Introduction
Bladder cancer (BC) is a potentially
life-threatening malignancy that is considered one of the most
expensive tumors in terms of treatment and medical care (1–3). After
prostate cancer, it is the second most common type of urological
cancer and ranks 10th among the most common types of cancer around
the globe (4). It has been estimated
that there were 549,393 new cases of BC and 199,922 deaths
resulting from this disease worldwide in 2018, according to a
report from the International Agency for Research on Cancer
(4). The primary histological subtype
of human BC is transitional cell carcinoma, which occurs at a high
rate of >90% (5). Moreover, 70–80%
of new cases are diagnosed as non-muscle invasive BC (NMIBC)
(6–8).
Despite undergoing transurethral resection of bladder tumor, up to
50% of patients with NMIBC can experience relapse, and 20% continue
to progress within 5 years (9). The
risk factors for NMIBC progression include tumor stage, grade,
size, number and recurrence rate (10). A multidisciplinary approach for the
reduction of the tumor recurrence rate, involving surgical
intervention combined with radiotherapy, chemotherapy or
immunotherapy, is a therapeutic option in patients with BC
(10,11). Nevertheless, such a treatment tactic
still has unsatisfactory clinical effects. Thus, the development of
novel therapies and enhancement of responses to current therapies
is urgently required, in order to improve clinical outcomes.
Autophagy, a fundamental evolutionary catabolic
process, plays an important role in the maintenance of cellular
environmental homeostasis by degrading and recycling damaged
cytoplasmic components, including macromolecules and organelles
(12). This process can be activated
by various cellular stress conditions, including nutrient
deprivation (13,14), organelle damage (15), radiotherapy or chemotherapy (15,16), to
satisfy cellular needs and promote cell survival. The role of
autophagy in cancer development and therapy appears to be
paradoxical depending on the context. During the initial stage of
cancer development, autophagy serves a major role in tumor
suppression by maintaining genomic integrity and preventing
proliferation and inflammation (17).
However, after the establishment of cancer, cancer cells may
utilize autophagy to survive cellular stresses in the adverse
microenvironment (18). In addition,
autophagy is considered a double-edged sword with regard to the
treatment of cancer. Cellular protective autophagy induced by
anticancer therapy plays an important role in therapy resistance
among cancer cells. Thus, autophagy inhibition can increase the
sensitivity of cancer cells to anticancer therapy. On the contrary,
autophagy promotion may induce type II programmed cell death, which
is referred to as autophagic cell death, similar to apoptosis
(19). In this review, the current
knowledge on autophagy modulation in BC development and treatment
is summarized, with the aim of exploring novel and potential
therapeutic targets.
Autophagy overview
Autophagy is a lysosomal degradation process in
which damaged, long-lived cytoplasmic proteins and organelles are
swallowed by double-membrane autophagic vesicles termed
autophagosomes (20). The fusion of
an autophagosome with a lysosome results in the formation of an
autolysosome, which provides an acidic environment for hydrolytic
enzymes to degrade the internalized cellular components (21). Autophagy is a biological phenomenon
widely occurring in eukaryotic cells. In addition, it is a
fascinating process regulated by multiple autophagy-related
proteins.
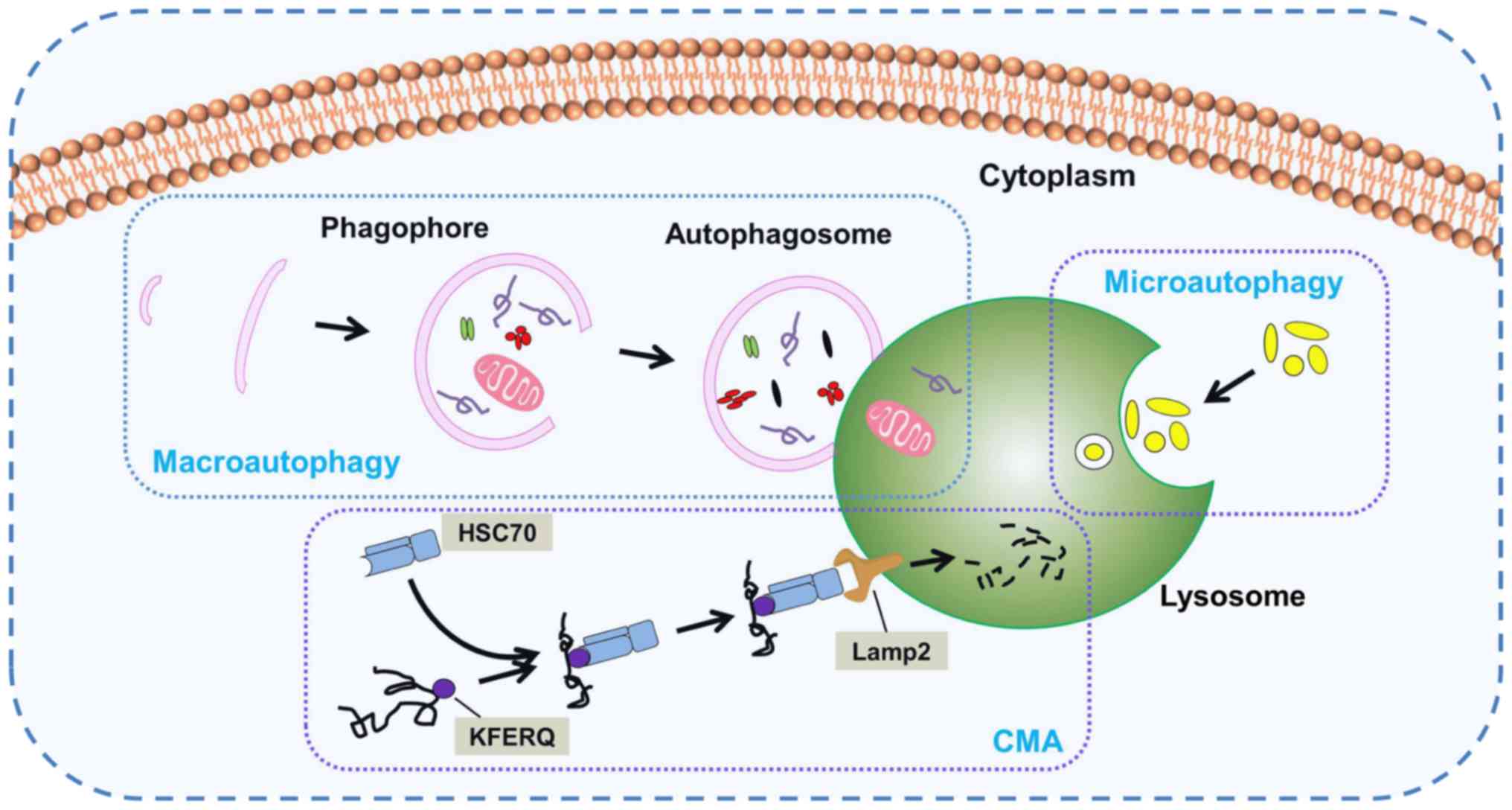
According to the mode of transport for intracellular
components to the lysosome, the following three important subtypes
of autophagy in mammals have been identified: Macroautophagy,
microautophagy and chaperone-mediated autophagy (CMA) (Fig. 1). Macroautophagy, which is usually
referred to as autophagy, is the main autophagy pathway. During
this pathway, the cell forms a double-membrane structure termed a
phagophore, which develops into an autophagosome. Subsequently,
cytoplasmic components are engulfed into autophagosomes and
delivered to lysosomes for fusion and degradation. Nevertheless,
there is a fundamental difference between micro- and
macroautophagy. In microautophagy, the lysosome membrane
autonomously changes shape via invagination or bulging to engulf
cytoplasmic components directly. CMA is a selective and unique
autophagic process that selectively discerns and degrades substrate
proteins containing the specific KFERQ pentapeptide sequence. Heat
shock cognate protein of 70 kDa, a cytosolic chaperone protein, is
a key component in CMA able to discern the KFERQ motif and deliver
it to lysosome associated membrane protein type 2 for degradation
(22,23). Owing to its easily recognizable
double-membrane vesicles, macroautophagy is the best-characterized
variant of autophagy. The unique morphological features of
macroautophagy have been the focus of numerous studies since 1950
(24). Moreover, macroautophagy is
the central hub of autophagy-lysosomal pathways. In consideration
of the large volume of studies existing on macroautophagy in
mammals, the rest of this review will focus on macroautophagy,
hereafter referred to as autophagy.
Autophagy is a dynamic process, and is referred to
as autophagic flux. The complete process of autophagic flux can be
divided into the following four steps: i) Activation and
elongation, ii) maturation, iii) lysosome fusion and iv)
degradation (Fig. 2) (25). The initial stage of phagophore
formation is the most complex step in the process of autophagic
flux, in which various functional units are involved, including the
mammalian uncoordinated-51-like protein kinase (ULK1/ATG1) complex,
the PI3K complex and two ubiquitin-like conjugation systems
[autophagy-related protein 12 homolog (ATG12) and ATG8] (Fig. 3) (26,27).
Presumably, ATG8-phosphatidylethanolamine (PE), a useful marker of
autophagic membranes, is involved in almost all steps of the
autophagic flux, particularly the later steps, including phagophore
expansion and lysosome fusion (26).
 | Figure 3.Signaling pathways of autophagy. mTOR
kinase is a pivotal molecule in the mTORC1 complex that plays an
important role in the regulation of autophagy. Autophagy activation
is triggered by decreased activity of the mTORC1 complex due to the
activation of AMPK or p53 signaling. The decreased activity of
mTORC1, an inhibitor of the mammalian ULK1 complex, leads to the
increase the activity of the ULK1 complex, which subsequently
initiates the formation of phagophore in conjunction with the PI3K
complex. The elongation and maturation of the phagophore is
dependent on two ubiquitin-like conjugation systems (ATG12 and
ATG8), which involve multiple autophagy proteins, including ATG5,
ATG16 and LC3. ATG, autophagy-related protein homolog; mTORC1, mTOR
complex 1; AMPK, AMP-activated protein kinase; ULK1,
uncoordinated-51-like protein kinase; LC3, microtubule-associated
protein light chain 3; PE, phosphatidylethanolamine; TTI1,
Tel2-interacting protein 1; TEL2, telomere length regulation
protein TEL2; DEPTOR, DEP domain-containing mTOR-interacting
protein; RAPTOR, regulatory-associated protein of mTOR; PRAS40,
proline-rich Akt substrate of 40 kDa; MLST8, mTOR-associated
protein LST8 homolog; MAPK, mitogen-activated protein kinase;
FIP200, fusion-inhibiting peptide 200. |
In addition, more than 30 autophagy-related genes
and homologous proteins have been identified to be essential for
autophagy (28). Therefore, the
activation of autophagy in response to various stresses leads to
alterations in the expression levels of autophagy-related proteins,
including microtubule-associated protein light chain 3 (LC3), p62
and Beclin1. As a member of the ATG8 family in mammals, LC3 is a
specific protein marker of autophagosome formation. In the ATG8
ubiquitin-like conjugation system, LC3 is first cleaved by ATG4, a
specific cysteine protease, to expose the C-terminal cysteine.
Subsequently, LC3 is conjugated to PE by ATG7 and ATG3,
facilitating the conversion of cytosolic LC3-I to membrane-bound
LC3-II (Fig. 3) (29). p62, also referred to as sequestosome
1, is a multi-functional signaling protein implicated in cell
proliferation, survival and death. Recent studies have revealed
that p62 plays a critical role in the autophagic proteolytic
cascade by binding to and delivering ubiquitinated contents to
autophagosomes. p62 interacts with membrane protein LC3 via its
LC3-interacting region motif (30–32).
Therefore, p62 degradation occurs in autolysosomes. On the
contrary, autophagy inhibition is often accompanied by upregulation
of p62 expression. Beclin1, a multi-domain protein, regulates the
cross talk between autophagy and apoptosis. Additionally, the class
III PI3K complex has been identified as an important signaling hub
that regulates the autophagy-flux and balances the processes of
autophagy and apoptosis. Therefore, as a PI3K-III regulatory
subunit, Beclin1 plays an important role in the regulation of
autophagy through phosphorylation and ubiquitination (33).
Autophagy signaling pathways
Autophagy is induced under conditions of cellular
stress, including hypoxia, oxidative stress, nutrient deprivation,
organelle damage and radiotherapy or chemotherapy in order to meet
cellular needs and promote cell survival. Moreover, autophagy
levels are regulated by these cellular stresses through different
signaling pathways. Subsequently, once these stresses are
eliminated by autophagy upregulation, levels return to normal.
mTOR signaling pathway
mTOR is a 300-kDa serine/threonine protein kinase
present in mTOR complex 1 (mTORC1) (34). It plays a crucial role in regulating
cellular growth, proliferation and protein synthesis. mTORC1 lies
upstream of the ULK1 complex and negatively regulates this complex,
resulting in autophagy suppression. Specifically, ULK1 is
phosphorylated at Ser757 by mTORC1 and then inactivated under
nutrient-enriched conditions. On the contrary, nutrient deficiency
or mTORC1 inhibitors can lead to the activation of ULK1. Activated
ULK1 phosphorylates ATG14 at Ser29 and Beclin1 at Ser14 to
stimulate the kinase activity of the class III PI3K, resulting in
phagophore and autophagosome formation (35). In addition, numerous upstream
signaling pathways that regulate mTORC1 activity may affect the
intracellular autophagy levels (Fig.
3).
Mitogen stimulation signaling
pathway
The mitogen stimulation signal pathway is
characterized by its dependence on serine/threonine kinase Akt.
Class I PI3K is stimulated by multiple mitogen signals, including
activated receptor tyrosine kinases, activated oncogene Ras and G
protein-coupled receptors (36).
Activated class I PI3Ks phosphorylate phosphatidylinositol
4,5-bisphosphate to phosphatidylinositol 3,4,5-trisphosphate
(PIP3). PIP3 recruits Akt and phosphoinositide-dependent kinases-1
(PDK1), which bind to the cell membrane. Akt serine/threonine
kinases are phosphorylated by PDK1 and mTORC2 at Thr308 and Ser473,
respectively, resulting in their activation. The downstream
effector of the PI3K/Akt pathway is mTOR, which forms two
macromolecular complexes, mTORC1 and mTORC2. Akt activates mTORC1
via the inhibition of tuberous sclerosis complex proteins 1/2
(TSC1/2), thereby promoting Rheb (37). Subsequently, active mTORC1 inhibits
autophagy by hindering ULK1.
Energy signaling pathway
AMP-activated protein kinase (AMPK), a heterotrimer
comprising of an α-catalytic subunit and regulatory β- and
γ-subunits, is an evolutionarily conserved regulator of cellular
energy homeostasis (38). When the
cellular AMP/ATP ratio increases under conditions of cellular
stress, such as hypoxia and nutrient deprivation, AMP binds
directly to the γ-subunit, thereby activating AMPK. TSC2 is
phosphorylated by activated AMPK at Ser1345 and Ser1227,
subsequently leading to the inhibition of mTORC1 activity (39).
Amino acid signaling pathway
Amino acids are not only substrates of various
metabolic pathways, but also signal molecules that regulate signal
transduction pathways. The regulation of mTOR by amino acids is
associated with the PI3K/Akt independent pathway. Mitogen-activated
protein kinase kinase kinase kinase 3 (MAP4K3), a Ste20-related
kinase, has been identified as an upstream regulator of mTORC1 in
response to amino acids (40). Amino
acids can upregulate the activity of MAP4K3, which is essential for
the activation of S6 kinase and induce the phosphorylation of
eukaryotic initiation factor 4E-binding protein, an mTOR-regulated
inhibitor (40). Amino acids are
capable of regulating autophagy via several signaling pathways,
including the Ras/Raf-1/ERK1/2 pathway (41), the integrin/p38 mitogen-activated
protein kinase pathway (42), and the
mTOR signaling pathway, which is dominant compared with the other
pathways.
p53 signaling pathway
Numerous studies have shown that p53, an important
tumor suppressor, plays an important role in regulating glucose
homeostasis (43,44). In the case of glucose deficiency, p53
is able to induce autophagy through the mTOR and damage regulated
autophagy modulator (DRAM) pathways (45). Further studies have indicated that p53
activation is able to suppress mTOR activity and that this
regulation involves p53-dependent AMPK activation with the
subsequent activation of the TSC1/TSC2 complex (46–48).
Moreover, DRAM is a p53 target gene and encodes a lysosomal
protein that induces autophagy. Crighton et al (49) showed that p53 was able to induce
autophagy in a DRAM-dependent manner.
Beclin 1-related regulatory
pathways
At present, mTOR and Beclin1 are considered
significant signaling hubs in the context of autophagy. Beclin1,
which is described as the mammalian homolog of yeast ATG6, plays an
important role in the process of autophagosome nucleation. It
recruits class III PIK3/vacuolar sorting protein-34 to form a
regulated complex that generates phosphatidylinositol 3-phosphate
[PI(3)P]. Subsequently, certain proteins, including ATG8 and ATG12
complex, bind with PI(3)P-binding domains to modulate autophagosome
formation (50). Anti-apoptotic
protein Bcl-2 is able to bind to the N-terminal Bcl-2 homology 3
domain of Beclin1, thus inhibiting autophagy (51).
Autophagy regulation in BC
During tumor formation, autophagy plays a major role
in suppressing tumor initiation and development by maintaining
genomic integrity and preventing proliferation and inflammation
(17). In the present review, we
hypothesize that this is also the case for BC. To the best of our
knowledge, no studies that prove this hypothesis have been
published to date. Once a tumor has become established, tumor cells
can utilize autophagy to survive cellular stresses in the adverse
microenvironment. Zhu et al (52) demonstrated that ATG7 was notably
overexpressed in invasive BC and knockdown of this protein was able
to markedly inhibit BC invasion, suggesting that ATG7 was involved
in the regulation of BC development.
The role of autophagy appears to be paradoxical in
cancer therapy depending on the context. On one hand, inhibition of
autophagy may be employed to increase the cytotoxic effect of
treatments, including chemotherapy and radiotherapy (53–55). On
the other hand, excessive activation of autophagy may lead to
autophagic cell death, also known as programmed cell death type II,
which is similar to apoptosis and is defined as cell death in the
presence of lysosomes (56–60). Therefore, it is essential to identify
the role of autophagy in cancer cells in order to develop new
therapeutic agents.
Inhibiting protective autophagy in
BC
BC is a malignant tumor associated with high
morbidity and mortality, and a significant economic burden
associated with it (4). A
comprehensive treatment approach involving surgery combined with
chemoradiotherapy or immunotherapy is a therapeutic option to
reduce the tumor recurrence rate in patients with BC (10,11). Yet,
in spite of effective therapy, the majority of patients still
experience disease relapse and ultimately die of tumor metastasis
(61). Poor prognosis is often
attributed to resistance to various therapeutic interventions,
which is a distinguishing feature of cancer. Numerous studies
suggest that cancer cells may achieve resistance through a wide
variety of mechanisms, including cell intrinsic and extrinsic
factors, such as genetic heterogeneity (62), autophagy (19,53), tumor
microenvironment (63) and cancer
stem cells factors (64).
Furthermore, autophagy may affect the tumor microenvironment by
supplying cellular energy demands and preventing cytotoxicity under
stressful conditions such as hypoxia, oxidative stress,
inflammation and cytokine release. In addition, autophagy may have
an impact on the regulation of cancer stem cell homeostasis by
contributing to the maintenance of stemness (65). Given the importance of these
mechanisms, increasing interest has arisen in the development of
efficient therapeutic approaches based on the autophagy
regulation.
Autophagy may be inhibited at any stage of the
autophagic flux. In the last decade, many studies involving
autophagy mechanisms have been performed to identify chemical
inhibitors of autophagy, including chloroquine (CQ) and
3-methyladenine (3-MA). Numerous studies have revealed that
inhibition of protective autophagy via various approaches,
including pharmaceutical inhibitors (53,55,66,67),
RNA-interference agents (66,68) and natural bioactive compounds
(69,70) (Table I),
is able to increase the sensitivity of BC to therapeutic
interventions.
 | Table I.Autophagy inhibitors in bladder
cancer. |
Table I.
Autophagy inhibitors in bladder
cancer.
| Inhibitor | Mechanism of
action | Treatments combined
with inhibitor | Bladder cell
line | (Refs.) |
|---|
| Chloroquine | Lysosomal lumen
alkalizer | Cisplatin,
radiotherapy, lapatinib or gefitinib | EJ, T24, RT-112,
5637, J82 | (53,55,66,68) |
|
3-Methyladenine | PI3K inhibitor | Cisplatin,
Fangchinoline, lapatinib or gefitinib | RT-112, T24,
J82 | (55,67,68) |
| Icaritin | Protein synthesis
inhibitor | Epirubicin | 5637, T24 | (69) |
| Frondoside A | Protein synthesis
inhibitor | Cisplatin and
gemcitabine | RT112 | (70) |
| shRNA | Knockdown of
Beclin1 and ATG7/ATG12 | Cisplatin | 5637, T24 | (66) |
| siRNA | Suppression of
ATG12 | Lapatinib or
gefitinib | T24, J82 | (68) |
CQ, an anti-malarial drug, is the most frequently
used and proficient agent for the inhibition of autophagy.
Currently, CQ and its derivative hydroxychloroquine are the only
clinically available autophagy inhibitors approved by the US Food
and Drug Administration (71). These
drugs are weak lipophilic bases that have the ability to accumulate
in lysosomes, increasing lysosomal pH. Subsequently, the
alkalization of lysosomes prevents their fusion with autophagosomes
and inactivates lysosomal acidic proteases, thereby preventing
cargo degradation in autolysosomes. Studies have indicated that
CQ-mediated lysosomal dysfunction is able to sensitize cancer cells
to chemotherapy and radiotherapy, thus enhancing the anticancer
effect of this therapeutic method. A recent study in which BC cells
were treated with CQ in combination with cisplatin, demonstrated
that inhibition of autophagy enhanced the cytotoxicity of cisplatin
(55). Another study revealed the
same result that inhibition of cisplatin-induced autophagy using CQ
significantly increased BC cell sensitivity to cisplatin, hence
enhancing its cytotoxicity (66).
These findings suggest that autophagy is induced by cisplatin as a
protective mechanism in BC cells and that inhibition of autophagy
is able to significantly enhance chemosensitivity in
cisplatin-resistant cells. Moreover, it has also been proved that
autophagy inhibition by CQ is able to enhance the
radiosensitization efficiency in BC cells. Wang et al
(53) reported that radiotherapy
activated autophagy in BC cells and that subsequent protective
autophagy was strongly associated with radioresistance. In
addition, the combination of radiation and CQ induced synergistic
anticancer effects, as confirmed by evidence of enhanced apoptosis
rate, indicating that inhibition of autophagy contributes to the
enhancement of radiosensitization (53).
The second most widely used autophagy inhibitor is
3-MA, a class III PI3K inhibitor. PI3Ks are a diverse family of
lipid kinases that play important roles in cellular processes,
including cell proliferation, metabolism and autophagy regulation
(72,73). Class III PI3K, which represents one of
the three classes of PI3Ks in mammalian cells, is an activator of
autophagy that plays an important role in the early stages of
autophagosome formation (25). Fan
et al (67) performed a study
that aimed to assess the antitumor effects of fangchinoline (Fcn),
which is a natural product found in Stephania tetrandra, on
BC. They reported that Fcn was able to induce autophagy and
apoptosis in BC cells and that inhibition of autophagy by 3-MA
resulted in the enhancement of Fcn-induced apoptosis, evident by
the increased cleavage of caspase-3 (67). These results suggest that inhibition
of protective autophagy may enhance the apoptotic efficiency of
anticancer drugs.
In addition to the pharmaceutical inhibitors listed
in Table I, natural bioactive
compounds have also been proved to possess synergistic anticancer
effects with other agents by inhibiting autophagy. Icaritin, a
flavonol glycoside isolated from the genus Epimedium, is a
hydrolysate of the traditional Chinese herb icariin (69). Previous studies have demonstrated that
icaritin has the potential to be an effective anticancer agent by
promoting apoptosis, inhibiting cell proliferation and inducing the
cell cycle. Pan et al (69)
treated human BC cells with icaritin and/or epirubicin (EPI) to
investigate how icaritin plays a synergistic role in suppressing BC
development. The results indicated that the half maximal inhibitory
concentration values with regard to the inhibition of both BT5637
and T24 cell proliferation were significantly higher with icaritin
or EPI alone than in combination. Western blot analysis revealed
that icaritin not only downregulated the expression levels of major
autophagy proteins (ATG3, ATG5, ATG7 and ATG12), but also induced a
significant decrease in the LC3-II/LC3-I ratio, suggesting that
icaritin was able to enhance BC cell sensitivity to EPI through the
inhibition of EPI-induced protective autophagy (69). In addition, another bioactive
compound, marine triterpene glycoside frondoside A, extracted from
the sea cucumber Cucumaria frondosa, has been demonstrated
to exert an anticancer effect. Dyshlovoy et al (70) used frondoside A in combination with
cisplatin and gemcitabine and demonstrated that frondoside A was
able to enhance the anticancer capabilities of the two standard
chemotherapeutic agents in BC RT112 cells by inhibiting protective
autophagy.
An increasing number of studies focusing on
RNA-interference have been performed to further explore the effects
of protective autophagy inhibition on anticancer therapy. Kang
et al (68) demonstrated that
genetic inhibition of autophagy by disabling ATG12, which is
one of the key transcription genes for autophagosome formation and
completion, was able to potentiate the anticancer effects of
epidermal growth factor receptor (EGFR) inhibitors on BC cells.
Small interfering RNA (siRNA) was transfected into BC cells to
suppress autophagy through blockage of ATG12. Subsequently,
the transfected cells were treated with the EGFR inhibitors
lapatinib or gefitinib. The results indicated that inhibition of
autophagy by ATG12-siRNA synergistically increased apoptotic
cell death when combined with EGFR inhibitors, as confirmed by flow
cytometry analysis, suggesting that autophagy acted as a protective
mechanism in BC cells (68). In
addition, Lin et al (66)
combined the autophagy inhibitor short hairpin RNA (shRNA)-based
lentivirus with cisplatin and demonstrated that inhibition of
autophagy through knockdown of ATG7/ATG12 or Beclin1 using
shRNA was able to synergistically enhance the anticancer
capabilities of cisplatin in BC 5637 and T24 cells. The results
showed that inhibition of autophagy by shRNA increased
cisplatin-induced apoptosis, which was evident by the enhanced
cleavage of caspase-3, indicating that the combination of cisplatin
with autophagy inhibitors was able to increase the sensitivity of
BC cells to cisplatin, thus enhancing the cytotoxicity of this
chemotherapeutic drug (66).
In summary, autophagy has been identified as a
critical mechanism contributing to cancer therapy resistance.
Moreover, autophagy may be regarded as a potential target for
therapies involving autophagy inhibitors in combination with
conventional therapeutics. The numerous studies mentioned above
indicate that inhibition of protective autophagy may be able to
increase the sensitivity of BC to chemotherapy or radiotherapy,
providing important information on the effective regulation of
autophagy during cancer treatment.
Activation of autophagic cell death in
BC
Autophagy appears to play a contradictory role in
cancer therapy depending on the context. Apart from cytoprotective
autophagy, the other primary and opposing form of autophagy, which
may facilitate cell death either alone or in association with
apoptosis, is cytotoxic autophagy. Functionally, cytotoxic
autophagy is capable of decreasing the number of viable cells
and/or reducing clonogenic survival upon treatment (74). Gewirtz (74) hypothesized that the contradictory
functions of autophagy may be associated with specific signaling
pathways and/or substrates for the autophagic machinery. Moreover,
it has been widely acknowledged that high autophagy levels may
induce type II programmed cell death, which is also known as
autophagic cell death (56). On the
basis of this concept, a number of studies have been performed to
investigate the therapeutic potential of autophagic cell death
activation in different fields of cancer treatment. In BC,
autophagic cell death has been noted in chemotherapy with pazopanib
(57), biological therapy with
Cheliensisine A-fluoride (ChlA-F) (58) and with alternative therapies (59,60,75)
(Table II).
| Table II.Activators of autophagic cell death
in bladder cancer. |
Table II.
Activators of autophagic cell death
in bladder cancer.
| Activator | Mechanism of
action | Signaling pathways
involved | Bladder cell
line | (Refs.) |
|---|
| Pazopanib | Increasing
cathepsin B activity | ERK1/2 | 5637, J82 | (57) |
| ChlA-F | Upregulating
Sestrin-2 expression | Sestrin-2 | RT4, T24T,
UMUC3 | (58) |
| Ubenimex | Akt agonist | Akt | 5637, RT112 | (75) |
| Salidroside | Suppressing PI3K
and p-Akt |
Autophagy/PI3K/Akt | T24 | (59) |
| Tetrandrine | Upregulating p-AMPK
and downregulating p-mTOR | AMPK/mTOR | T24, 5637 | (60) |
A better understanding of the potential role of
vascular endothelial growth factor (VEGF) in tumor formation and
progression has led to the addition of more effective agents to the
therapeutic field of multiple tumor types. Among these
anti-angiogenic agents, pazopanib, an oral tyrosine kinase
inhibitor that targets the VEGF receptor, has been approved for
metastatic renal cell carcinoma and soft tissue sarcoma (57). However, to the best of our knowledge,
the development of pazopanib for BC is still in the initial stages
of clinical research. A previous study, in which BC cells were
treated with pazopanib, indicated that this anti-angiogenic agent
was able to induce autophagy by increasing ERK1/2 phosphorylation,
as evaluated by LC3-II/LC3-I ratio increase, acidic vesicular
organelle formation, p62 protein degradation and autophagic flux
(57). In addition, pazopanib has
been proven to induce autophagic cell death, which is markedly
reverted by 3-MA. The aforementioned study confirmed that
autophagic cell death induced by pazopanib was associated with
increased cathepsin B activity. Finally, comparative gene
expression analysis in BC cells at the molecular level indicated
that pazopanib induced cytotoxic autophagy by affecting the
expression of autophagic genes, such as the upregulation of
ATG9B and downregulation of the tumor protein p73 gene
(57).
ChlA-F, a novel derivative of Cheliensisin A
isolated from Goniothalamus cheliensis Hu, has been
identified as a potential anticancer drug due to its enhanced water
solubility and chemical stability. Hua et al (58) treated human BC cells and the normal
urothelial cell line UROtsa with ChlA-F to investigate the
anticancer activity and molecular mechanisms of its biological
effects. The results revealed that ChlA-F significantly inhibited
BC cell growth and led to a significant increase in the
LC3-II/LC3-I ratio, indicating that ChlA-F was able to activate
autophagy in human BC cells and may possess antitumor properties.
Interestingly, the inhibition of human BC cell growth by ChlA-F was
significantly reversed by combining treatment with bafilomycin A1,
a fusion inhibitor of autophagosomes and lysosomes (58). Moreover, Hua et al (58) evaluated the effects of ChlA-F on
autophagy-related protein expression to illustrate the molecular
mechanisms involved in ChlA-F-induced autophagy. They demonstrated
that ChlA-F treatment notably increased Sestrin-2 (SESN2) protein
expression; however, no prominent effects on the expression levels
of other autophagy-related proteins were reported. Further studies
confirmed that ChlA-F treatment specifically induced SESN2
expression by increasing its transcription and mRNA stability
(58). These findings suggest that
the activation of cytotoxic autophagy via specific signaling
pathways contributes to the anticancer effects of ChlA-F, therefore
providing new information for therapeutic alternatives against
human BC.
With the exception of pazopanib and ChlA-F, a number
of alternative BC therapies have also been proven to provide
anticancer effects through autophagy induction. One example is
ubenimex, a broad-spectrum antitumor agent that has been used in
adjuvant therapy. Aminopeptidase N (APN), known as the cell surface
molecule CD13, is involved in several cell life activities,
including cell survival, blood pressure regulation, angiogenesis,
invasion and metastasis of tumor cells (76). Therefore, as an APN inhibitor,
ubenimex is a promising agent for cancer treatment. Ubenimex has
been shown to inhibit the proliferation, migration and invasion of
BC cells by downregulating APN expression levels and inducing
autophagy through inhibition of the Akt signaling pathway (75). On the contrary, autophagy inhibition
with 3-MA reversed the antiproliferative properties of ubenimex,
indicating that ubenimex was capable of inducing autophagic cell
death in BC cells (75).
Salidroside, a bioactive tyrosine-derived phenolic
compound isolated from Rhodiola rosea, possesses properties
against fatigue, anoxia, cardiovascular disease and cancer
(77). Furthermore, salidroside has
been proven to induce autophagic cell death along with apoptosis in
BC cells. It has been demonstrated that salidroside causes
apoptosis through the autophagy/PI3K/Akt and matrix
metalloproteinase-9 signaling pathways (59). Kou et al (60) indicated that autophagy induced by
tetrandrine may synergistically enhance apoptosis in human BC cells
by regulating the AMPK/mTOR signaling pathway.
Conclusions
Although a large number of studies have been
performed to confirm novel treatment methods for BC over the past
few decades, the management and long-term survival rate of patients
with BC have remained relatively stagnant without any significant
improvement in clinical outcomes. Autophagy has been shown to be a
complex cellular process with contrasting effects in the treatment
of BC. Undoubtedly, the aforementioned findings prove that the
application of autophagy activators and inhibitors provides further
insight for the development of novel therapeutic options against
human BC. To the best of our knowledge, since the multidisciplinary
approach of surgery combined with radiotherapy, or chemotherapy is
usually considered in patients with BC, autophagy inhibitors,
including CQ and 3-MA, may be more beneficial in BC treatment due
to their ability to increase cancer cell sensitivity to
chemotherapy or radiotherapy. Therefore, a better understanding of
the role of autophagy in BC treatment is crucial for the selection
of effective drugs to target the autophagic pathway.
Acknowledgments
Not applicable.
Funding
This work was supported by Graduate Innovation Fund
of Jilin University (grant no. 101832018C068).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
FL and HZ conceived and designed the study and
prepared the manuscript. HG, MF and XR were responsible for the
literature search, data visualization and analysis. YY and BL
retrieved the relevant literature and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Perlis N, Krahn MD, Boehme KE, Alibhai
SMH, Jamal M, Finelli A, Sridhar SS, Chung P, Gandhi R, Jones J, et
al: The bladder utility symptom scale: A novel patient reported
outcome instrument for bladder cancer. J Urol. 200:283–291. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steurer S, Singer JM, Rink M, Chun F,
Dahlem R, Simon R, Burandt E, Stahl P, Terracciano L, Schlomm T, et
al: MALDI imaging-based identification of prognostically relevant
signals in bladder cancer using large-scale tissue microarrays.
Urol Oncol. 32:1225–1233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson DC, Greene PS and Nielsen ME:
Surgical advances in bladder cancer: At what cost? Urol Clin North
Am. 42:235–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen C, Hu L, Chen Y and Hou J: The
prognostic value of histological subtype in patients with
metastatic bladder cancer. Oncotarget. 8:28408–28417.
2017.PubMed/NCBI
|
|
6
|
Chandrasekar T and Evans CP: Autophagy and
urothelial carcinoma of the bladder: A review. Investig Clin Urol.
57 (Suppl 1):S89–S97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kelly JD, Tan WS, Porta N, Mostafid H,
Huddart R, Protheroe A, Bogle R, Blazeby J, Palmer A, Cresswell J,
et al: BOXIT-A randomised phase III placebo-controlled trial
evaluating the addition of celecoxib to standard treatment of
transitional cell carcinoma of the bladder (CRUK/07/004). Eur Urol.
75:593–601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan WS, Tan WP, Tan MY, Khetrapal P, Dong
L, deWinter P, Feber A and Kelly JD: Novel urinary biomarkers for
the detection of bladder cancer: A systematic review. Cancer Treat
Rev. 69:39–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cambier S, Sylvester RJ, Collette L,
Gontero P, Brausi MA, van Andel G, Kirkels WJ, Silva FC,
Oosterlinck W, Prescott S, et al: EORTC nomograms and risk groups
for predicting recurrence, progression, and disease-specific and
overall survival in non-muscle-invasive stage Ta-T1 urothelial
bladder cancer patients treated with 1–3 years of maintenance
bacillus calmette-guérin. Eur Urol. 69:60–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderson B: Bladder cancer: Overview and
management. Part 2: Muscle-invasive and metastatic bladder cancer.
Br J Nurs. 27:S8–S20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anderson B: Bladder cancer: Overview and
disease management. Part 1: Non-muscle-invasive bladder cancer. Br
J Nurs. 27:S27–S37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dower CM, Wills CA, Frisch SM and Wang HG:
Mechanisms and context underlying the role of autophagy in cancer
metastasis. Autophagy. 14:1110–1128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Long M and McWilliams TG: Monitoring
autophagy in cancer: From bench to bedside. Semin Cancer Biol. Jul
15–2019.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zachari M, Gudmundsson SR, Li Z, Manifava
M, Shah R, Smith M, Stronge J, Karanasios E, Piunti C,
Kishi-Itakura C, et al: Selective autophagy of mitochondria on a
ubiquitin-endoplasmic-reticulum platform. Dev Cell. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng Y, Ren X, Hait WN and Yang JM:
Therapeutic targeting of autophagy in disease: Biology and
pharmacology. Pharmacol Rev. 65:1162–1197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bishop E and Bradshaw TD: Autophagy
modulation: A prudent approach in cancer treatment? Cancer
Chemother Pharmacol. 82:913–922. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizumura K, Cloonan S, Choi ME, Hashimoto
S, Nakahira K, Ryter SW and Choi AM: Autophagy: Friend or foe in
lung disease? Ann Am Thorac Soc. 13 (Suppl 1):S40–S47.
2016.PubMed/NCBI
|
|
21
|
Jang M, Park R, Kim H, Namkoong S, Jo D,
Huh YH, Jang IS, Lee JI and Park J: AMPK contributes to
autophagosome maturation and lysosomal fusion. Sci Rep.
8:126372018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mukherjee A, Patel B, Koga H, Cuervo AM
and Jenny A: Selective endosomal microautophagy is
starvation-inducible in Drosophila. Autophagy. 12:1984–1999. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marinković M, Šprung M, Buljubašić M and
Novak I: Autophagy modulation in cancer: Current knowledge on
action and therapy. Oxid Med Cell Longev. 2018:80238212018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Galluzzi L, Baehrecke EH, Ballabio A, Boya
P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P,
Colombo MI, et al: Molecular definitions of autophagy and related
processes. EMBO J. 36:1811–1836. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taylor MA, Das BC and Ray SK: Targeting
autophagy for combating chemoresistance and radioresistance in
glioblastoma. Apoptosis. 23:563–575. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang S, Shin KD, Kim JH and Chung T:
Autophagy-related (ATG) 11, ATG9 and the phosphatidylinositol
3-kinase control ATG2-mediated formation of autophagosomes in
Arabidopsis. Plant Cell Rep. 37:653–664. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vicencio JM, Ortiz C, Criollo A, Jones AW,
Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al:
The inositol 1,4,5-trisphosphate receptor regulates autophagy
through its interaction with Beclin 1. Cell Death Differ.
16:1006–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Döring T, Zeyen L, Bartusch C and Prange
R: Hepatitis B virus subverts the autophagy elongation complex
Atg5-12/16L1 and does not require Atg8/LC3 lipidation for viral
maturation. J Virol. 92:2018. View Article : Google Scholar
|
|
30
|
Zaffagnini G, Savova A, Danieli A, Romanov
J, Tremel S, Ebner M, Peterbauer T, Sztacho M, Trapannone R,
Tarafder AK, et al: Phasing out the bad-How SQSTM1/p62 sequesters
ubiquitinated proteins for degradation by autophagy. Autophagy.
14:1280–1282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cha-Molstad H, Yu JE, Feng Z, Lee SH, Kim
JG, Yang P, Han B, Sung KW, Yoo YD, Hwang J, et al:
p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule
pathway which modulates autophagosome biogenesis. Nat Commun.
8:1022017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kwon DH, Park OH, Kim L, Jung YO, Park Y,
Jeong H, Hyun J, Kim YK and Song HK: Insights into degradation
mechanism of N-end rule substrates by p62/SQSTM1 autophagy adapter.
Nat Commun. 9:32912018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Menon MB and Dhamija S: Beclin 1
phosphorylation-at the center of autophagy regulation. Front Cell
Dev Biol. 6:1372018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grunwald DS, Otto NM, Park JM, Song D and
Kim DH: GABARAPs and LC3s have opposite roles in regulating ULK1
for autophagy induction. Autophagy. 1–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ersahin T, Tuncbag N and Cetin-Atalay R:
The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. 11:1946–1954.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Menon S, Dibble CC, Talbott G, Hoxhaj G,
Valvezan AJ, Takahashi H, Cantley LC and Manning BD: Spatial
control of the TSC complex integrates insulin and nutrient
regulation of mTORC1 at the lysosome. Cell. 156:771–785. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dite TA, Ling NXY, Scott JW, Hoque A,
Galic S, Parker BL, Ngoei KRW, Langendorf CG, O'Brien MT, Kundu M,
et al: The autophagy initiator ULK1 sensitizes AMPK to allosteric
drugs. Nat Commun. 8:5712017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen W, Pan Y, Wang S, Liu Y, Chen G, Zhou
L, Ni W, Wang A and Lu Y: Cryptotanshinone activates AMPK-TSC2 axis
leading to inhibition of mTORC1 signaling in cancer cells. BMC
Cancer. 17:342017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Findlay GM, Yan L, Procter J, Mieulet V
and Lamb RF: A MAP4 kinase related to Ste20 is a nutrient-sensitive
regulator of mTOR signalling. Biochem J. 403:13–20. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pattingre S, Bauvy C and Codogno P: Amino
acids interfere with the ERK1/2-dependent control of macroautophagy
by controlling the activation of Raf-1 in human colon cancer HT-29
cells. J Biol Chem. 278:16667–16674. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Prick T, Thumm M, Köhrer K, Häussinger D
and Vom Dahl S: In yeast, loss of Hog1 leads to osmosensitivity of
autophagy. Biochem J. 394:153–161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawauchi K, Araki K, Tobiume K and Tanaka
N: p53 regulates glucose metabolism through an IKK-NF-kappaB
pathway and inhibits cell transformation. Nat Cell Biol.
10:611–618. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Goldstein I, Yizhak K, Madar S, Goldfinger
N, Ruppin E and Rotter V: p53 promotes the expression of
gluconeogenesis-related genes and enhances hepatic glucose
production. Cancer Metab. 1:92013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Itahana Y and Itahana K: Emerging roles of
p53 family members in glucose metabolism. Int J Mol Sci.
19:2018.PubMed/NCBI
|
|
46
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dando I, Cordani M and Donadelli M: Mutant
p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life.
68:722–726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Budanov AV and Karin M: p53 target genes
sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling.
Cell. 134:451–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang
Y, Lee JH, Hawke D, Wang Y, Xia Y, et al: Phosphoglycerate kinase 1
phosphorylates Beclin1 to induce autophagy. Mol Cell.
65:917.e6–931.e6. 2017. View Article : Google Scholar
|
|
51
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhu J, Tian Z, Li Y, Hua X, Zhang D, Li J,
Jin H, Xu J, Chen W, Niu B, et al: ATG7 promotes bladder cancer
invasion via autophagy-mediated increased ARHGDIB mRNA stability.
Adv Sci (Weinh). 6:18019272019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang F, Tang J, Li P, Si S, Yu H, Yang X,
Tao J, Lv Q, Gu M, Yang H and Wang Z: Chloroquine enhances the
radiosensitivity of bladder cancer cells by inhibiting autophagy
and activating apoptosis. Cell Physiol Biochem. 45:54–66. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Piya S, Andreeff M and Borthakur G:
Targeting autophagy to overcome chemoresistance in acute
myleogenous leukemia. Autophagy. 13:214–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schlütermann D, Skowron MA, Berleth N,
Böhler P, Deitersen J, Stuhldreier F, Wallot-Hieke N, Wu W, Peter
C, Hoffmann MJ, et al: Targeting urothelial carcinoma cells by
combining cisplatin with a specific inhibitor of the
autophagy-inducing class III PtdIns3K complex. Urol Oncol.
36:160.e1–160.e13. 2018. View Article : Google Scholar
|
|
56
|
Chiao MT, Cheng WY, Yang YC, Shen CC and
Ko JL: Suberoylanilide hydroxamic acid (SAHA) causes tumor growth
slowdown and triggers autophagy in glioblastoma stem cells.
Autophagy. 9:1509–1526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Santoni M, Amantini C, Morelli MB,
Liberati S, Farfariello V, Nabissi M, Bonfili L, Eleuteri AM,
Mozzicafreddo M, Burattini L, et al: Pazopanib and sunitinib
trigger autophagic and non-autophagic death of bladder tumour
cells. Br J Cancer. 109:1040–1050. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hua X, Xu J, Deng X, Xu J, Li J, Zhu DQ,
Zhu J, Jin H, Tian Z, Huang H, et al: New compound ChlA-F induces
autophagy-dependent anti-cancer effect via upregulating Sestrin-2
in human bladder cancer. Cancer Lett. 436:38–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li T, Xu K and Liu Y: Anticancer effect of
salidroside reduces viability through autophagy/PI3K/Akt and MMP-9
signaling pathways in human bladder cancer cells. Oncol Lett.
16:3162–3168. 2018.PubMed/NCBI
|
|
60
|
Kou B, Liu W, Xu X, Yang Y, Yi Q, Guo F,
Li J, Zhou J and Kou Q: Autophagy induction enhances
tetrandrine-induced apoptosis via the AMPK/mTOR pathway in human
bladder cancer cells. Oncol Rep. 38:3137–3143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Alfred Witjes J, Lebret T, Compérat EM,
Cowan NC, De Santis M, Bruins HM, Hernández V, Espinós EL, Dunn J,
Rouanne M, et al: Updated 2016 EAU guidelines on muscle-invasive
and metastatic bladder cancer. Eur Urol. 71:462–475. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jamal-Hanjani M, Quezada SA, Larkin J and
Swanton C: Translational implications of tumor heterogeneity. Clin
Cancer Res. 21:1258–1266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sun Y: Tumor microenvironment and cancer
therapy resistance. Cancer Lett. 380:205–215. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Carnero A, Garcia-Mayea Y, Mir C, Lorente
J, Rubio IT and LLeonart ME: The cancer stem-cell signaling network
and resistance to therapy. Cancer Treat Rev. 49:25–36. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sharif T, Martell E, Dai C, Kennedy BE,
Murphy P, Clements DR, Kim Y, Lee PW and Gujar SA: Autophagic
homeostasis is required for the pluripotency of cancer stem cells.
Autophagy. 13:264–284. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lin JF, Lin YC, Tsai TF, Chen HE, Chou KY
and Hwang TI: Cisplatin induces protective autophagy through
activation of BECN1 in human bladder cancer cells. Drug Des Devel
Ther. 11:1517–1533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fan B, Zhang X, Ma Y and Zhang A:
Fangchinoline induces apoptosis, autophagy and energetic impairment
in bladder cancer. Cell Physiol Biochem. 43:1003–1011. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kang M, Lee KH, Lee HS, Jeong CW, Kwak C,
Kim HH and Ku JH: Concurrent autophagy inhibition overcomes the
resistance of epidermal growth factor receptor tyrosine kinase
inhibitors in human bladder cancer cells. Int J Mol Sci. 18:2017.
View Article : Google Scholar
|
|
69
|
Pan XW, Li L, Huang Y, Huang H, Xu DF, Gao
Y, Chen L, Ren JZ, Cao JW, Hong Y and Cui XG: Icaritin acts
synergistically with epirubicin to suppress bladder cancer growth
through inhibition of autophagy. Oncol Rep. 35:334–342. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dyshlovoy SA, Madanchi R, Hauschild J,
Otte K, Alsdorf WH, Schumacher U, Kalinin VI, Silchenko AS, Avilov
SA, Honecker F, et al: The marine triterpene glycoside frondoside A
induces p53-independent apoptosis and inhibits autophagy in
urothelial carcinoma cells. BMC Cancer. 17:932017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yu X, Long YC and Shen HM: Differential
regulatory functions of three classes of phosphatidylinositol and
phosphoinositide 3-kinases in autophagy. Autophagy. 11:1711–1728.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang X, Liu Y, Liu W, Zhang Y, Guo F,
Zhang L, Cui M, Liu S and Wu R: Ubenimex, an APN inhibitor, could
serve as an anti-tumor drug in RT112 and 5637 cells by operating in
an Akt-associated manner. Mol Med Rep. 17:4531–4539.
2018.PubMed/NCBI
|
|
76
|
Amin SA, Adhikari N and Jha T: Design of
aminopeptidase N inhibitors as anti-cancer agents. J Med Chem.
61:6468–6490. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Torrens-Spence MP, Pluskal T, Li FS,
Carballo V and Weng JK: Complete pathway elucidation and
heterologous reconstitution of rhodiola salidroside biosynthesis.
Mol Plant. 11:205–217. 2018. View Article : Google Scholar : PubMed/NCBI
|