Introduction
Bladder cancer (BC) is the fifth most common
malignancy occurring worldwide and a significant cause of
cancer-related morbidity and mortality (1). Experimental data (2) indicated that differentially expressed
genes and microRNAs (miRNAs) are crucial in the development,
metastasis and therapy of BC. For example, mutations in the TP53
gene may be useful in predicting whether BC cells are likely to
proliferate and spread to nearby tissues, and whether the disease
may recur after treatment. The tumor suppressor gene PTEN, which is
mutated or homozygously deleted in various types of cancer, maps to
a region of 10q within the reported region of minimal loss in
bladder tumors (3). Although there
has been a focus on differentially expressed genes and miRNAs, the
signaling pathway and mechanisms that elucidate miRNA
transcriptional regulation remain unclear.
Transcription factors (TFs) and miRNAs are notable
regulators for gene expression (4).
TFs are proteins capable of activating or repressing transcription
by binding to cis-regulatory elements located in the upstream
regions of genes. They are capable of regulating gene expression at
the transcriptional level individually or in combination with other
proteins.
miRNAs are 21–24 nt regulatory, non-coding RNAs that
regulate gene expression at the post-transcriptional level and are
therefore important cell components. miRNAs are involved in various
biological processes, including cell proliferation, differentiation
and apoptosis. Calin et al(5) first confirmed that differentially
expressed miRNAs are associated with BC.
miRNAs are located within genes designated as host
genes. Rodriguez et al(6)
suggested that miRNAs are transcribed in parallel with their host
transcripts, and the two different transcription classes of miRNAs
(exonic and intronic) identified in that study may require slightly
different mechanisms of biogenesis. Baskerville et
al(7) identified that intronic
miRNA and its host gene are closely associated. Intronic miRNAs and
their host genes are usually equally expressed in biological
progression, usually acting as a potential partner in order to
achieve biological function and affect the alteration of pathways
(8). The above-mentioned results
suggested that miRNAs together with their host gene or separately
were capable of contributing to cancer progression. In this study,
when miRNA was differentially expressed, we considered that the
host genes were also differentially expressed and were involved in
cancer progression.
The aim of the present study was to determine the
associations of among genes, miRNAs and their host genes.
Additionally, these transcriptional relations were regarded as a
penetration point to build the regulatory network of the genes and
miRNAs in human BC. The topology networks derived included the
differentially expressed, BC-associated and global networks. The
global network revealed experimentally validated pathways with
regard to genes and miRNAs, however, this network is extremely
complex and BC-associated pathways could not be determined.
Therefore, we focused on the remaining two networks. Pathways
regarding differentially expressed elements were extracted. The
differentially expressed network partially revealed BC formation,
and the topology network of BC was identified. Similarities and
differences among the three networks were compared and analyzed in
order to identify the key nodes and pathways. The network of
differentially expressed elements partially revealed the mechanism
of BC. The findings of this study may be useful for future
investigations into the mechanism of action, as well as the
pathogenesis and treatments of BC.
Materials and methods
Material collection and data
processing
miRNA-target interactions were extracted from
TarBase 5.0 (9) and miRTarBase
(10). TarBase 5.0 is a
comprehensive database of experimentally supported animal miRNA
targets, while miRTarBase is a database of experimentally validated
miRNA-target interactions. Different databases use varying symbols
to represent miRNAs and genes. In order to systemize the symbolic
representation method, official symbols from the National Center
for Biotechnology Information (NCBI) database, which can be
accessed online at http://www.ncbi.nlm.nih.gov/gene/, were used. These
experimentally validated data strongly support our study. The
complete data can be considered as
setU1.
TF-miRNA interactions from TransmiR were extracted
(11). TransmiR is a TF-miRNA
regulation database. Data of TransmiR are extracted from public
literature and biological experiments. The complete data of
TF-miRNA interactions can be considered as
setU2.
miRNAs and their host genes were extracted from
miRBase (12) and NCBI. miRBase
provides a collection of confirmed human miRNAs. Official symbols
and IDs from NCBI were used to verify host genes and their miRNAs.
The complete data can be considered as
setU3.
Differentially expressed genes of BC were extracted
from the KEGG pathway database (13) and the Cancer Genetics Web, which can
be accessed online at: http://www.cancerindex.org. The KEGG pathway database
comprises graphical diagrams of biochemical pathways as well as
some of the known regulatory pathways, and can be accessed online
at: http://www.kegg.com/kegg/pathway.html. For this study,
the BC pathway map was employed which indicates the validated
mutated BC genes. Similar methods were employed to elicit mutated
BC genes from the Cancer Genetics Web. In order to make the data
collected comprehensive, related SCI studies on BC-mutated genes
were manually obtained. Similarly, BC-related genes including
differentially expressed genes of BC and other genes associated
with BC were manually obtained. In the present study,
differentially expressed genes were considered as part of the
related genes in the BC-associated network. Additionally, the
P-match method was used to extract TFs that may involve in BC
(14). P-match is a new tool used
to identify TF-binding sites in DNA sequences. It combines pattern
matching and weight matrix approaches thus providing higher
accuracy of recognition than each of the methods separately. TFs
were regarded as BC-related genes and the focus was on the TFs
setU2. We downloaded 1,000
nt (5,000 nt) promoter region sequences of targets that were
targeted by mutated miRNAs from the UCSC database (15). The P-match method was used to map
TF-binding sites (TFBSs) onto the promoter region of targets.
P-match utilizes the matrix library as well as sets of known
TF-binding sites collected in TRANSFAC, allowing the possibility to
search for varying TF-binding sites (16). The vertebrate matrix and restricted
high-quality criterion were used. The latter is used to indicate
the matrix with a cut-off value allowing a false-negative rate of
50%, while the false-positive rate is required to be reduced below
a certain threshold (16). Thus,
the matrices that produce the highest number of false-positive
matches are defined as low-quality matrices. The complete data of
differentially expressed genes and related genes can be considered
as setU4.
Differentially expressed miRNAs were elicited from
mir2Disease (17). mir2Disease is a
manually curated database that aims to provide a comprehensive
resource of miRNA deregulation in various human diseases. To ensure
data were comprehensive, related SCI studies on BC were manually
obtained. The complete differentially expressed miRNAs and related
miRNAs of BC can be considered as
setU5.
Three networks construction
Transcriptional network of BC is a very complex
regulatory network. Differentially expressed genes and miRNAs are
important in this network as they are involved in every process of
cancer formation. Thus, the core regulation network of BC was
utilized using the following method: Differentially expressed data
from setU4 and
setU5 were mapped onto
setU1,
U2 and
U3, then regulatory
relations of TF-miRNA, miRNA-targets and hostgene-miRNA were
determined. The relations were then combined to determine the core
network.
Besides differentially expressed genes and miRNAs,
BC-related genes and miRNAs are also involved in each process of
cancer formation. Thus, a BC-related network was determined to
further demonstrate the regulatory network of BC. This network
included both the core network and more complex regulatory
relations. The regulation network in BC was obtained using the
method mentioned above.
The former two networks present are crucial in
regulatory relations. Besides the experimentally validated genes
and miRNAs included in the former two networks, there may be some
genes and miRNAs that have not been validated experimentally to be
involved in the progression of BC. In the third network, TFs and
miRNAs present in the related network were mapped
ontoU1,
U2 and
U3, then the regulatory
relations of TF-miRNA, miRNA-targets and hostgene-miRNA were
extracted. Following the combination of all relations, the expanded
global network was derived.
Results
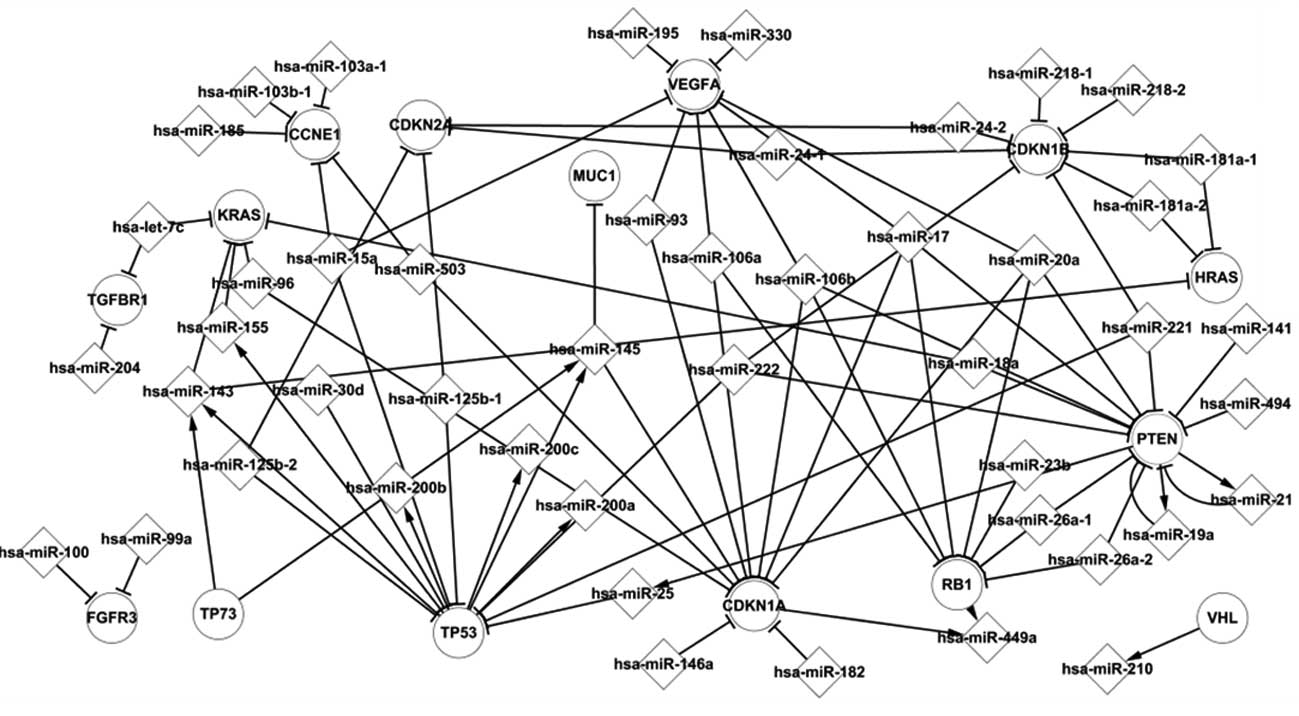
Core transcriptional network of BC
Through the statistical analysis, we derived a core
transcriptional network that reported the regulation mechanisms in
human BC. Five important TFs, i.e., TP53, TP73, CDKN1A, RB1 and
PTEN, had more relations with other elements in this network and
therefore were regarded as essential regulatory elements.
Fig. 1 shows some of
the regulation pathways, such as PTEN which regulates hsa-miR-25a,
which in turn targets TP53. TP53 regulates the miRNAs, hsa-miR-143,
hsa-miR-145, hsa-miR-155, hsa-miR-200a, hsa-miR-200b and
hsa-miR-200c, which target the genes KRAS, HRAS, MUC1 and CDKN1A.
At the end of the pathway, hsa-miR-449a was targeted by CDKN1A.
These genes and miRNAs are important elements in the progression of
BC. The TP53 gene provides instructions for generating tumor
protein p53. This protein acts as a tumor suppressor, thus it
regulates cell division by keeping cells from growing and dividing
too rapidly or in an uncontrolled manner. Mutations in the TP53
gene may be useful in predicting whether BC is likely to
proliferate and spread to nearby tissues and whether the disease
may recur following treatment. HRAS is the oncogene of BC, and it
is the starting point of the MAPK signaling pathway. Mucins are a
group of high molecular weight glycoproteins present on epithelial
surfaces. In the bladder, they have a protective role, inhibiting
bacterial and stone adhesion. Nine human mucin genes have been
identified, one of which is located on chromosome 1 (18) encoding for MUC1 mucin. This mucin is
upregulated (19) and abnormally
glycosylated (20) in several
common types of cancer, including BC (21).
The tumor suppressor gene PTEN, which is mutated or
homozygously deleted in various types of cancer, maps to a region
of 10q within the reported region of minimal loss in bladder tumors
(19). PTEN is also involved in two
feedback loops by targeting hsa-miR-21 and hsa-miR-19a, and vice
versa.
In the present study, we utilized the core
transcriptional network to explore these mechanisms of BC.
BC-associated network
The related regulatory network of BC comprises
differentially expressed genes and miRNAs, related genes and
miRNAs, targets of miRNAs and host genes of miRNAs, with the
BC-associated network including the core network. Fig. 2 shows more complex regulatory
relations as compared to Fig.
1.
Fig. 1 shows six
TFs, i.e., CDKN1A, RB1, PTEN, TP53, TP73, and VHL in the regulation
network, which may be considered essential regulatory elements. In
Fig. 2, there are 37 TFs in the
regulation network, which include the six essential TFs (Fig. 1) and which regulate 59 mutated
miRNAs. Among the TFs in the network BRCA1, PTEN and NFKB1 regulate
more miRNA expression and therefore have a high potential of being
more influential than others TFs. BRCA1 regulates hsa-miR-146a
expression, which targets EGFR, NFKB1, CDKN1A and BRCA1. Thus,
BRCA1 and hsa-miR-146a constitute a feedback loop.
Fig. 2 shows some
additional pathways that affect the progression of BC with regard
to differentially expressed TFs and additional miRNAs. PTEN is a
mutated gene that regulates hsa-miR-19a, hsa-miR-21 and hsa-miR-25
expression. These three miRNAs target 19 relevant genes including
PTEN, ESR1 and PDCD4. It has been demonstrated that ESR1 mediated a
decrease in hsa-miR-21 expression correlated with an increased
protein expression of endogenous hsa-miR-21 targets such as PDCD4,
PTEN and BCL2 (22). Another
important TF is NFKB1 which regulates the expression of 11 miRNAs
including hsa-miR-21, hsa-miR-155 and hsa-miR-146a. Hsa-mir-155
targets 13 relevant genes, including KRAS (22).
In our network, 32 feedback loops were identified
consisting of 35 genes and 32 miRNAs. Fourteen differentially
expressed genes are involved in the feedback loops. Fig. 2 shows a regulation network of the
interactions of 35 relevant TFs, 100 relevant miRNAs and their 161
relevant targets.
Global network of BC
The global regulatory network of BC provides more
comprehensive regulatory relations. Furthermore, it has additional
TFs, targets, miRNAs and host genes of miRNA compared to the
related network. The global network includes more comprehensive
regulatory relations including all relations in
U1,
U2 and
U3. This network includes
both the differentially expressed and BC-associated network.
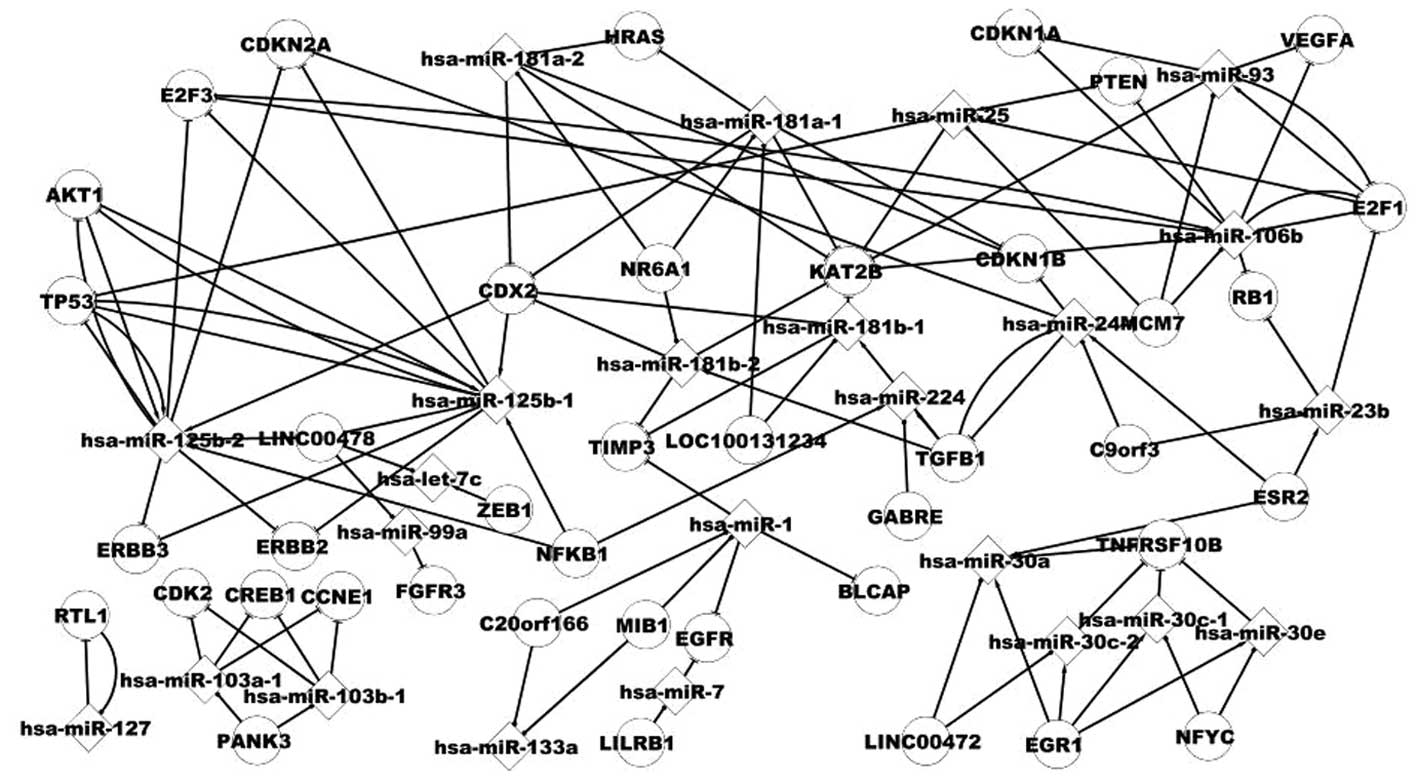
Analysis of the host gene and its miRNA
in BC
The host gene and its miRNA demonstrate some
important features in this study. Alhough these host genes are not
differentially expressed genes of BC, we considered them as
differentially expressed genes when miRNAs they regulate are
differentially expressed miRNAs. Fig.
3 shows some pathways of host genes and miRNAs, for example,
NR6A1 includes hsa-miR-181a-2, which targets HRAS. The association
between hsa-miR-93 and E2F1 is noteworthy. E2F1 includes hsa-miR-93
and is targeted by hsa-miR-93. A host gene includes several miRNAs,
for example, MIB1 includes hsa-miR-1 and hsa-miR-133a. A miRNA is
located in several genes, for example, hsa-miR-133a includes the
host genes MIB1 and C20orf166.
It is therefore suggested that host genes and their
miRNAs may be useful in our understanding of the pathogenesis of
BC.
Transcriptional network of TFs and
differentially expressed miRNAs
There are 82 miRNAs in the differentially expressed
network, of which 47 are included in Fig. 4. These miRNAs and popular TFs
constitute a transcriptional network. They present some significant
characteristics in BC formation. TFs and miRNAs present several
types of regulatory relations and affect the expression of their
target elements. Fig. 4 shows that
PTEN regulates three miRNAs, while being targeted by 12 miRNAs.
These miRNAs, i.e., hsa-miR-19a, hsa-miR-21 and hsa-miR-25, are
regulated by CDKN1A, but also target CDKN1A. CDKN1A regulates
hsa-miR-25, which in turn targets TP53. TP53, RB1, TP73 and VHL are
similar to CDKN1A and together with differentially expressed miRNAs
interact with each other to influence the progression of BC.
Fig. 4 also shows that one
differentially expressed miRNA is regulated by several TFs, one TF
is targeted by several differentially expressed miRNAs, one TF
indirectly influenced another TF by differentially expressed miRNA
and one differentially expressed miRNA indirectly influenced
another miRNA by TF.
Regulatory pathways of differentially
expressed genes
To describe the BC network more clearly, we
extracted the upstream and downstream information of the important
elements: differentially expressed genes, differentially expressed
miRNAs and popular TFs from the P-match method.
Nodes were classified according to the regulatory
relations, with adjacent nodes for the three-level networks in
order to compare and analyze the interactions of each
differentially expressed gene. Of the genes investigated, CDKN1A,
PTEN, TP53 and RB1 demonstrated the special feature involving each
gene regulating miRNA(s) and being regulated by the miRNA(s).
Discussion
In the present study, we initially focused on TFs.
The first class of TF has six types of adjacent nodes (three types
of successors and three types of predecessors). This class of TFs
includes CDKN1A, PTEN, TP53 and RB1.
Table I provides
evidence of the relationship of PTEN and miRNAs in each network. In
the core network, 12 miRNAs targeting PTEN were identified, with
PTEN regulating three miRNAs. In the BC-associated network 14
miRNAs (2 additional miRNAs: hsa-miR-19b-1 and hsa-miR-19b-2) were
identified to target PTEN, with PTEN regulating three miRNAs. In
the global network, 17 miRNAs (3 additional miRNAs: hsa-miR-214,
hsa-miR-217 and hsa-miR-216a) were identified to target PTEN, with
PTEN regulating 9 miRNAs (6 additional miRNAs: hsa-miR-22,
hsa-miR-302a, hsa-miR-302b, hsa-miR-302c, hsa-miR-302d and
hsa-miR-302f). These predecessors indirectly affected the
successors through PTEN. Of all the miRNAs, we observed that
hsa-miR-19a targets PTEN and PTEN regulates hsa-miR-19a in all
three networks. Additionally, they form self-adapting associations
and are both differentially expressed elements in BC.
 | Table IRegulatory relation between microRNAs
(miRNA) and PTEN. |
Table I
Regulatory relation between microRNAs
(miRNA) and PTEN.
| PTEN | |
|---|
|
|---|
| miRNAs that target
genes | miRNAs that are
regulated by genes |
|---|
|
|
|---|
| Differentially
expressed miRNAs | Related miRNAs | Global miRNAs | Differentially
expressed miRNAs | Related miRNAs | Global miRNAs |
|---|
| | hsa-miR-106b | | | |
| | hsa-miR-141 | | | |
| hsa-miR-106b | hsa-miR-17 | | | |
| hsa-miR-106b | hsa-miR-141 | hsa-miR-18a | | | |
| hsa-miR-141 | hsa-miR-17 | hsa-miR-19a | | | hsa-miR-19a |
| hsa-miR-17 | hsa-miR-18a | hsa-miR-20a | | | hsa-miR-25 |
| hsa-miR-18a | hsa-miR-19a | hsa-miR-21 | | | hsa-miR-21 |
| hsa-miR-19a | hsa-miR-20a | hsa-miR-221 | hsa-miR-19a | hsa-miR-19a | hsa-miR-22 |
| hsa-miR-20a | hsa-miR-21 | hsa-miR-222 | hsa-miR-21 | hsa-miR-21 | hsa-miR-302f |
| hsa-miR-21 | hsa-miR-221 | hsa-miR-26a-1 | hsa-miR-25 | hsa-miR-25 | hsa-miR-302a |
| hsa-miR-221 | hsa-miR-222 | hsa-miR-26a-2 | | | hsa-miR-302b |
| hsa-miR-222 | hsa-miR-26a-1 | hsa-miR-494 | | | hsa-miR-302c |
| hsa-miR-26a-1 | hsa-miR-26a-2 | hsa-miR-19b-1 | | | hsa-miR-302d |
| hsa-miR-26a-2 | hsa-miR-494 | hsa-miR-19b-2 | | | |
| hsa-miR-494 | hsa-miR-19b-1 | hsa-miR-216a | | | |
| hsa-miR-19b-2 | hsa-miR-217 | | | |
| | hsa-miR-214 | | | |
The second class of TFs has five types of adjacent
nodes (two types of successor and three types of predecessor), for
example, KRAS. Two differentially expressed miRNAs target KRAS,
however, KRAS does not regulate any miRNA.
The third class of TFs has four types of adjacent
nodes (three types of successors and a type of predecessor, a type
of successor and three types of predecessors), such as CDKN1A. Four
miRNAs target CDKN1A, which, however, does not regulate any miRNA
in the differentially expressed or BC-associated networks.
The fourth class of TFs has three types of adjacent
nodes (three types of successors), for example VHL. No miRNA
targets VHL, which only regulates hsa-miR-210.
We also focused on the genes that do not regulate
any miRNA. The first class of genes has three types of adjacent
nodes (three types of predecessors), including KRAS and HRAS.
Although they are targeted by some miRNAs, they do not regulate any
miRNA, possibly due to their being the last node in the pathway.
Oncogenic HRAS directly results in the development of BC (18).
The second class of genes has a type of adjacent
node (a type of predecessor), for example GRB2. GRB2 is targeted by
two miRNAs in the global network but does not regulate any miRNA.
It is suggested that GRB2 has the least influence compared to any
other differentially expressed genes.
In conclusion, we collected all currently validated
genes and miRNAs associated with BC and derived three regulatory
networks to analyze complex regulatory relations of differentially
expressed elements in BC. We extracted and compared the
similarities and differences of all differentially expressed
elements in the three networks to distinguish the key nodes and
pathways that contribute to understanding the tumorigenic mechanism
and therapy of BC. We observed that some pathways of differentially
expressed elements have been validated in BC, while other pathways
have not been validated in BC although they have an impact on other
types of cancer. Additionally, some related pathways in the
BC-associated network and additional pathways in the global network
also affect the progression of other types of cancer. Pathways of
differentially expressed elements must be involved in BC, however,
the majority of their mechanisms remain unclear. Consequently,
these pathways which have not been validated in BC potentially
affect the progression of cancer. However, their exact role in BC
remains to be clarified and additional investigations on these
pathways in BC should be conducted. The present study has provided
comprehensive data associated with BC which is useful in
investigations regarding the mechanism of differentially expressed
genes and miRNAs in BC. In the subsequent studies, transcriptional
co-factors and the interaction of proteins in our network are to be
further investigated in order to derive a more comprehensive and
extensive network to obtain greater insights into the pathogenesis
and treatments of BC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 60973091 and
60905022).
References
|
1
|
Jemal A, Murray T, Ward E, et al: Cancer
statistics, 2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar
|
|
2
|
Pashos CL, Botteman MF, et al: Bladder
cancer: epidemiology, diagnosis, and management. Cancer Pract.
10:311–322. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang DS, Rieger-Christ K, Latini JM, et
al: Molecular analysis of PTEN and MXI1 in primary bladder
carcinoma. Int J Cancer. 88:620–625. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Calin GA, Dumitru CD, Shimizu M, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao GJ, Huang BC, Liu ZH, et al: Intronic
miR-301 feedback regulates its host gene, ska2, in A549 cells by
targeting MEOX2 to affect ERK/CREB pathways. Biochem Biophys Res
Commun. 396:978–982. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sethupathy P, Corda B and Hatzigeorgiou
AG: TarBase: a comprehensive database of experimentally supported
animal microRNA targets. RNA. 12:192–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hsu SD, Lin FM, Wu WY, et al: miRTarBase:
a database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39:D163–D169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Lu M, Qiu CX and Cui QH: TransmiR:
a transcription factor-microRNA regulation database. Nucleic Acids
Res. 38:D119–D122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Griffiths-Jones S, Saini HK, van Dongen S
and Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids
Res. 36:D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanehisa M, Goto S, Furumichi M, Tanabe M
and Hirakawa M: KEGG for representation and analysis of molecular
networks involving diseases and drugs. Nucleic Acids Res.
38:D355–D360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chekmenev DS, Haid C and Kel AE: P-Match:
transcription factor binding site search by combining patterns and
weight matrices. Nucleic Acids Res. 33:W432–W437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dreszer TR, Karolchik D, Zweig AS, et al:
The UCSC Genome Browser database: extensions and updates 2011.
Nucleic Acids Res. 40:D918–D923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kao A, Poteet SR, Jones DH, et al:
P-MATCH: Identifying Part Name in Noisy Text Data.
Cross-Disciplinary Advances in Applied Natural Language Processing:
Issues and Approaches. Boonthum-Denecke C, McCarthy PM and Lamkin
T: IGI Global; Hershey: pp. 172–184. 2011
|
|
17
|
Jiang QH, Wang YD, Hao YY, et al:
miR2Disease: a manually curated database for microRNA deregulation
in human disease. Nucleic Acids Res. 37:D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Swallows DM, Gendler S, Gribths B, et al:
The hypervariable gene locus PUM, which encodes for the tumour
associated epithelial mucins, is located on chromosome 1, within
the region 1q21–24. Ann Hum Genet. 51:289–294. 1987.PubMed/NCBI
|
|
19
|
Zotter S, Hageman PC, Lossnitzer A, Mooi
WJ and Hilgers J: Tissue and tumour distribution of human
polymorphicepithelial mucin. Cancer Rev. 11:55–79. 1988.
|
|
20
|
Girling A, Bartkova J, Burchell J, et al:
A core protein epitope of the polymorphic epithelial mucin detected
by the monoclonal antibody SM-3 is selectively exposed in a range
of primary carcinomas. Int J Cancer. 43:1072–1076. 1989. View Article : Google Scholar
|
|
21
|
Simms MS, Hughes ODM, Limb M, Price MR and
Bishop MC: MUC1 mucin as a tumour marker in bladder cancer. BJU
Int. 84:350–352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kompier LC, Lurkin I, van der Aa MN, van
Rhijn BW, van der Kwast TH and Zwarthoff EC: FGFR3, HRAS, KRAS,
NRAS and PIK3CA mutations in bladder cancer and their potential as
biomarkers for surveillance and therapy. PloS One. 5:e138212010.
View Article : Google Scholar : PubMed/NCBI
|