Introduction
Congenital nephrogenic diabetes insipidus (CNDI) is
a rare disorder caused by mutations of the arginine vasopressin V2
receptor (AVPR2) or aquaporin 2 (AQP2) genes
(1,2). As patients with this disorder are
unable to concentrate urine effectively, polyuria with compensatory
polydipsia, nocturia and enuresis are commonly observed. The
well-known clinical manifestations of NDI in extremely early
infancy include poor weight gain with recurrent dehydration,
irritability, constipation, mild-intermittent fever, failure to
thrive and hypernatremia (3). A
prolonged period of insufficient water replacement or delay in the
diagnosis of NDI may lead to growth or mental retardation (4).
CNDI is divided into three types according to the
patterns of inheritance. The X-linked recessive form (OMIM 304800)
is caused by a mutation of the AVPR2 gene and is the most
commonly inherited pattern, accounting for ~90% of CNDI cases
(1). The autosomal recessive form
(OMIM 222000) is caused by a mutation in the AQP2 gene and
accounts for <10% of CNDI cases (2). The third type of CNDI is the extremely
rare autosomal dominant form (5).
The current study presented the case of CNDI in a
male infant with a novel mutation detected through the molecular
gene analysis of the AQP2 gene.
Case report
A 1-month-old male was referred to the Department of
Pediatrics (Eulji University Hospital, Daejeon, Korea) due to the
occurrence of hypernatremia and mild-intermittent fever for several
days. The patient had suffered from uncontrollable irritability
since birth, and was born at 39 weeks of gestation without asphyxia
with a birth weight of 3,310 g. The past medical history was
non-specific and the patient was the younger child among two
siblings. The patient had an unremarkable family history.
Vaccinations were administered as scheduled and on admission to the
department the height of the patient was 57 cm (25–75th
percentile), weight was 5.4 kg (50–75th percentile) and head
circumference was 39 cm (25–50th percentile). The patient was
frequently irritable and had a large urine volume output. The blood
pressure of the patient was 90/60 mmHg, with a regular heart rate
of 148 beats/min and respiratory rate of 32 breaths/min. The body
temperature of the patient was 36.6°C. On physical examination, the
patient was lethargic and his tongue and oral mucosa were mildly
dry. There was no evidence of pharyngeal or ear infection. The
chest, abdomen, genitourinary system and musculoskeletal system
showed no abnormalities.
The initial complete blood counts were as follows:
White blood cell and platelet counts of 8,940/μl and 418,000/μl,
respectively, with hemoglobin levels of 10.3 g/dl. The blood
chemistry results showed the following levels: Serum sodium, 154
mEq/l; serum potassium, 5.3 mEq/l; serum chloride, 122 mEq/l; blood
urea nitrogen, 19 mg/dl; creatinine, 0.5 mg/dl; protein, 6.1 mg/dl;
albumin, 3.9 mg/dl; and serum osmolality, 317 mOsm/kg. The urine
sodium concentration, specific gravity and osmolality were 12
mEq/l, 1.005, and 107 mOsm/kg, respectively. The total urine volume
output was 10.0 ml/kg/h on the first hospital day. Diabetes
insipidus was suspected and an arginine vasopressin (AVP)
stimulation test was performed (Table
I). The test results were compatible with NDI. The study was
reviewed and approved by the Institutional review board (IRB) of
Eulji University Hospital (Daejeon, Korea). Informed consent was
obtained for a molecular genetic study.
 | Table IArginine vasopressin (AVP) stimulation
test. |
Table I
Arginine vasopressin (AVP) stimulation
test.
| Time | Ur Osm (mosm/kg) | Serum Osm
(mosm/kg) |
|---|
| Basal | 116 | 285 |
| 1 h | 84 | 292 |
| 2 h | 78 | 297 |
| 3 h | 81 | 299 |
| 4 h | 80 | 303 |
Genomic DNA was extracted from nucleated cells in
the peripheral blood using a commercial kit (QIAamp DNA Blood Mini
Kit; Qiagen, Hilden, Germany). The 4 exons flanking the introns on
both sides of the AQP2 gene were amplified individually from
the genomic DNA using polymerase chain reaction (PCR) and were
directly sequenced. The sequences of the PCR primers were: sense,
5′-CATCCTGGCCCTGAGACA-3′ and antisense, 5′-GGATGGCAAAGTTGTGGC-3′
(exon 1); sense, 5′-CAGGAAGATGGAGCCAGAGA-3′ and antisense,
5′-TGGAGTGGTCTGTGTGTCTG-3′ (exon 2); sense,
5′-GGACTTCCTGCCCTGTCC-3′ and antisense, 5′-CCAGCTCTTGTTCTCCCT-3′
(exon 3); and sense, 5′-GCAGCTGGCGTTGTCGTTGT-3′ and antisense,
5′-TTCTGCCTCGGGCCTCACCC-3′ (exon 4).
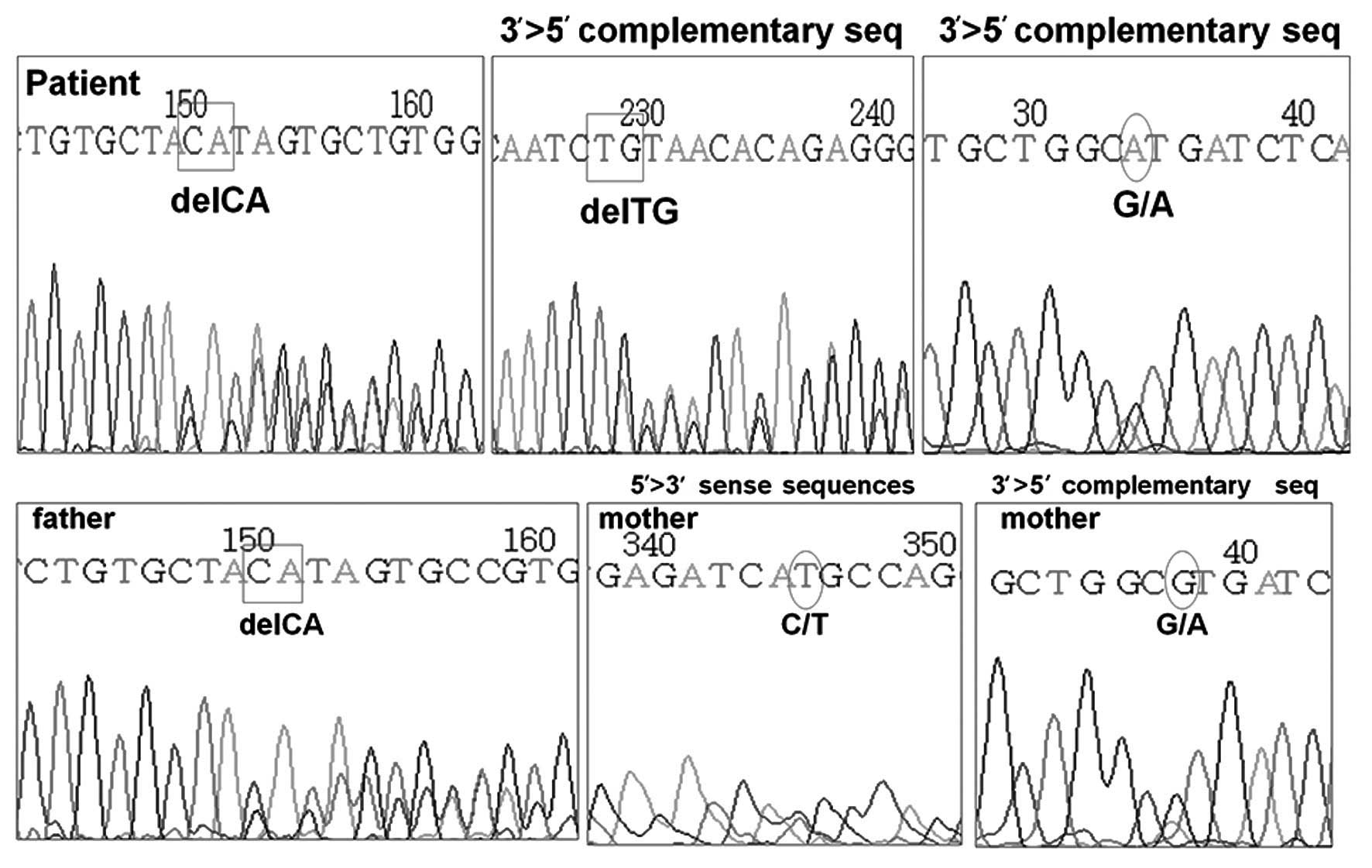
DNA analysis of the AVPR2 gene was normal,
however, the AQP2 gene revealed two heterozygous mutations
located in exon 1. One of the mutations was a C to T substitution
at the nucleotide position 108, leading to a missense mutation,
with the methionine being substituted for threonine. The other was
a frame-shift mutation resulting from a deletion of CA at
nucleotide position 127–128, with glutamine in position 43 at codon
CAG as the first affected amino acid, and the new reading frame
ending in a termination codon at position 62. The former mutation
was confirmed to be a heterozygous mutation from the mother of the
patient, and the latter was confirmed to be from the father
(Fig. 1). The treatment of the
patient commenced with hydrochlorothiazide and amiloride for the
management of NDI, but he continued to present with poor weight
gain while the polyuria and polydipsia were partially relieved.
Discussion
The current study presents the case of a 1-month-old
male with CNDI that was caused by novel mutations in the
AQP2 gene. DNA analysis of the AQP2 gene showed that
the patient was a compound heterozygote. The molecular study
revealed that the missense mutation was inherited maternally and
the deletion mutation was inherited paternally. The parents showed
no signs and symptoms of diabetes insipidus suggesting autosomal
recessive inheritance. The brother of the patient did not undergo
molecular genetic analysis as the parents did not consent to
further study.
A review of the literature indicated that there were
44 AQP2 gene mutations associated with CNDI (5–11). The
missense mutation of 108Thr (ACG) to Met (ATG) was found
to be a novel mutation. A study by Tajima et al (5) described a young Japanese male with a
deletion mutation in exon 1 of the AQP2 gene, which was the
same as the patient of the present study (127, 128 delCA). In
Korea, three novel mutations of two patients have been reported.
Cheong et al (6) reported a
Korean female CNDI patient with two novel missense AQP2 gene
mutations (A70D, R187H). The patient had polyuria and polydipsia,
and was diagnosed with CNDI at 3 years. Moon et al (7) reported a Korean male CNDI patient with
a novel missense AQP2 mutation (S216F). The 18-year-old
patient was diagnosed with CNDI and was suffering from severe
polyuria and polydipsia. Although the patients showed typical
symptoms of NDI at the time of diagnosis, both of the patients
suffered from non-specific symptoms, such as recurrent episodes of
fever, in their infancy.
NDI is a condition that results from vasopressin
insensitivity at the level of the kidney, leading to polyuria,
polydipsia and dehydration that can result in failure to thrive or
mental retardation. In early infants with CNDI, polyuria and
polydipsia cannot be detected easily. Since non-specific symptoms,
including vomiting, anorexia, failure to thrive, fever and
constipation, are the most commonly reported clinical symptoms of
CNDI infants (3), it is often
challenging to diagnose the patients with CNDI in infancy. The
patient of the present study was misdiagnosed as having a fever
without a focus on his first visit to a local clinic. When the
patient visited a local clinic again, a serum electrolyte study
revealed hypernatremia, following which the patient was referred to
the Eulji University Hospital (Daejeon, Korea) with suspected
CNDI.
Similar to the patient of the present study, the
majority of infants with CNDI are not diagnosed until severe
hypernatremia occurs, which predisposes them to a risk of severe
dehydration. Recurrent episodes of severe dehydration are the cause
of growth delay and mental retardation in CNDI patients. In
contrast to a previous study, the prevalence of mental retardation
in CNDI patients appears lower than expected (4). However, growth delay develops in the
majority of CNDI patients immediately following birth, and catch-up
growth occurs subsequent to the initiation of treatment (12). As infants with CNDI are unable to
meet their high fluid demands, this age group is the most likely to
experience severe dehydration and its consequences, such as growth
delay and mental retardation. Therefore, the early diagnosis of
CNDI and prompt initiation of treatment are crucial.
The AQP2 gene is a small gene with four exons
and 5 kb of genomic DNA (13),
which enables direct sequencing of the gene to identify CNDI in
clinical practice. The molecular identification of the AQP2
gene has immediate clinical significance, as early recognition of
CNDI in infants, showing only non-specific symptoms, can be
facilitated. Thus, repeated episodes of dehydration, which may
cause physical and mental retardation, can be avoided (14). The accumulating spectrum of the
AQP2 gene mutations and their inheritance patterns not only
aids early recognition of the disease, but also enables genetic
counseling and prenatal diagnosis in high-risk populations. As
growth retardation in CNDI patients begins immediately following
birth (12), anticipation of the
disease prenatally and the prompt initiation of treatment may be
beneficial to the patients.
In conclusion, the present study reports a patient
with CNDI caused by compound heterozygote mutations in the
AQP2 gene. The study revealed a novel mutation in the
AQP2 gene, 108Thr (ACG) to Met (ATG), which has
not been previously described, and thus expands the spectrum of the
AQP2 gene mutations.
References
|
1
|
Birnbaumer M, Seibold A, Gilbert S, Ishido
M, Barberis C, Antaramian A, Brabet P and Rosenthal W: Molecular
cloning of the receptor for human antidiuretic hormone. Nature.
357:333–335. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fushimi K, Uchida S, Hara Y, Hirata Y,
Marumo F and Sasaki S: Cloning and expression of apical membrane
water channel of rat kidney collecting tubule. Nature. 361:549–552.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Lieburg AF, Knoers NV and Monnens LA:
Clinical presentation and follow-up of 30 patients with congenital
nephrogenic diabetes insipidus. J Am Soc Nephrol. 10:1958–1964.
1999.PubMed/NCBI
|
|
4
|
Hoekstra JA, van Lieburg AF, Monnens LA,
Hulstijn-Dirkmaat GM and Knoers VV: Cognitive and psychosocial
functioning of patients with congenital nephrogenic diabetes
insipidus. Am J Med Genet. 61:81–88. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tajima T, Okuhara K, Satoh K, Nakae J and
Fujieda K: Two novel aquaporin-2 mutations in a sporadic Japanese
patient with autosomal recessive nephrogenic diabetes insipidus.
Endocr J. 50:473–476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheong HI, Cho SJ, Zheng SH, Cho HY, Ha IS
and Choi Y: Two novel mutations in the aquaporin 2 gene in a girl
with congenital nephrogenic diabetes insipidus. J Korean Med Sci.
20:1076–1078. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moon SS, Kim HJ, Choi YK, Seo HA, Jeon JH,
Lee JE, Lee JY, Kwon TH, Kim JG, Kim BW and Lee IK: Novel mutation
of aquaporin-2 gene in a patient with congenital nephrogenic
diabetes insipidus. Endocr J. 56:905–910. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loonen AJ, Knoers NV, van Os CH and Deen
PM: Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin
Nephrol. 28:252–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liberatore RD Junior, Carneiro JG, Leidenz
FB, Melilo-Carolino R, Sarubi HC and De Marco L: Novel compound
aquaporin 2 mutations in nephrogenic diabetes insipidus. Clinics
(Sao Paulo). 67:79–82. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sahakitrungruang T, Wacharasindhu S,
Sinthuwiwat T, Supornsilchai V, Suphapeetiporn K and Shotelersuk V:
Identification of two novel aquaporin-2 mutations in a Thai girl
with congenital nephrogenic diabetes insipidus. Endocrine.
33:210–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robben JH, Knoers NVAM and Deen PM: Cell
biological aspects of the vasopressin type-2 receptor and aquaporin
2 water channel in nephrogenic diabetes insipidus. Am J Physiol
Renal Physiol. 291:F257–270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lejarraga H, Caletti MG, Caino S and
Jiménez A: Long term growth of children with nephrogenic diabetes
insipidus. Pediatr Nephrol. 23:2007–2012. 2008. View Article : Google Scholar
|
|
13
|
Knoers NV and Deen PM: Molecular and
cellular defects in nephrogenic diabetes insipidus. Pediatr
Nephrol. 16:1146–1152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bichet DG: Nephrogenic diabetes insipidus.
Adv Chronic Kidney Dis. 13:96–104. 2006. View Article : Google Scholar
|