Introduction
De novo or recurrent metastatic prostate
cancers are initially amenable to androgen deprivation therapy
(ADT). However, the majority of these lesions inevitably relapse
and evolve into incurable and lethal castration-resistant prostate
cancer (CRPC) (1–4). CRPC is extremely resistant to all
types of currently available therapeutic regimens, posing a
formidable clinical challenge (5,6).
Therefore, it is imperative to identify promising druggable targets
that yield significant clinical benefits and to develop effective
treatment strategies that overcome therapeutic resistance.
Cyclosporin A (CsA) is a potent immunosuppressive
agent that has been widely used in organ transplantation (7,8).
Previous studies have demonstrated that CsA exerts antitumor or
chemosensitizing activity against different types of cancer,
including prostate cancer (9,10). In
addition, CsA has been investigated in clinical trials for its
potential to treat several cancers (11–13).
However, the mechanism of antitumor action of CsA is poorly
understood, particularly in the context of prostate cancer.
Therefore, further research is needed to elucidate the mechanism of
action of CsA in prostate cancer, which may lead to identifying
potential targets for therapeutic intervention.
E2F8 is a member of the atypical E2F family that
plays a crucial role in embryonic development (14). Emerging evidence demonstrates that
E2F8 functions as an oncogene by mediating the hallmarks of cancer,
including sustaining proliferative signaling and resisting cell
death (15–17). However, it remains to be elucidated
whether E2F8 is a promising therapeutic target for the treatment of
prostate cancer. In addition, the signaling mechanism regulating
E2F8 expression remains elusive.
The present study performed transcriptomic analyses
to assess the mechanism of the antitumor action of CsA in prostate
cancer. E2F8 was identified as a master transcription factor that
induced oncogenic phenotypes and determined clinical outcome in
prostate cancer. The results will provide insight into the
development of E2F8-targeted therapy for the treatment of prostate
cancer.
Materials and methods
Cell culture and reagents
PC-3 (cat. no. CRL-1435), LNCaP (cat. no. CRL-1740),
DU145 (cat. no. HTB-81) and 22Rv1 (cat. no. CRL-2505) prostate
cancer cell lines were purchased from the American Type Culture
Collection. Cells were cultured in RPMI 1640 (PC-3, LNCaP and
22Rv1) or DMEM (DU145) containing 10% fetal bovine serum,
penicillin (100 U/ml) and streptomycin (100 µg/ml). All cell
culture reagents were obtained from HyClone (Cytiva). All other
reagents not specified were supplied by MilliporeSigma. All cell
lines tested negative for mycoplasma contamination using the
Mycoplasma PCR Detection kit (Intron Biotechnology, Inc.). These
cell lines have been authenticated in the three years using short
tandem repeat analysis.
Tumor xenograft experiments
Male Balb/C nude mice (4–5 weeks old; 18–20 g; n=10)
were purchased from Charles River Laboratories Japan, Inc. Mice
were housed in laminar flow cabinets under specific pathogen-free
condition (37°C; 12-h light/dark cycle; 60% relative humidity and
free access to food and water). Mice were anesthetized by the
inhalation of isoflurane (Terrell; Piramal Critical Care Inc.) in
oxygen (2 l/min): Induction with 4% isoflurane for 2 min in an
anesthetic induction chamber, followed by maintenance with 2%
isoflurane for 1–2 min after transferring to a nose cone. The 22Rv1
cells (5×106 cells; 100 µl of cell suspension) were
subcutaneously injected into the right flank of each mouse. The
humane endpoints were when the largest tumor size was >20 mm in
diameter. None of the mice reached the endpoints of the present
study. When the tumor reached ~180 mm3, mice were
randomly divided into two groups (five in each group) and
intraperitoneally injected with 20 mg/kg CsA or DMSO, every other
day for 14 days. After 14 days, mice were humanely sacrificed under
overdosed isoflurane. Mice were placed into a chamber filled with
vapor of the anesthetic isoflurane until respiration ceased (within
2 min) and the tumors were excised. The tumor volume was calculated
using the following formula: V=(L × W2) × 0.5 (V, volume
of tumor; L, length of tumor; W, width of tumor). All animal
experiments were performed in accordance with protocols approved by
the Institutional Animal Care and Use Committee of the Asan
Institute for Life Sciences at the ASAN Medical Center, University
of Ulsan College of Medicine, Seoul (approval no. 2021-13-234).
MTT assay
After cells were transfected with the short
interfering (si)RNAs for 48 h, MTT assay was performed to assess
cell growth according to the manufacturer's instructions
(MilliporeSigma). The assay was quantitated by measuring the
absorbance at 570 nm on a BioTek SynergyMx microplate reader
(BioTek Instruments, Inc.).
Colony formation assay
PC-3 (2–5×103 cells/well), LNCaP
(8×103 cells/well), DU145 (1–3×103
cells/well) and 22Rv1 (2–5×103 cells/well) cells were
seeded into 6-well plates. Cells were treated with CsA (10 µM) or
siRNAs for 9 days (once every 3 days). The cells were fixed with
3.7% paraformaldehyde and stained with 0.5% crystal violet
(MilliporeSigma) for 15 min at room temperature. The number of
colonies, defined as >50 cells/colony, was counted using ImageJ
(1.8.0 172; National Institutes of Health).
Microarray experiment
PC-3 cells were treated with 10 µM CsA in 0.1%
ethanol (vehicle) for 24 h. Microarray experiments were performed
as we previously described (18,19).
The microarray data are available through the Gene Expression
Omnibus database (GSE109505; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109505).
Rigorous data preprocessing and single channel array normalization
(SCAN) were performed and microarray probes were mapped to gene
symbols as previously described (20–22).
Of the 20,661 mapped genes, 17,629 protein-coding genes were
selected for further analyses. The internal clusters were validated
through hierarchical clustering and principal component analysis
(PCA) (20,21).
Collection of public microarray data
and analysis of the prostate cancer-specific transcriptional
interactome
The GSE67157 and GSE109505 datasets were used to
construct the PC-3 cell-specific interactome. Rigorous
preprocessing, such as quality control testing, normalization and
batch effect adjustment, was conducted as previously described
(21,23,24).
Of the common 11,506 genes from two datasets, 5,000 genes of high
variance were selected for implementing the Algorithm for the
Reconstruction of Gene Regulatory Networks (ARACNe) (20,21).
The list of human transcription factors (TFs) was obtained from the
Animal Transcription Factor Database 2.0 (AnimalTFDB 2.0) (23) and used for Master Regulator Analysis
(MRA). ARACNe preprocessing and MRA-Fisher's exact test (FET)
analysis were run in geWorkbench software version 2.6.0 (http://wiki.c2b2.columbia.edu/workbench/i-ndex.php/Home)
as described in our previous reports (20,21,25,26).
GSE3325 and GSE35988 data were used to assess the changes in gene
expression profiles during prostate cancer progression. A more
detailed description is provided in Fig. S1 and its legend.
Significance analysis of microarrays
(SAM) and gene set enrichment analysis (GSEA)
SAM was used to identify differentially expressed
genes (DEGs) from the GSE67157, GSE109505, GSE3325 and GSE35988
data (27). A tuning parameter,
delta of 0.4, was optimized to give the cutoff for significance
with the estimation of the false discovery rate (FDR)
q-value threshold of 0.01. GSEA (Hallmark Gene Set from the
Molecular Signature Database) was performed to obtain a biological
interpretation of clinically significant CsA-specific DEGs
(28). Universal concept signature
scores were calculated for E2F8 target genes and concept signature
enrichment analysis performed for deep functional assessment of the
pathways. A more detailed description is given in Fig. S1 and its legend.
Reverse transcription (RT)PCR and
western blotting
PC-3, LNCaP, DU145 and 22Rv1 cells were treated with
CsA at the indicated concentrations for various times. Total RNA
was extracted using the RNeasy Mini Kit (Qiagen GmbH) and reverse
transcribed with SuperScriptIII First-Strand (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturers'
instructions. RT-PCR and real-time PCR were performed using
specific primers for E2F8 (16,21),
MELK (29), β-actin (30) or 18S (31). The primer sequences are provided in
Table SI. The thermocycling
conditions for PCR were: 95°C for 5 min, followed by 31 cycles of
95°C for 45 sec, 51°C (MELK and β-actin) or 54°C (E2F8) for 45 sec,
and 72°C for 45 sec. PCR products were separated by 1% agarose gel
electrophoresis and visualized with SYBR Safe DNA Gel Stain
(Invitrogen; Thermo Fisher Scientific, Inc.). Agarose gel
electrophoresis images were acquired with Gel Documentation XR
System (Bio-Rad Laboratories, Inc.). Real-time PCR was conducted
using StepOne Real-Time PCR system (Thermo Fisher Scientific
Inc.).
For western blotting, the crude extracts were
prepared by incubation with RIPA buffer (50 mM Tris-HCl, pH 7.4,
150 mM NaCl, 1% triton X-100, 0.5% sodium deoxycholate, 0.1% SDS,
0.5 M EDTA) containing protease and phosphatase inhibitor cocktails
(MilliporeSigma). The protein concentrations were determined by BCA
assay kit (Thermo Fisher Scientific, Inc.). The samples (10–30 µg
for each) were resolved in 7% (E2F8), 8% (MELK), or 10% (E2F1 and
β-tubulin) SDS-PAGE gels and transferred onto NC membranes (Bio-Rad
Laboratories, Inc.). The membranes were blocked using 5% w/v
skimmed milk (BD Difco; BD Biosciences) in Tris-buffered saline
containing 0.1% Tween-20 (TBST) for 1 h at room temperature. The
membranes were probed with the anti-E2F8 (1:2,000; cat. no.
A303-039A; Bethyl Laboratories, Inc.), anti-MLEK (1:1,000; cat. no.
2274S; Cell Signaling Technology, Inc.), anti-E2F1 (1:500; cat. no.
sc-193; Santa Cruz, Inc.), and anti-β-tubulin (1:5,000; cat. no.
T4026; MilliporeSigma, Inc.) antibodies for 1 h at room
temperature. After washing three times with TBST, the membranes
were incubated with a goat anti-Rabbit IgG-HRP antibody (1:5,000;
cat. no. A120-101P, Bethyl Laboratories, Inc.) or a goat anti-Mouse
IgG-HRP antibody (1:5,000; cat. no. A90-116P, Bethyl Laboratories,
Inc.) for 1 h at room temperature. The signals were determined by
the enhanced chemiluminescence reaction (ECL; Amersham; Cytiva).
X-ray films were scanned and analyzed using EPSON Scan Software
(EPSON Expression 11000XL, Seiko Epson Corporation). The data shown
are representative of at least four independent experiments. Full
scan images are shown in Appendix
S1.
siRNA transfection
Cells were transfected with 50 nM siControl
(21) or siRNAs against E2F8
[siE2F8-1 and siE2F8-2 (21)] or
MELK [siMELK-1 (29) and siMELK-2
(32)] for 48 h at 37°C using
Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). The siRNAs were obtained from Genolution
Pharmaceuticals Inc. (Seoul, Korea). The siRNA sequences are
provided in Table SII. The time
interval between transfection and subsequent experiments was 48 h
or 72 h for MTT assay, 9 days for colony formation assay (see also
Figure legends).
Bayesian network analysis
The Cancer Genome Atlas (TCGA)-Prostate
Adenocarcinoma (PRAD) gene expression data were obtained from Xena
Functional Genomics Explorer (https://tcga.xenahubs.net/download/TCGA.PRAD.sampleMap/HiSeqV2.gz).
The E2F8 gene signature, which was inferred in MRA-FET analysis,
was extracted and the continuous value of expression levels in each
gene was discretized to equal-width bins (8 bins) using the
unsupervised discretization method available in the
Information-Theoretic Measures (Infotheo) R version 4.1.1
(http://www.R-project.org/) (11). The fast greedy equivalence search
(FGES)-discrete algorithm was used to identify E2F8-interacting
genes (33). A more detailed
description is given in Fig. S1
and its legend.
Coexpression analysis
The gene expression data of TCGA-primary tumor
samples were downloaded using the TCGA Biolinks package (ver.
2.14.1, http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html).
Before normalization, duplicated FFPE samples were removed. Gene
reads were normalized in counts per million (CPM) using the edgeR
package (ver. 3.28.1, http://bioconductor.org/packages/release/bioc/html/edgeR.html)
and log2 transformation (CPM+1). The Ensembl gene IDs
were mapped into HGNC symbols using the biomaRt package (ver.
2.42.1, http://bioconductor.org/packages/release/bioc/html/biomaRt.html)
and the expression data of androgen receptor (AR), MELK and E2F8
extracted for coexpression analysis.
The coordinated TF activities
associated with therapeutic response
TCGA-PRAD RNA-seq raw bam files (n=554) were
downloaded from NCI Genomic Data Commons (GDC) data portal
(https://portal.gdc.cancer.gov/). Patient
response data for chemotherapy or radiotherapy were provided by
Xena Functional Genomics Explorer (https://xenabrowser.net/hub/) GDC Hub (34). When the therapeutic response data
were mapped to the available PRAD RNA-seq samples, 313 patients
were identified as having a complete response (CR), 34 patients as
partial response (PR), 29 patients as progressive disease (PD) and
27 patients as stable disease (SD). Integrated System for Motif
Activity Response Analysis (ISMARA, http://ismara.unibas.ch/mara/) was used to infer
regulatory networks and sample-specific TF motif activities from
gene expression data (35). ISMARA
allowed the mapping of transcriptomic profiles to a
lower-dimensional inferred TF activity space, largely preserving
the relationships between samples. The TF activities driving
expression changes were calculated and the TF activity differences
between therapeutic response and non-response patients were
examined. The TF activities significantly associated with therapy
response were defined by P-value <0.05 and absolute mean
activity difference >0.008.
Statistical analysis
The Kaplan-Meier survival curve and log-rank test
were used to determine overall survival curves as previously
described (36,37). Using median gene expression values
from cBioPortal transcriptomic data as a bifurcating point, the
samples were divided into high- and low-expression groups and the
survival rates were compared between the two groups. The Cox
proportional hazards model was applied to estimate hazard ratios
(HRs) and 95% confidence intervals (CIs). Pearson correlation was
used to determine the correlations of the expression levels among
E2F8, MELK, or AR from the TCGA-PRAD and cBioPortal data. A
comparison of means among experimental groups was performed using
one-way ANOVA followed by Bonferroni's multiple comparison test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CsA inhibits prostate cancer growth in
vitro and in vivo
To determine the antitumor activity of CsA in
prostate cancer, MTT and colony formation assays were first
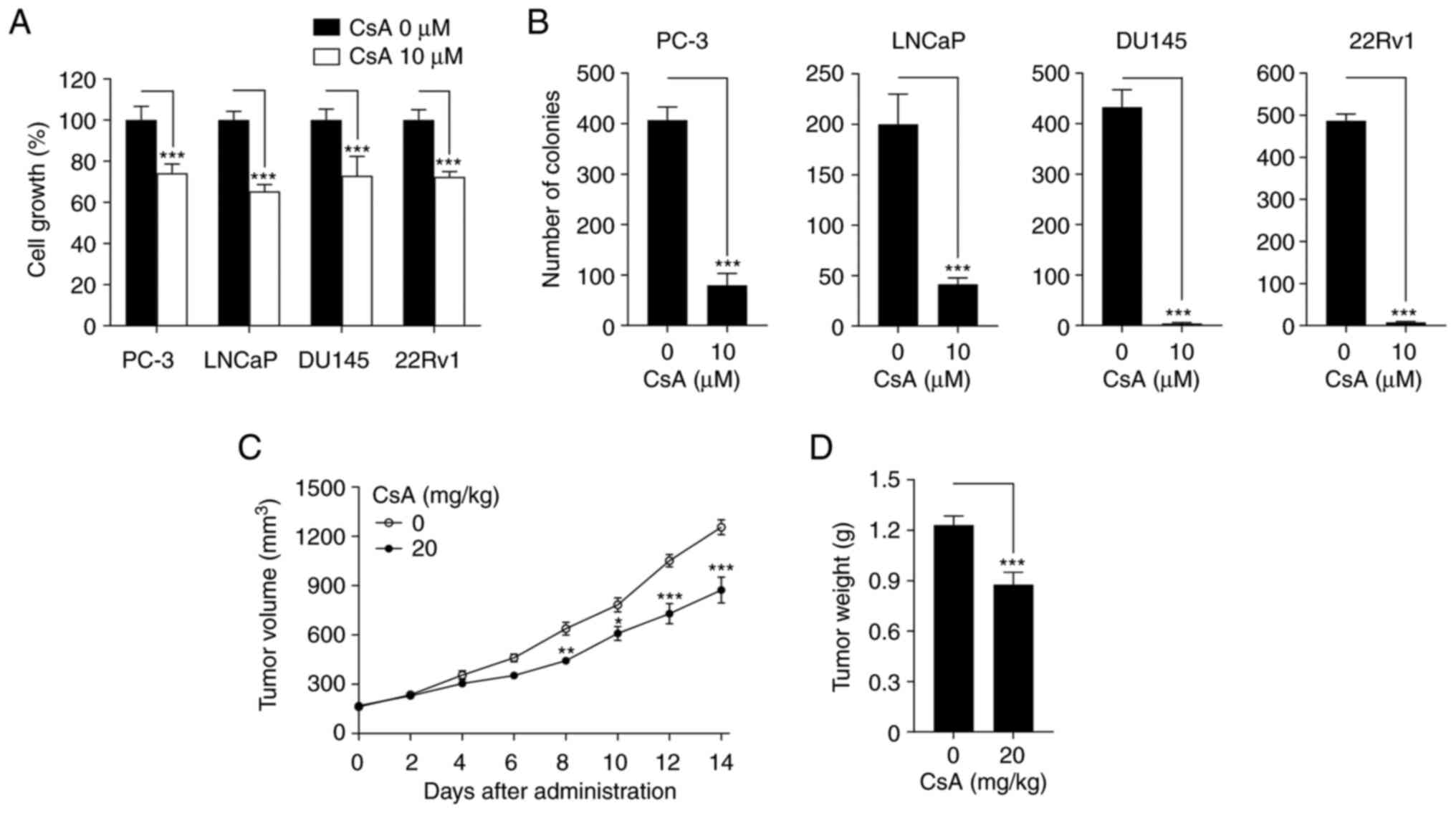
performed. CsA suppressed cell growth and colony formation in PC-3,
LNCaP, DU145 and 22Rv1 prostate cancer cells (Fig. 1A and B). In addition, CsA inhibited
tumor volume and weight in 22Rv1 cell xenograft mouse model
(Fig. 1C and D). These results
demonstrate that CsA has an anticancer activity against prostate
cancer.
To understand the antitumor mechanism of action of
CsA, microarray experiments we performed using CsA-treated PC-3
cells (GSE109505). Hierarchical clustering analysis and PCA showed
that CsA-treated and untreated cells were clustered into two
discrete groups (Figs. S1,
S2A and S2B), indicating that CsA induces a
distinct change in gene expression profiles. Analysis using SAM
found that CsA significantly affected the expression levels of
3,319 genes (Fig. S2C). Among
these 3,319 differentially expressed genes (DEGs), 2,500
(593+1,907) genes were downregulated in CsA-treated cells, whereas
819 (278+541) genes were upregulated in CsA-treated PC-3 cells
(Fig. S2D).
To assess the clinical significance of the 3,319
DEGs, they were compared with the DEGs derived from the
transcriptomic data of patients with prostate cancer (GSE3325).
First 3,654 DEGs between metastatic and nonmetastatic (benign or
primary) cancer were obtained: 1,398 genes were upregulated and
2,256 genes were downregulated in patients with metastatic cancer
(Fig. S2D; leftmost circles). It
was identified that 871 (593+278) DEGs showed anti-similarity (or
inverse correlation) between the 3,319 DEGs from GSE109505 and the
3,654 DEGs from GSE3325 (Fig. S2C and
D). These 871 anti-similar DEGs were termed the ‘clinically
significant CsA-induced gene expression (CCI) signature’. GSEA
revealed that this CCI signature is associated with 19 hallmark
pathways (Fig. S2E). In
particular, CsA activates cell death-related pathways and inhibits
cell cycle-related pathways, providing the mechanistic explanation
for the anticancer activity of CsA (Fig. 1).
E2F8 is identified as a master
regulator that is associated with a poor prognosis in patients with
prostate cancer
To elucidate the molecular mechanism by which CsA
regulates the CCI signature, the prostate cancer cell-specific
transcriptional interactome was analyzed using ARACNe and MRA
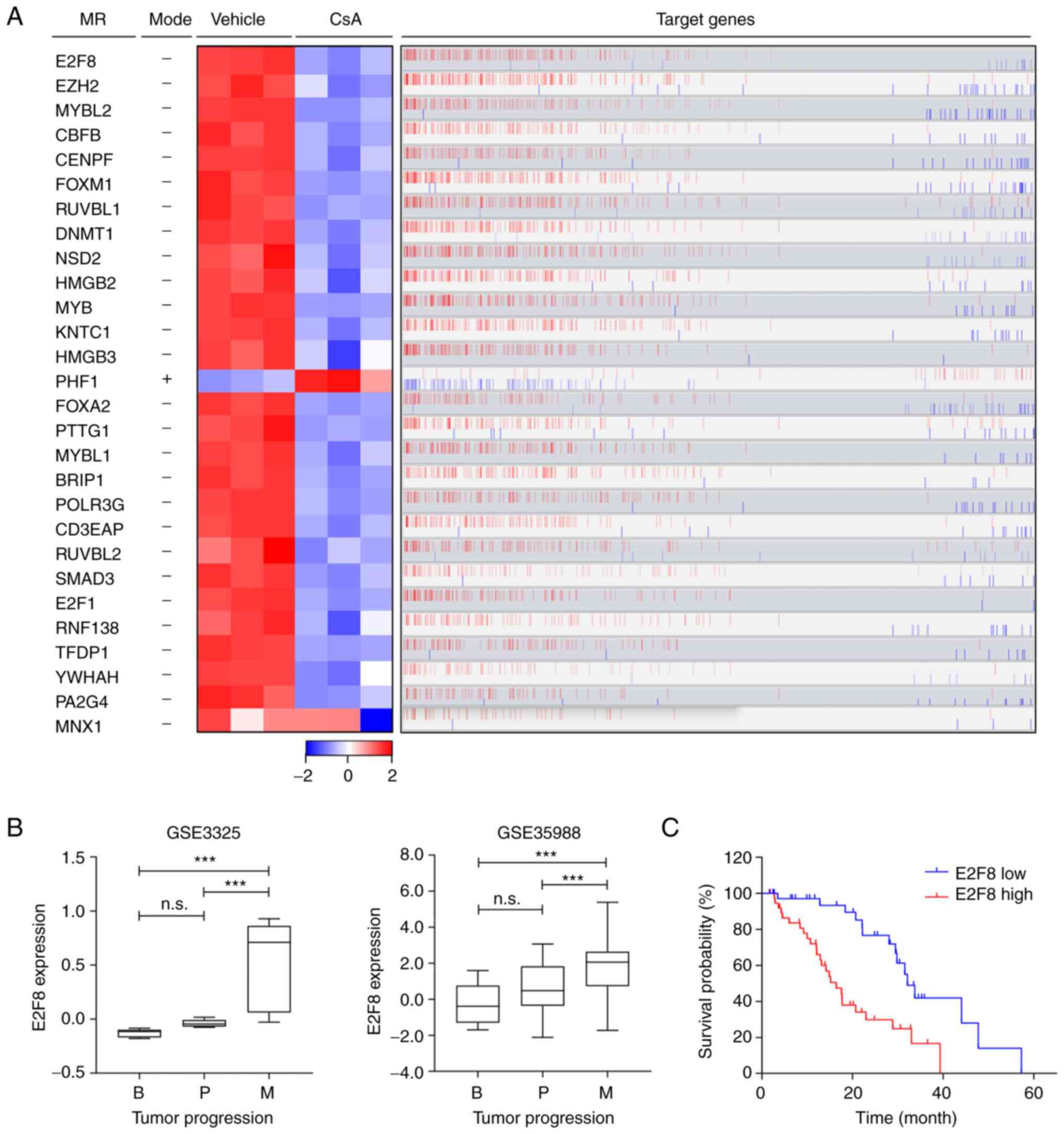
algorithms. These identified 28 transcription factors as master
regulators (MRs) that control the CCI signature (Fig. 2A and Table I): 27 MRs were markedly
downregulated in CsA-treated PC-3 cells, whereas only one MR (PHF1)
was upregulated (Fig. 2A; heatmap).
Overall, the expression patterns of these MRs correlated with those
of their target genes (Fig. 2A; red
bars). Based on ‘markers in the intersection set’ (Table I), E2F8 was identified as a top MR
that affected the majority of genes belonging to the CCI signature
(216 target genes out of 871; Figs.
2A and S3). These 216 target
genes of E2F8 were mainly associated with cell cycle or
proliferation pathways (Fig. S3).
CsA markedly reduced E2F8 expression (Fig. 2A), which correlated positively with
the expression levels of the most target genes (bar plot in
Fig. 2A). These results suggested
that E2F8 mainly acts as a transcriptional activator.
 | Table I.List of 28 MRs that control the CCI
signature. |
Table I.
List of 28 MRs that control the CCI
signature.
| GeneID | Symbol | Description | Markers in
intersection seta | FET
P-valueb | Markers in
regulonc | Moded | Fold Change | q-value |
|---|
| 79733 | E2F8 | E2F transcription
factor 8 | 216 |
4.11×10−56 | 439 | - | 0.71 |
1.00×10−16 |
| 2146 | EZH2 | enhancer of zeste 2
polycomb repressive complex 2 subunit | 198 |
1.09×10−29 | 509 | - | 0.78 |
1.00×10−16 |
| 4605 | MYBL2 | MYB proto-oncogene
like 2 | 196 |
9.37×10−44 | 434 | - | 0.73 |
1.00×10−16 |
| 865 | CBFB | core-binding factor
beta subunit | 190 |
4.32×10−30 | 498 | - | 0.82 |
1.00×10−16 |
| 1063 | CENPF | centromere protein
F | 183 |
9.56×10−36 | 429 | - | 0.66 |
1.00×10−16 |
| 2305 | FOXM1 | forkhead box
M1 | 183 |
1.63×10−31 | 445 | - | 0.73 |
1.00×10−16 |
| 8607 | RUVBL1 | RuvB like AAA
ATPase 1 | 173 |
7.29×10−7 | 656 | - | 0.83 |
1.00×10−16 |
| 1786 | DNMT1 | DNA
methyltransferase 1 | 170 |
2.87×10−26 | 440 | - | 0.74 |
1.00×10−16 |
| 7468 | NSD2 | nuclear receptor
binding SET domain protein 2 | 162 |
1.39×10−16 | 512 | - | 0.83 |
1.00×10−16 |
| 3148 | HMGB2 | high mobility group
box 2 | 161 |
5.27×10−23 | 422 | - | 0.66 |
1.00×10−16 |
| 4602 | MYB | MYB proto-oncogene,
transcription factor | 158 |
4.24×10−16 | 487 | - | 0.72 |
1.00×10−16 |
| 9735 | KNTC1 | kinetochore
associated 1 | 151 |
2.48×10−36 | 319 | - | 0.75 |
1.00×10−16 |
| 3149 | HMGB3 | high mobility group
box 3 | 135 |
3.35×10−23 | 335 | - | 0.83 |
1.00×10−16 |
| 5252 | PHF1 | PHD finger protein
1 | 135 |
2.33×10−11 | 394 | + | 1.16 |
1.07×10−5 |
| 3170 | FOXA2 | forkhead box
A2 | 132 |
9.60×10−6 | 513 | - | 0.76 |
1.00×10−16 |
| 9232 | PTTG1 | pituitary
tumor-transforming 1 | 131 |
3.00×10−9 | 395 | - | 0.73 |
1.00×10−16 |
| 4603 | MYBL1 | MYB proto-oncogene
like 1 | 127 |
8.34×10−31 | 272 | - | 0.75 |
1.00×10−16 |
| 83990 | BRIP1 | BRCA1 interacting
protein C-terminal helicase 1 | 126 |
1.10×10−6 | 479 | - | 0.73 |
1.00×10−16 |
| 10622 | POLR3G | RNA polymerase III
subunit G | 123 |
4.26×10−7 | 457 | - | 0.82 |
1.00×10−16 |
| 10849 | CD3EAP | CD3e molecule
associated protein | 121 |
4.19×10−15 | 333 | - | 0.84 |
1.00×10−16 |
| 10856 | RUVBL2 | RuvB like AAA
ATPase 2 | 120 |
5.93×10−7 | 403 | - | 0.89 |
1.06×10−6 |
| 4088 | SMAD3 | SMAD family member
3 | 116 |
2.87×10−7 | 407 | - | 0.82 |
1.00×10−16 |
| 1869 | E2F1 | E2F transcription
factor 1 | 109 |
3.46×10−15 | 304 | - | 0.82 |
1.00×10−16 |
| 51444 | RNF138 | ring finger protein
138 | 98 |
6.15×10−7 | 343 | - | 0.80 |
1.00×10−16 |
| 7027 | TFDP1 | transcription
factor Dp-1 | 98 |
3.28×10−12 | 291 | - | 0.77 |
1.00×10−16 |
| 7533 | YWHAH | tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein
eta | 89 |
7.85×10−9 | 277 | - | 0.83 |
1.00×10−16 |
| 5036 | PA2G4 |
proliferation-associated 2G4 | 80 |
3.40×10−7 | 254 | - | 0.84 |
1.00×10−16 |
| 3110 | MNX1 | motor neuron and
pancreas homeobox 1 | 49 |
1.01×10−7 | 128 | - | 0.77 |
1.00×10−16 |
To assess the clinical relevance of the MRs, the
data of patients with prostate cancer (GSE3325 and GSE35988) were
analyzed. Among these 28 MRs, the expression levels of 10 MRs were
markedly upregulated during prostate cancer progression (E2F8 in
Fig. 2B and other nine MRs in
S4). Kaplan-Meier analysis showed
that high expression levels of these 10 MRs are associated with
worse prognosis in patients with prostate cancer (E2F8 in Fig. 2C and other nine MRs in S5). In particular, E2F8 showed the
highest hazard ratio (HR=3.028; P=0.0002; Figs. 2C and S5B). These results indicated that E2F8
acts as a clinically significant MR crucial for governing a large
portion of the CCI signature. In addition, the findings suggested
that E2F8 serves as a useful prognosis marker for prostate
cancer.
E2F8 serves as a therapeutic target
for prostate cancer
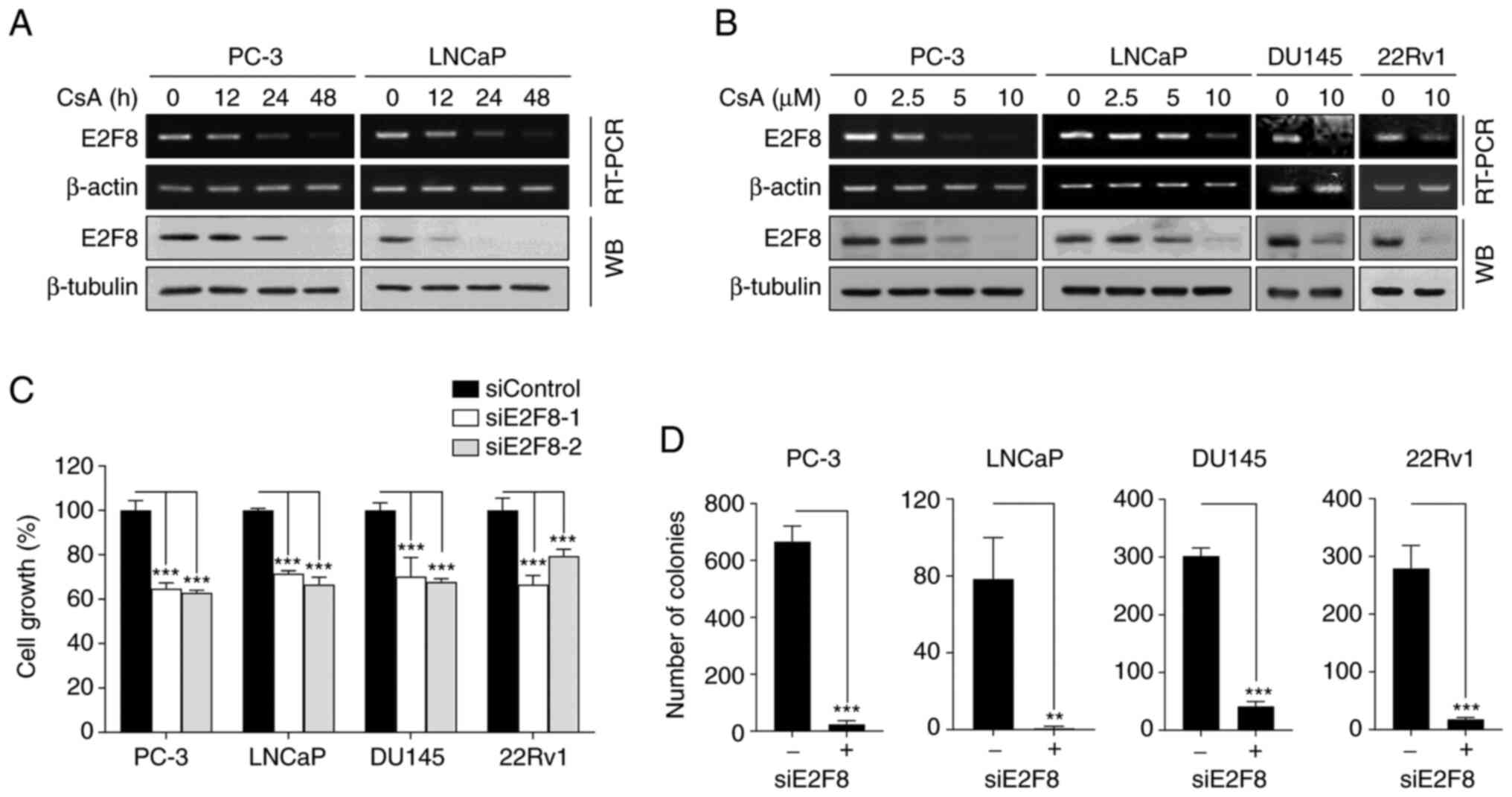
To confirm whether CsA suppressed E2F8 expression,
RT-PCR, real-time PCR and western blot analysis we performed in
various types of prostate cancer cells. CsA inhibited E2F8 mRNA
(Figs. 3A and B and S6A) and protein expression (Fig. 3A and B) in a time- and
concentration-dependent manner. As PC-3 and DU145 cells do not
express functional AR, the results suggested that CsA suppressed
E2F8 expression through an AR-independent mechanism.
To assess the role of E2F8 in prostate cancer, the
effect of siRNAs against E2F8 (siE2F8-1 and −2) were examined using
MTT and colony formation assays. The siE2F8s effectively inhibited
E2F8 expression in all tested cell lines (Fig. S6B). It was found that E2F8
knockdown suppressed cell growth and colony formation in all tested
cells (Fig. 3C and D). These
results suggested that E2F8 represents an attractive therapeutic
target for prostate cancer.
MELK is crucial for regulating E2F8
expression in prostate cancer
To elucidate the molecular mechanism by which CsA
suppresses E2F8 expression, Bayesian network analysis was employed,
which is an effective approach to model causal relationships
between observed biological data and gene expression data (38,39).
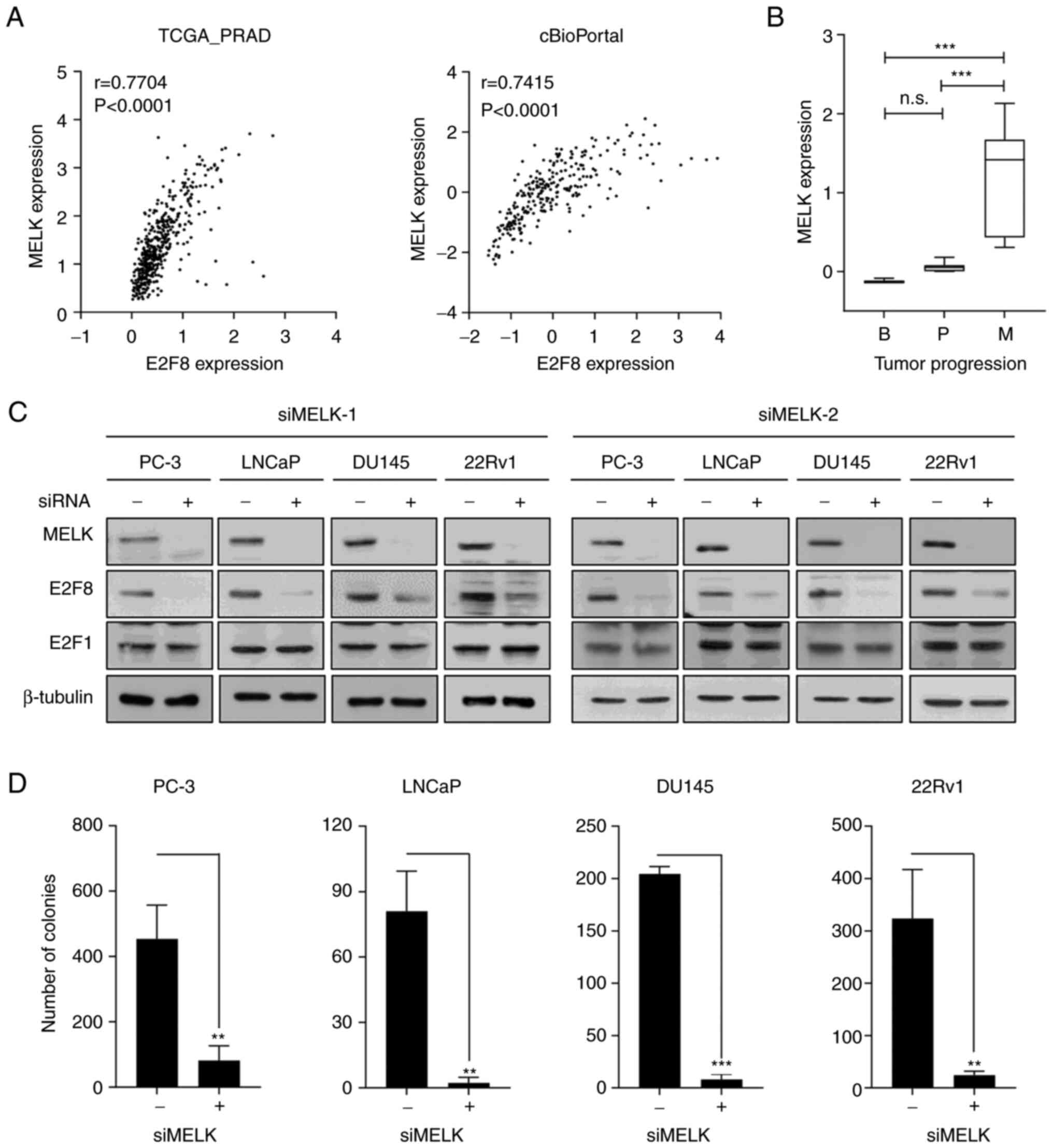
The FGES-discrete algorithm identified MELK as an E2F8-interacting
gene. Analysis of TCGA-PRAD and cBioPortal transcriptomic data
showed the strong positive correlation between E2F8 and MELK
expression in patients with prostate cancer (r=0.7704, P<0.0001
and r=0.7415, P<0.0001, respectively; Fig. 4A). In addition, analysis of the
GSE3325 data showed that MELK is markedly upregulated during
prostate cancer progression (Fig.
4B). Whether MELK causally regulates E2F8 expression was then
examined. The siRNA-based knockdown of MELK (siMELK-1 and −2)
markedly reduced E2F8 expression in all tested cells (Fig. 4C). In contrast to E2F8, siMELKs did
not affect the expression of E2F1, one of the identified MRs
(Fig. 2 and Table I), which plays a crucial role in
prostate cancer growth (20,40).
Altogether, these results indicate that MELK is a crucial upstream
signaling molecule for controlling E2F8 expression in prostate
cancer cells.
To determine whether CsA suppresses MELK expression,
RT-PCR, real-time PCR and western blot analysis were performed in
various prostate cancer cells. CsA inhibited MELK expression at the
protein level (Fig. S7A), but not
at the mRNA level (Fig. S7A and
B), suggesting that CsA regulated MELK at the
posttranscriptional or translational level. To assess the
functional importance of MELK in prostate cancer cells, whether
forced suppression of MELK expression affects prostate cancer
growth was investigated using MTT and colony formation assays. The
siMELKs inhibited cell growth and colony formation in various
prostate cancer cells (Figs. 4D,
S7C and S7D). Altogether, these results indicate
that the MELK-E2F8 signaling axis plays a crucial role in prostate
cancer biology.
E2F8-targeted therapy demonstrates
clinical significance in prostate cancer
AR is a crucial driver of CRPC progression and its
expression is frequently upregulated during prostate cancer
progression (41–44). The present study showed that E2F8
expression increases during prostate cancer progression and exerts
oncogenic activity (Figs. 2 and
3). Based on these observations,
the interplay between AR and E2F8 signaling was examined. The
analysis of TCGA-PRAD and cBioPortal transcriptomic data found that
there was no significant coexpression correlation between E2F8 and
AR or between MELK and AR expression levels (Fig. 5A and C).
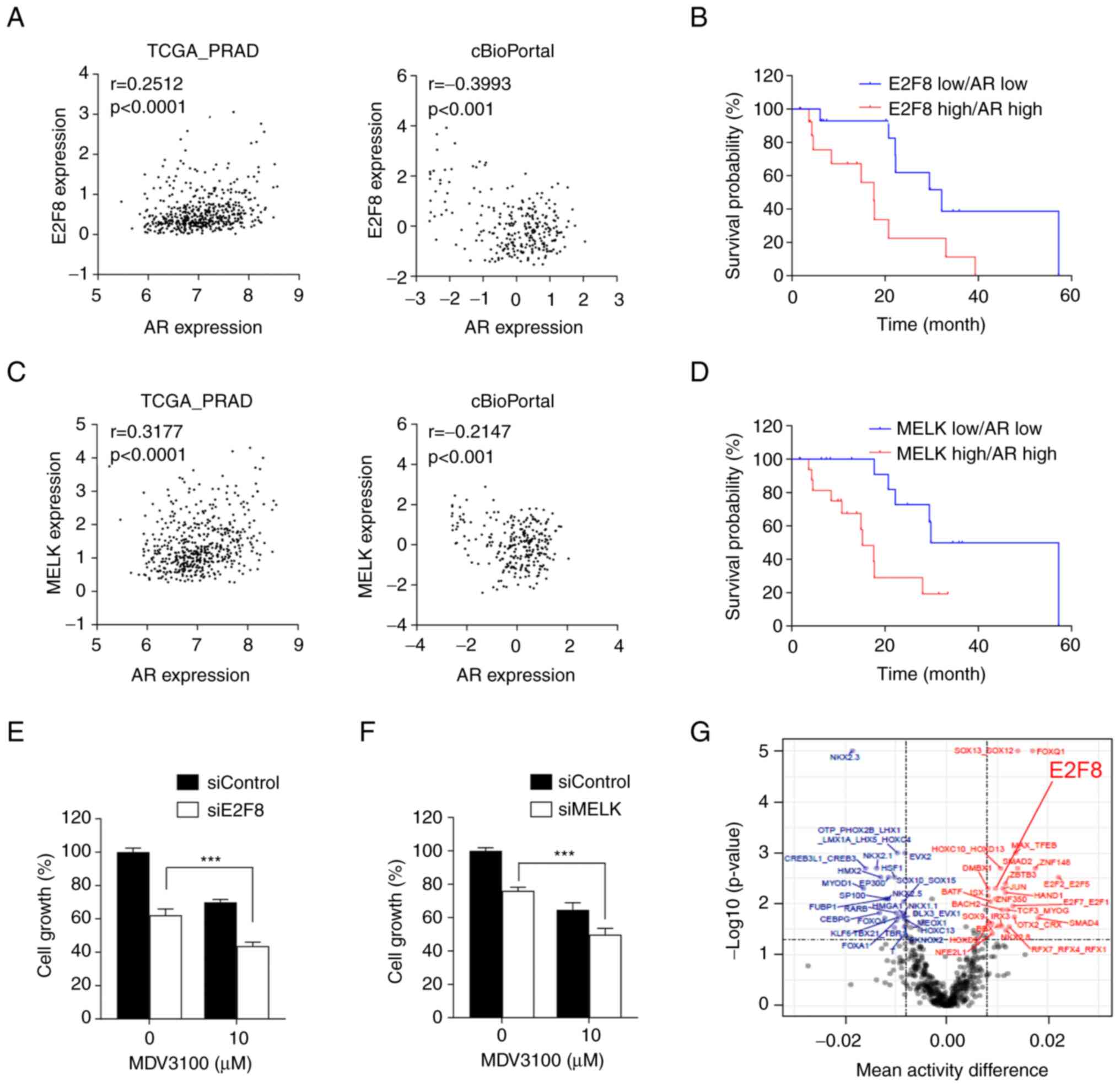 | Figure 5.Clinical significance of E2F8 in
prostate cancer. (A and C) Coexpression correlation between E2F8
and AR or between MELK and AR in TCGA-PRAD and cBioPortal
transcriptomic data. (B and D) Survival curve for patients with
prostate cancer based on the expression levels of E2F8 and AR or
those of MELK and AR from cBioPortal data. (E) LNCaP cells were
treated with 50 nM siE2F8-1, 10 µM MDV300, or both for 72 h prior
to MTT assay. The data were expressed as the mean ± SEM (n=4-6).
***P<0.005. (F) LNCaP cells were treated with 50 nM siMELK-1, 10
µM MDV300, or both for 72 h prior to MTT assay. The data were
expressed as the mean ± SEM (n=4-6) ***P<0.005. (G) Inferred TF
motif activity differences between patients with CR (highlighted in
blue) and SD (highlighted in red). The x-axis denotes the mean TF
activity differences and the y-axis indicates-log10 (P-values). The
vertical dotted line represents an absolute mean TF activity
difference of 0.008 and the horizontal dotted line means the
P-value 0.05 for significant TFs. AR, androgen receptor; TCGA-PRAD,
The Cancer Genome Atlas-Prostate Adenocarcinoma; r, Pearson
correlation coefficient; TF, transcription factor; si, short
interfering; TCGA-PRAD, The Cancer Genome Atlas-Prostate
Adenocarcinoma. |
Nonetheless, Kaplan-Meier analysis showed that
overall survival was greatly reduced in patients with prostate
cancer with high levels of both E2F8 and AR expression, compared
with those with low levels of both genes (HR=4.33, P=0.0019;
Fig. 5B). Similar results were
obtained from patients with prostate cancer with high levels of
both MELK and AR expression, compared with those with low levels of
both genes (HR=3.3, P=0.0041; Fig.
5D). Altogether, these results suggest that the MELK-E2F8 axis
and AR signaling act independently, but their additive effects
worsen the clinical outcomes of prostate cancer. Hence.
simultaneous inhibition of E2F8 and AR could be a promising
therapeutic strategy for patients with prostate cancer with
concurrent overexpression of these molecules.
To investigate the translatability of the findings,
LNCaP cells (AR-positive, AR-V7-negative cells) were treated with
siE2F8 and the AR antagonist MDV3100. The treatment with siE2F8
increased the sensitivity of prostate cancer cells to MDV3100
(Fig. 5E), suggesting a potential
therapeutic strategy for improving the efficacy anti-androgen
therapy. In addition, siE2F8 had a similar effect as siMELK in
enhancing the efficacy of MDV3100 (Fig.
5F). Finally, the ISMARA algorithm showed that E2F8 activity is
markedly higher in patients with SD than in those with CR (Fig. 5G), suggesting that E2F8 is a
predictive marker for therapeutic response in patients with
prostate cancer.
Discussion
The present study described five main findings: i)
CsA exhibits antitumor properties against prostate cancer in
cultured cell and xenograft models; ii) E2F8 is a master regulator
in controlling the CCI signature; iii) E2F8 is upregulated during
tumor progression and the high expression levels of E2F8 are
associated with a poor prognosis in patients with prostate cancer;
iv) MELK is a crucial upstream regulator of E2F8 expression; and v)
the inhibition of E2F8 or MELK sensitizes prostate cancer cells to
AR blockade therapy. Considering that CsA has been investigated in
clinical trials (11–13), the results provided a promising
basis for future research aimed at the development of prostate
cancer therapy and prognosis in clinic.
The present study demonstrated that CsA reduces E2F8
expression levels, although the pattern of reduction differs among
prostate cancer cell lines. CsA reduced E2F8 protein expression in
LNCaP cells faster than in PC-3 cells (Fig. 3A). These findings suggest that the
cellular rewiring determining E2F8 expression levels is cell type-
or context-dependent. Furthermore, the mechanism underlying E2F8
overexpression, which is commonly observed in patients with
prostate cancer, may vary among the individuals. Further research
into the various regulatory mechanisms of E2F8 expression can
contribute to an improved understanding of prostate cancer and the
development of diagnostic and therapeutic strategies toward
precision oncology.
Accumulating evidence indicates that MELK is
upregulated in various types of human cancer, including advanced
prostate cancer (45) and acts as
an oncogenic driver, making it a potential therapeutic target
(46). In line with these findings,
the present study showed that knockdown of MELK suppressed cell
growth and colony formation in prostate cancer cells. In addition,
it found that high expression levels of both MELK and AR are
associated with a poor prognosis in patients with prostate cancer,
similar to those of both E2F8 and AR. In addition, siE2F8 had a
similar effect to siMELK in enhancing the efficacy of MDV3100.
Collectively, these findings suggested that the MELK-E2F8 signaling
axis has oncogenic potential and cooperates with AR signaling to
promote prostate carcinogenesis.
Survival analysis revealed that the combined high
expression of both E2F8 and AR predicted a poor prognosis in
patients with prostate cancer. However, there was no significant
coexpression correlation between E2F8 and AR. In addition, when the
overlap between the 216 target genes of E2F8 and the 149 target
genes of AR (47) were compared, it
was found that only six genes (CDK1, CHAF1A, IGF2BP3, MCM2, RPA3
and STIL) were commonly shared, suggesting that both TFs act
independently in prostate cancer. These results suggested that in
significant portion of patient with prostate cancer, E2F8 and AR
signaling acted independently. However, in certain cases where both
genes are concurrently overexpressed, their combined effects
exacerbate the clinical outcomes of prostate cancer. Exploring
further investigations into the interaction between E2F8 and AR
signaling may provide to new avenues for prostate cancer research.
In particular, considering the findings that CsA regulates E2F8
expression in an AR-independent manner, it is plausible to
investigate the possibility that E2F8 mediates the non-AR-driven
evolution of CRPC.
In conclusion, the present study demonstrated that
CsA suppressed MELK-mediated E2F8 expression, leading to its
antitumor activity against prostate cancer. High expression level
of E2F8 was associated with a poor prognosis in patients with
prostate cancer and inhibiting E2F8 enhanced AR blockade therapy.
Therefore, CsA may serve as an effective anticancer agent for
prostate cancer, while E2F8 represents a promising target for
diagnosis and treatment of this disease.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the National
Research Foundation of Korea grant funded by the Korea government
(MSIT; grant nos. 2018R1A4A1023822 and 2020R1A2C1102574), a grant
from the Education and Research Encouragement Fund of Seoul
National University Hospital (grant no. 0320210340).
Availability of data and materials
All data supporting the findings of this study are
included in this published article. All additional information are
available from the corresponding author on reasonable request.
Authors' contributions
SL, JNC, and JHJ designed the present study. DYL,
SL, YSK, SP, SMB, EAC performed the experiments. DYL, SL, JNC, and
JHJ confirm the authenticity of all the raw data. JNC, EJP, HHP,
SYK and IS contributed to data analysis and interpretation. SL,
DYL, SP, SYK, JNC, and JHJ wrote the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with protocols approved by the Institutional Animal Care and Use
Committee of the Asan Institute for Life Sciences at the ASAN
Medical Center, University of Ulsan College of Medicine, Seoul
(approval no. 2021-13-234).
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Carceles-Cordon M, Kelly WK, Gomella L,
Knudsen KE, Rodriguez-Bravo V and Domingo-Domenech J: Cellular
rewiring in lethal prostate cancer: The architect of drug
resistance. Nat Rev Urol. 17:292–307. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Attard G, Parker C, Eeles RA, Schröder F,
Tomlins SA, Tannock I, Drake CG and de Bono JS: Prostate cancer.
Lancet. 387:70–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Litwin MS and Tan HJ: The diagnosis and
treatment of prostate cancer: A review. JAMA. 317:2532–2542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sandhu S, Moore CM, Chiong E, Beltran H,
Bristow RG and Williams SG: Prostate cancer. Lancet. 398:1075–1090.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yap TA, Smith AD, Ferraldeschi R,
Al-Lazikani B, Workman P and de Bono JS: Drug discovery in advanced
prostate cancer: Translating biology into therapy. Nat Rev Drug
Discov. 15:699–718. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ku SY, Gleave ME and Beltran H: Towards
precision oncology in advanced prostate cancer. Nat Rev Urol.
16:645–654. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsuda S and Koyasu S: Mechanisms of
action of cyclosporine. Immunopharmacology. 47:119–125. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tedesco D and Haragsim L: Cyclosporine: A
review. J Transplant. 2012:2303862012.PubMed/NCBI
|
|
9
|
Periyasamy S, Hinds T Jr, Shemshedini L,
Shou W and Sanchez ER: FKBP51 and Cyp40 are positive regulators of
androgen-dependent prostate cancer cell growth and the targets of
FK506 and cyclosporin A. Oncogene. 29:1691–1701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee CR, Chun JN, Kim SY, Park S, Kim SH,
Park EJ, Kim IS, Cho NH, Kim IG, So I, et al: Cyclosporin A
suppresses prostate cancer cell growth through CaMKKβ/AMPK-mediated
inhibition of mTORC1 signaling. Biochem Pharmacol. 84:425–431.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krishnamurthy A, Dasari A, Noonan AM,
Mehnert JM, Lockhart AC, Leong S, Capasso A, Stein MN, Sanoff HK,
Lee JJ, et al: Phase Ib results of the rational combination of
selumetinib and cyclosporin A in advanced solid tumors with an
expansion cohort in metastatic colorectal cancer. Cancer Res.
78:5398–5407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Flores C, Fouquet G, Moura IC, Maciel TT
and Hermine O: Lessons to learn from low-dose cyclosporin-A: A new
approach for unexpected clinical applications. Front Immunol.
10:5882019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Isshiki Y, Tanaka H, Suzuki Y and Yoshida
Y: Cyclosporine is a potential curative treatment option for
advanced thymoma. Exp Hematol Oncol. 6:132017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li J, Ran C, Li E, Gordon F, Comstock G,
Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al:
Synergistic function of E2F7 and E2F8 is essential for cell
survival and embryonic development. Dev Cell. 14:62–75. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park SA, Platt J, Lee JW, López-Giráldez
F, Herbst RS and Koo JS: E2F8 as a novel therapeutic target for
lung cancer. J Natl Cancer Inst. 107:djv1512015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX,
Wang KS, Teng XM, Zhang X, Huang J and Han ZG: E2F8 contributes to
human hepatocellular carcinoma via regulating cell proliferation.
Cancer Res. 70:782–791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee DY, Chun JN, Cho M, So I and Jeon JH:
Emerging role of E2F8 in human cancer. Biochim Biophys Acta Mol
Basis Dis. 1869:1667452023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SH, Lee S, Piccolo SR, Allen-Brady K,
Park EJ, Chun JN, Kim TW, Cho NH, Kim IG, So I and Jeon JH: Menthol
induces cell-cycle arrest in PC-3 cells by down-regulating G2/M
genes, including polo-like kinase 1. Biochem Biophys Res Commun.
422:436–441. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chun JN, Kim SY, Park EJ, Kwon EJ, Bae DJ,
Kim IS, Kim HK, Park JK, Lee SW, Park HH, et al: Schisandrin B
suppresses TGFβ1-induced stress fiber formation by inhibiting
myosin light chain phosphorylation. J Ethnopharmacol. 152:364–371.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee S, Chun JN, Kim SH, So I and Jeon JH:
Icilin inhibits E2F1-mediated cell cycle regulatory programs in
prostate cancer. Biochem Biophys Res Commun. 441:1005–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee S, Park YR, Kim SH, Park EJ, Kang MJ,
So I, Chun JN and Jeon JH: Geraniol suppresses prostate cancer
growth through down-regulation of E2F8. Cancer Med. 5:2899–2908.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chun JN, Park S, Lee S, Kim JK, Park EJ,
Kang M, Kim HK, Park JK, So I and Jeon JH: Schisandrol B and
schisandrin B inhibit TGFβ1-mediated NF-κB activation via a
Smad-independent mechanism. Oncotarget. 9:3121–3130. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Johnson WE, Li C and Rabinovic A:
Adjusting batch effects in microarray expression data using
empirical Bayes methods. Biostatistics. 8:118–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee S, Piccolo SR and Allen-Brady K:
Robust meta-analysis shows that glioma transcriptional subtyping
complements traditional approaches. Cell Oncol (Dordr). 37:317–329.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Floratos A, Smith K, Ji Z, Watkinson J and
Califano A: geWorkbench: An open source platform for integrative
genomics. Bioinformatics. 26:1779–1780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Margolin AA, Nemenman I, Basso K, Wiggins
C, Stolovitzky G, Dalla Favera R and Califano A: ARACNE: An
algorithm for the reconstruction of gene regulatory networks in a
mammalian cellular context. BMC Bioinformatics. 7 (Suppl 1):S72006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kig C, Beullens M, Beke L, Van Eynde A,
Linders JT, Brehmer D and Bollen M: Maternal embryonic leucine
zipper kinase (MELK) reduces replication stress in glioblastoma
cells. J Biol Chem. 292:127862017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim SH, Kim SY, Park EJ, Kim J, Park HH,
So I, Kim SJ and Jeon JH: Icilin induces G1 arrest through
activating JNK and p38 kinase in a TRPM8-independent manner.
Biochem Biophys Res Commun. 406:30–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuang J, Yan X, Genders AJ, Granata C and
Bishop DJ: An overview of technical considerations when using
quantitative real-time PCR analysis of gene expression in human
exercise research. PLoS One. 13:e01964382018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Wang Y, Shen F, Xu Y, Zhang Y, Zou
X, Zhou J and Chen Y: Maternal embryonic leucine zipper kinase: A
novel biomarker and a potential therapeutic target of cervical
cancer. Cancer Med. 7:5665–5678. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ramsey J, Glymour M, Sanchez-Romero R and
Glymour C: A million variables and more: the fast greedy
equivalence search algorithm for learning high-dimensional
graphical causal models, with an application to functional magnetic
resonance images. Int J Data Sci Anal. 3:121–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Balwierz PJ, Pachkov M, Arnold P, Gruber
AJ, Zavolan M and van Nimwegen E: ISMARA: Automated modeling of
genomic signals as a democracy of regulatory motifs. Genome Res.
24:869–884. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park YR, Chun JN, So I, Kim HJ, Baek S,
Jeon JH and Shin SY: Data-driven analysis of TRP channels in
cancer: Linking variation in gene expression to clinical
significance. Cancer Genomics Proteomics. 13:83–90. 2016.PubMed/NCBI
|
|
37
|
Park S, Lim JM, Chun JN, Lee S, Kim TM,
Kim DW, Kim SY, Bae DJ, Bae SM, So I, et al: Altered expression of
fucosylation pathway genes is associated with poor prognosis and
tumor metastasis in non-small cell lung cancer. Int J Oncol.
56:559–567. 2020.PubMed/NCBI
|
|
38
|
Yu J, Smith VA, Wang PP, Hartemink AJ and
Jarvis ED: Advances to Bayesian network inference for generating
causal networks from observational biological data. Bioinformatics.
20:3594–3603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang YX and Huang H: Review on statistical
methods for gene network reconstruction using expression data. J
Theor Biol. 362:53–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chun JN, Cho M, Park S, So I and Jeon JH:
The conflicting role of E2F1 in prostate cancer: A matter of cell
context or interpretational flexibility? Biochim Biophys Acta Rev
Cancer. 1873:1883362020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taplin ME, Bubley GJ, Shuster TD, Frantz
ME, Spooner AE, Ogata GK, Keer HN and Balk SP: Mutation of the
androgen-receptor gene in metastatic androgen-independent prostate
cancer. N Engl J Med. 332:1393–1398. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shafi AA, Yen AE and Weigel NL: Androgen
receptors in hormone-dependent and castration-resistant prostate
cancer. Pharmacol Ther. 140:223–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard G, et al: Integrative clinical genomics of advanced prostate
cancer. Cell. 161:1215–1228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kuner R, Fälth M, Pressinotti NC, Brase
JC, Puig SB, Metzger J, Gade S, Schäfer G, Bartsch G, Steiner E, et
al: The maternal embryonic leucine zipper kinase (MELK) is
upregulated in high-grade prostate cancer. J Mol Med (Berl).
91:237–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jurmeister S, Ramos-Montoya A, Sandi C,
Pértega-Gomes N, Wadhwa K, Lamb AD, Dunning MJ, Attig J, Carroll
JS, Fryer LG, et al: Identification of potential therapeutic
targets in prostate cancer through a cross-species approach. EMBO
Mol Med. 10:e82742018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sharma NL, Massie CE, Ramos-Montoya A,
Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY,
Mills IG and Neal DE: The androgen receptor induces a distinct
transcriptional program in castration-resistant prostate cancer in
man. Cancer Cell. 23:35–47. 2013. View Article : Google Scholar : PubMed/NCBI
|