO,
Harguindey S, et al: Resistance to cancer chemotherapy: Failure in
drug response from ADME to P-gp. Cancer Cell Int. 15:712015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu H, Lv L and Yang K: Chemotherapy
targeting cancer stem cells. Am J Cancer Res. 5:880–893.
2015.PubMed/NCBI
|
|
5
|
Levitzki A: Targeting the immune system to
fight cancer using chemical receptor homing vectors carrying
polyinosine/cytosine (PolyIC). Front Oncol. 2:42012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen ZY, Ma F, Huang H and He CY:
Synthetic immunity to break down the bottleneck of cancer
immunotherapy. Sci Bull. 60:977–985. 2015. View Article : Google Scholar
|
|
7
|
Smith AJ, Oertle J, Warren D and Prato D:
Chimeric antigen receptor (CAR) T cell therapy for malignant
cancers: Summary and perspective. J Cell Immunother. 2:59–68. 2016.
View Article : Google Scholar
|
|
8
|
Alberts B, Johnson A, Lewis J, Raff M,
Roberts K and Walter P: Lymphocytes and the cellular basis of
adaptive immunity. New York: Garland Science; 2002
|
|
9
|
Mak TW and Saunders ME: The immune
response: Basic and clinical principles. Academic Press; 2005
|
|
10
|
Dembic Z: The cytokines of the immune
system: The role of cytokines in disease related to immune
response. Academic Press; 2015, View Article : Google Scholar
|
|
11
|
Pennock ND, White JT, Cross EW, Cheney EE,
Tamburini BA and Kedl RM: T cell responses: Naive to memory and
everything in between. Adv Physiol Educ. 37:273–283. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hassanipour-Azgomi S,
Mohammadian-Hafshejani A, Ghoncheh M, Towhidi F, Jamehshorani S and
Salehiniya H: Incidence and mortality of prostate cancer and their
relationship with the human development index worldwide. Prostate
Int. 4:118–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haas GP, Delongchamps N, Brawley OW, Wang
CY and de la Roza G: The worldwide epidemiology of prostate cancer:
Perspectives from autopsy studies. Can J Urol. 15:3866–3871.
2008.PubMed/NCBI
|
|
15
|
Brawley OW: Trends in prostate cancer in
the United States. J Natl Cancer Inst Monogr. 2012:152–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cotter K, Konety B and Ordonez MA:
Contemporary management of prostate cancer. F1000Res. 5:2016.
|
|
17
|
Lynch JH, Batuello JT, Crawford ED,
Gomella LG, Kaufman J, Petrylak DP and Joel AB: Therapeutic
strategies for localized prostate cancer. Rev Urol. 3 Suppl
2:S39–S48. 2001.PubMed/NCBI
|
|
18
|
Mottet N, Bellmunt J, Bolla M, Briers E,
Cumberbatch MG, De Santis M, Fossati N, Gross T, Henry AM, Joniau
S, et al: EAU-ESTRO-SIOG guidelines on prostate cancer. Part 1:
Screening, diagnosis, and local treatment with curative intent. Eur
Urol. 71:618–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bander NH, Nanus DM, Milowsky MI,
Kostakoglu L, Vallabahajosula S and Goldsmith SJ: Targeted systemic
therapy of prostate cancer with a monoclonal antibody to
prostate-specific membrane antigen. Semin Oncol. 30:667–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morrissey KM, Yuraszeck T, Li CC, Zhang Y
and Kasichayanula S: Immunotherapy and novel combinations in
oncology: Current landscape, challenges, and opportunities. Clin
Transl Sci. 9:89–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khalil DN, Budhu S, Gasmi B, Zappasodi R,
Hirschhorn-Cymerman D, Plitt T, De Henau O, Zamarin D, Holmgaard
RB, Murphy JT, et al: The new era of cancer immunotherapy:
Manipulating T-cell activity to overcome malignancy. Adv Cancer
Res. 128:1–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu S, Li A, Liu Q, Li T, Yuan X, Han X and
Wu K: Chimeric antigen receptor T cells: A novel therapy for solid
tumors. J Hematol Oncol. 10:782017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mirzaei HR, Rodriguez A, Shepphird J,
Brown CE and Badie B: Chimeric antigen receptors T cell therapy in
solid tumor: Challenges and clinical applications. Front Immunol.
8:18502017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mohammed S, Sukumaran S, Bajgain P,
Watanabe N, Heslop HE, Rooney CM, Brenner MK, Fisher WE, Leen AM
and Vera JF: Improving chimeric antigen receptor-modified T cell
function by reversing the immunosuppressive tumor microenvironment
of pancreatic cancer. Mol Ther. 25:249–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abate-Daga D and Davila ML: CAR models:
Next-generation CAR modifications for enhanced T-cell function. Mol
Ther Oncolytics. 3:160142016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai
P, Li B, Lin S, Wang S, Wu Q, et al: Incorporation of a hinge
domain improves the expansion of chimeric antigen receptor T cells.
J Hematol Oncol. 10:682017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sommermeyer D, Hill T, Shamah SM, Salter
AI, Chen Y, Mohler KM and Riddell SR: Fully human CD19-specific
chimeric antigen receptors for T-cell therapy. Leukemia.
31:2191–2199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Monnier PP, Vigouroux RJ and Tassew NG: In
vivo applications of single chain Fv (variable domain) (scFv)
fragments. Antibodies. 2:193–208. 2013. View Article : Google Scholar
|
|
29
|
Whitlow M, Bell BA, Feng S-L, Filpula D,
Hardman KD, Hubert SL, Rollence ML, Wood JF, Schott ME, Milenic DE,
et al: An improved linker for single-chain Fv with reduced
aggregation and enhanced proteolytic stability. Protein Eng.
6:989–995. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alfthan K, Takkinen K, Sizmann D,
Söderlund H and Teeri TT: Properties of a single-chain antibody
containing different linker peptides. Protein Eng. 8:725–731. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zidovetzki R, Rost B and Pecht I: Role of
transmembrane domains in the functions of B-and T-cell receptors.
Immunol Lett. 64:97–107. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dotti G, Gottschalk S, Savoldo B and
Brenner MK: Design and development of therapies using chimeric
antigen receptor-expressing T cells. Immunol Rev. 257:107–126.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Janeway CA Jr, Travers P, Walport M and
Shlomchik M: Principles of innate and adaptive immunity
immunobiologyGarland Science. New York: 2001
|
|
34
|
Alberts B, Johnson A, Lewis J, Raff M,
Roberts K and Walter P: Molecular Biology of the Cell. 4th edition.
Garland Science; New York, NY: 2002
|
|
35
|
Artyomov MN, Lis M, Devadas S, Davis MM
and Chakraborty AK: CD4 and CD8 binding to MHC molecules primarily
acts to enhance Lck delivery. Proc Natl Acad Sci USA.
107:16916–16921. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choi YE, Yu HN, Yoon CH and Bae YS:
Tumor-mediated down-regulation of MHC class II in DC development is
attributable to the epigenetic control of the CIITA type I
promoter. Eur J Immunol. 39:858–868. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Porter DL, Levine BL, Kalos M, Bagg A and
June CH: Chimeric antigen receptor-modified T cells in chronic
lymphoid leukemia. New Engl J Med. 365:725–733. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Porter DL, Levine BL, Bunin N, Stadtmauer
EA, Luger SM, Goldstein S, Loren A, Phillips J, Nasta S, Perl A, et
al: A phase 1 trial of donor lymphocyte infusions expanded and
activated ex vivo via CD3/CD28 costimulation. Blood. 107:1325–1331.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jacobson CA and Ritz J: Time to put the
CAR-T before the horse. Blood. 118:4761–4762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cartellieri M, Bachmann M, Feldmann A,
Bippes C, Stamova S, Wehner R, Temme A and Schmitz M: Chimeric
antigen receptor-engineered T cells for immunotherapy of cancer. J
Biomed Biotechnol. 2010:9563042010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Haji-Fatahaliha M, Hosseini M, Akbarian A,
Sadreddini S, Jadidi-Niaragh F and Yousefi M: CAR-modified T-cell
therapy for cancer: An updated review. Artif Cells Nanomed
Biotechnol. 44:1339–1349. 2016.PubMed/NCBI
|
|
42
|
Kulemzin S, Kuznetsova V, Mamonkin M and
Taranin A: Engineering chimeric antigen receptors. Acta Naturae.
9:6–14. 2017.PubMed/NCBI
|
|
43
|
Chmielewski M and Abken H: TRUCKs: The
fourth generation of CARs. Expert Opin Biol Ther. 15:1145–1154.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chmielewski M, Kopecky C, Hombach AA and
Abken H: IL-12 release by engineered T cells expressing chimeric
antigen receptors can effectively Muster an antigen-independent
macrophage response on tumor cells that have shut down tumor
antigen expression. Cancer Res. 71:5697–5706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Scarfò I and Maus MV: Current approaches
to increase CAR T cell potency in solid tumors: Targeting the tumor
microenvironment. J Immunother Cancer. 5:282017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Anassi E and Ndefo UA: Sipuleucel-T
(provenge) injection: The first immunotherapy agent (vaccine) for
hormone-refractory prostate cancer. P T. 36:197–202.
2011.PubMed/NCBI
|
|
47
|
Westdorp H, Sköld AE, Snijer BA, Franik S,
Mulder SF, Major PP, Foley R, Gerritsen WR and de Vries IJM:
Immunotherapy for prostate cancer: Lessons from responses to
tumor-associated antigens. Front Immunol. 5:1912014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ge C, Li R, Song X and Qin S: Advances in
evidence-based cancer adoptive cell therapy. Chin Clin Oncol.
6:182017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kiessling A, Wehner R, Füssel S, Bachmann
M, Wirth MP and Schmitz M: Tumor-associated antigens for specific
immunotherapy of prostate cancer. Cancers. 4:193–217. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Murphy GP, Greene TG, Tino WT, Boynton AL
and Holmes EH: Isolation and characterization of monoclonal
antibodies specific for the extracellular domain of prostate
specific membrane antigen. J Urol. 160:2396–2401. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sokoloff RL, Norton KC, Gasior CL, Marker
KM and Grauer LS: A dual-monoclonal sandwich assay for
prostate-specific membrane antigen: Levels in tissues, seminal
fluid and urine. Prostate. 43:150–157. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Saeki N, Gu J, Yoshida T and Wu X:
Prostate stem cell antigen: A Jekyll and Hyde molecule? Clin Cancer
Res. 16:3533–3538. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Silver DA, Pellicer I, Fair WR, Heston W
and Cordon-Cardo C: Prostate-specific membrane antigen expression
in normal and malignant human tissues. Clin Cancer Res. 3:81–85.
1997.PubMed/NCBI
|
|
54
|
NCBI: PSCA prostate stem cell antigen.
NCBI. 2010.
|
|
55
|
Kloss CC, Condomines M, Cartellieri M,
Bachmann M and Sadelain M: Combinatorial antigen recognition with
balanced signaling promotes selective tumor eradication by
engineered T cells. Nat Biotechnol. 31:71–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Duong CP, Westwood JA, Berry LJ, Darcy PK
and Kershaw MH: Enhancing the specificity of T-cell cultures for
adoptive immunotherapy of cancer. Immunotherapy. 3:33–48. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Feldmann A, Arndt C, Bergmann R, Loff S,
Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F,
Albert S, et al: Retargeting of T lymphocytes to PSCA- or PSMA
positive prostate cancer cells using the novel modular chimeric
antigen receptor platform technology ‘UniCAR’. Oncotarget.
8:31368–31385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bühler P, Molnar E, Dopfer EP, Wolf P,
Gierschner D, Wetterauer U, Schamel WW and Elsässer-Beile U:
Target-dependent T-cell activation by coligation with a PSMA × CD3
diabody induces lysis of prostate cancer cells. J Immunother.
32:565–573. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bühler P, Wolf P, Gierschner D, Schaber I,
Katzenwadel A, Schultze-Seemann W, Wetterauer U, Tacke M, Swamy M,
Schamel W, et al: A bispecific diabody directed against
prostate-specific membrane antigen and CD3 induces T-cell mediated
lysis of prostate cancer cells. Cancer Immunol, Immunother.
57:43–52. 2008. View Article : Google Scholar
|
|
60
|
Ma Q, Safar M, Holmes E, Wang Y, Boynton
AL and Junghans RP: Anti-prostate specific membrane antigen
designer T cells for prostate cancer therapy. Prostate. 61:12–25.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dai H, Wang Y, Lu X and Han W: Chimeric
antigen receptors modified T-cells for cancer therapy. J Natl
Cancer Inst. 108:djv4392016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kiniwa Y, Miyahara Y, Wang HY, Peng W,
Peng G, Wheeler TM, Thompson TC, Old LJ and Wang RF:
CD8+ Foxp+ regulatory T cells mediate
immunosuppression in prostate cancer. Clin Cancer Res.
13:6947–6958. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vergati M, Cereda V, Madan RA, Gulley JL,
Huen NY, Rogers CJ, Hance KW, Arlen PM, Schlom J and Tsangsa KY:
Analysis of circulating regulatory T cells in patients with
metastatic prostate cancer pre- versus post-vaccination. Cancer
Immunol Immunother. 60:197–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cao Z and Kyprianou N: Mechanisms
navigating the TGF-β pathway in prostate cancer. Asian J Urol.
2:11–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lopez-Bujanda Z and Drake CG:
Myeloid-derived cells in prostate cancer progression: Phenotype and
prospective therapies. J Leukocyte Biol. 102:393–406. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Koehler H, Kofler D, Hombach A and Abken
H: CD28 costimulation overcomes transforming growth
factor-β-mediated repression of proliferation of redirected human
CD4+ and CD8+ T cells in an antitumor cell
attack. Cancer Res. 67:2265–2273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Loskog A, Giandomenico V, Rossig C, Pule
M, Dotti G and Brenner M: Addition of the CD28 signaling domain to
chimeric T-cell receptors enhances chimeric T-cell resistance to T
regulatory cells. Leukemia. 20:1819–1828. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ha S, Ruoff R, Kahoud N, Franke TF and
Logan SK: Androgen receptor levels are upregulated by Akt in
prostate cancer. Endocr Relat Cancer. 18:245–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mikhailova M, Wang Y, Bedolla R, Lu XH,
Kreisberg JI and Ghosh PM: AKT regulates androgen
receptor-dependent growth and PSA expression in prostate cancer.
Adv Exp Med Biol. 617:397–405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gosmann C, Frazer IH, Mattarollo SR and
Blumenthal A: IL-18, but not IL-12, induces production of IFN-γ in
the immunosuppressive environment of HPV16 E7 transgenic
hyperplastic skin. J Invest Dermatol. 134:2562–2569. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jiang T, Zhou C and Ren S: Role of IL-2 in
cancer immunotherapy. Oncoimmunology. 5:e11634622016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhao J, Zhao J and Perlman S: Differential
effects of IL-12 on Tregs and non-Treg T cells: Roles of IFN-γ,
IL-2 and IL-2R. PLoS One. 7:e462412012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rivière I and Sadelain M: Chimeric antigen
receptors: A cell and gene therapy perspective. Mol Ther.
25:1117–1124. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sadelain M, Brentjens R and Rivière I: The
basic principles of chimeric antigen receptor (CAR) design. Cancer
Discov. 3:388–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang C, Liu J, Zhong JF and Zhang X:
Engineering CAR-T cells. Biomark Res. 5:222017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bonifant CL, Jackson HJ, Brentjens RJ and
Curran KJ: Toxicity and management in CAR T-cell therapy. Mol Ther
Oncolytics. 3:160112016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Grupp SA, Kalos M, Barrett D, Aplenc R,
Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et
al: Chimeric antigen receptor-modified T cells for acute lymphoid
leukemia. New Engl J Med. 368:1509–1518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lee DW, Gardner R, Porter DL, Louis CU,
Ahmed N, Jensen M, Grupp SA and Mackall CL: Current concepts in the
diagnosis and management of cytokine release syndrome. Blood.
124:188–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Curran KJ, Pegram HJ and Brentjens RJ:
Chimeric antigen receptors for T cell immunotherapy: Current
understanding and future directions. J Gene Med. 14:405–415. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tey SK: Adoptive T-cell therapy: Adverse
events and safety switches. Clin Transl Immunol. 3:e172014.
View Article : Google Scholar
|
|
81
|
Ligtenberg MA, Mougiakakos D, Mukhopadhyay
M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H and Kiessling
R: Coexpressed catalase protects chimeric antigen
receptor-redirected t cells as well as bystander cells from
oxidative stress-induced loss of antitumor activity. J Immunol.
196:759–766. 2016. View Article : Google Scholar : PubMed/NCBI
|














