Introduction
Hypertension is one of the most significant public
issues with a high worldwide prevalence and its treatment can lead
to reduced incidences of complications, such as stroke, myocardial
infarction and renal disease (1).
There are a number of antihypertensive drugs of various categories,
including angiotensin-converting enzyme (ACE) inhibitors, calcium
channel blockers, β-blockers and angiotensin II receptor
antagonists. Thus far, clinical trials have indicated that
monotherapy, the use of a single drug, is insufficient for
achieving the goal blood pressure in patients with hypertension and
ongoing trials have provided guidance on the appropriate
combination regimens using ≥2 antihypertensive drugs for increasing
the synergic effects and reducing the unexpected adverse effects
(2,3). Furthermore, the combination regimens
targeting functional improvement, as well as antihypertension,
enhance the therapeutic effects even when the monotherapy is not
evident in patients with renal dysfunction (4).
Perindopril is a long-acting ACE inhibitor that
results in preventing the generation of angiotensin II in the
renin-angiotensin-aldosterone system and subsequently lowering
blood pressure. Numerous studies have revealed that perindopril is
useful for treatment of hypertension (5), chronic heart failure (6) and diabetic nephropathies (7). Perindopril has good preclinical
profiles with an LD50 at relatively high doses in
various experimental animals (8),
and clinical introduction and post-marketing surveillance studies
have shown that perindopril is well-tolerated in a wide range of
patients with hypertension (9).
However, perindopril has a risk of severe hypotension despite low
incidences and possible fetal and neonatal morbidity and mortality
when used during pregnancy (9),
indicating that the use of perindopril requires caution to avoid
the unexpected adverse effects.
Natural products have received increasing attention
in the development of novel drug materials. There are a number of
natural herbal products based on Korean medicine that have been
adjusted from traditional Chinese medicine and the commercially
available herbal drugs have been evaluated for novel combination
regimens as an adjunctive medication (10). Wu Ling San (Oryungsan, ORS) known as
a five-ingredient formula with poria, is the most famous
nephroprotective Korean traditional polyherbal formula (11). The accumulated clinical trials have
shown that ORS is useful for various diseases involved in
hypertension, such as kidney diseases, cardiac edema, ascites,
diabetes, liver cirrhosis and hydrocephalus. In addition, the
therapeutic improvement has been revealed in experimental animal
models of renal damage (12),
nephrotic syndrome (13) and renal
dysfunction (14).
Chungsinoryungsan (CSORS) is based on the materials of ORS and 20
types of herb exhibiting nephroprotective effects are also added
additionally (15). CSORS is
indicated to possibly be useful in combination with
antihypertensive drugs as an adjunctive medication. Therefore, the
aim of the present study was to examine the drug-drug interactions
between CSORS and perindopril via comprehensive pharmacokinetic
analyses.
Materials and methods
Animals
Six-week-old male Sprague-Dawley rats (170–190 g)
were obtained from Japan SLC, Inc. (Shizuoka, Japan). A total of 20
rats were separated randomly to five per polycarbonate cage and
acclimatized in a room controlled at 20–25°C and 40–45% humidity
for 2 weeks. The rats were maintained on a 12-h light/dark cycle
with free access to standard rodent chow and water. All the
experimental procedures were approved by the Institutional Animal
Care and Use Committee at Daegu Haany University (Gyeongsan,
Korea).
Drugs and treatment
Perindopril was purchased from Panaaya Pharma
Private, Ltd. (Hyderabad, India). CSORS was prepared at the
Department of Herbology (College of Korean Medicine, Daegu Haany
University). For producing CSORS, 25 types of herb were purchased
from Jecheon Hanbang Yakcho (Jecheon, Korea) following confirmation
of the complete morphology under microscopy (Table I). The herbs (1,420 g) were boiled
in 2 l distilled water for 3 h, three times at 80°C and
subsequently filtered. The resultant filtrate was decompressed with
a rotary vacuum evaporator (Rotavapor R-144; Buchi, Flawil,
Switzerland) and lyophilized in a programmable freeze dryer
(FreeZone 1 Liter Benchtop; Labconco Corporation, Kansas City, MO,
USA). Eventually, the acquired CSORS extract volume was 173.24 g as
a light brown powder (yield, 12.2%). The perindopril and CSORS
drugs were stored as a powder at 4°C in the dark until
required.
 | Table ITwenty five types of herb consisting
of Chungsinoryungsan aqueous extracts. |
Table I
Twenty five types of herb consisting
of Chungsinoryungsan aqueous extracts.
| Herbs | Scientific
names/produce region | Amounts, g |
|---|
| Alismatis
rhizoma | Alisma
orientale (Sam.) Juz./Chunnam | 100 |
| Tokoro rhizoma | Dioscorea
tokoro Makino/China | 100 |
| Alpiniae
fructus | Alpinia
oxyphylla Miquel/China | 80 |
| Polyporus | Dendropolyporus
umbellatus (Pers.:Fr.) Jülich/China | 80 |
| Hoelen | Poria cocos
Wolf//China | 80 |
| Dioscoreae
rhizoma | Dioscorea
batatus Decne./Kyungbuk | 80 |
| Astragali
radix | Astragalus
membranaceus Bunge/Chungbuk | 60 |
| Mantidis
ootheca | Paratenodera
sinensis De Saussure/China | 60 |
| Atractylodis
rhizoma alba | Atractylodes
ovata (Thunb.) DC./China | 60 |
| Nelumbinis
semen | Nelumbo
nucifera Gaertn./China | 60 |
| Acori Gramineri
rhizoma | Acorus
gramineus Soland./China | 60 |
| Artemisiae
capillaris herba | Artemisia
capillaris Thunberg/Kyungbuk | 60 |
| Plantaginis
semen | Plantago
asiatica L./China | 60 |
| Amomi fructus | Amomum
villosum Loureiro var. xanthioides T.L.Wu et
Senjen/China | 60 |
| Remotiflori
radix | Adenophora
remotiflora (Siebold and Zucc.) Miq./China | 60 |
| Citri unshii
pericarpium | Citrus
unshiu S.Marcov./Cheju | 40 |
| Fossilia ossis
mastodi | Fossilia ossis
mastodi/China | 40 |
| Terminaliae
fructus | Terminalia
chebula Retz./China | 40 |
| Ginseng radix
alba | Panax
ginseng C.A.Meyer/Chungnam | 40 |
| Cimicifugae
rhizoma | Cimicifuga
heracleifolia Kom./China | 40 |
| Aurantii immaturus
fructus | Citrus
aurantium L./China | 40 |
| Myristicae
semen | Myristica
fragrans Houtt./China | 40 |
| Pulvis ostreae
testa | Crassostrea
gigas Thunberg/China | 30 |
| Cinnamomi
cortex | Cinnamomum
cassia J. Presl./China | 30 |
| Mume fructus | Prunus mume
Siebold et Zuccarini/China | 20 |
One batch of 10 rats received single oral dosing of
perindopril combination with CSORS (combination group) or
perindopril with distilled water (control) and another batch of 10
rats received repeated oral dosing of combination and control once
a day for a week. The co-administration with CSORS or distilled
water was performed by the single dosing within 5 min after
perindopril, or the repeated dosing at a 2-h interval after
perindopril. The drug dosing was a volume of 5 ml/kg at 100 ml/kg
CSORS and 50 mg/kg perindopril, based on its toxicity and clinical
database (8). Body weights were
measured prior to every administration using an automatic
electronic balance (Precisa Instruments AG, Dietikon,
Switzerland).
Collection of blood samples and sample
preparation
The rats were fasted overnight a day before
collection of the blood sample to avoid dietary effects. The blood
sample via the retro-orbital route was collected in anticoagulant
tubes, including 50 IU heparin, at 0.5 h prior to the
administration and 0.5, 1, 2, 3, 4, 6, 8 and 24 h
post-administration. The plasma sample was centrifuged at 11,400 ×
g for 10 min and the supernatant aliquot was stored at −70°C until
pharmacokinetic analyses.
Sample preparation and calibrations
For a calibration, 1.0 mg/ml perindopril (Sigma, St.
Louis, MO, USA) diluted with 50% acetonitrile was used as a primary
stock solution and 500 ng/ml carbamazepine (Sigma) in acetonitrile
was used as an internal standard (IS) solution. The working
standard solutions were prepared by dilution of the primary stock
solution with acetonitrile and stored in the dark at −20°C. The 100
μl working standard solutions were mixed with 100 μl blank plasma
and IS solutions in 100 μl acetonitrile for the perindopril
concentration standard curve. The 100-μl plasma sample was prepared
as a mixture with 100-μl IS solution in 200 μl acetonitrile. The
mixtures were centrifuged at 9,700 × g for 10 min at 4°C and the
supernatant was transferred to injection vials for liquid
chromatography-mass spectrometry/mass spectrometry (LC-MS/MS).
LC-MS/MS conditions
Chromatographic analysis was performed using an
Agilent 1100 series HPLC (Agilent Technologies, Santa Clara, CA,
USA) equipped with online degasser, binary pump, auto-sampler,
column compartment and column oven at 30°C. Separation of the
analyte from potentially interfering material was achieved using
Waters XTerra MS C18 columns (2.1×50 mm, 3.5 μm) (Waters
Corporation, Milford, MA, USA). The mobile phase for
chromatographic separation was composed of 5–95% acetonitrile,
including 0.1% formic acid, and it was delivered isocratically at a
flow rate of 0.3 ml/min. The column effluent was monitored using an
API 2000 triple quadrupole mass-spectrometer detector (Applied
Biosystems, Foster City, CA, USA). The instrument was equipped with
an electrospray interface in positive-ion mode, which was
controlled by the Analyst version 1.4.2 software (Applied
Biosystems). The samples were introduced to the interface through
turbo ionspray at 400°C. A high positive voltage of 5.0 kV was
applied to the ion spray. Nitrogen was used as nebulizer gas,
curtain gas and collision gas with sets of 12, 6 and 8 PSI,
respectively. The multiple reaction monitoring detection method was
employed for the detection of perindopril; the transitions
monitored were carbamazepine (IS): m/z 237>194 (retention time,
2.7 min); and perindopril: 369>172 (retention time, 2.5 min).
Calibration curves of perindopril were linear over the ranges with
r2>0.999. The lower limit of quantification was 0.1
ng/ml.
Pharmacokinetic analyses
The perindopril concentration in plasma was analyzed
using a non-compartmental method on the commercial pharmacokinetics
data analyzer program (PK Solutions 2.0; Summit Research Services,
Montrose, CO, USA) (16). The
elimination rate constant (Kel) was calculated by
log-linear regression of perindopril concentration data during the
elimination phase, and the terminal half-life (t1/2) was
calculated by 0.693/Kel. The peak concentration
(Cmax) of plasma perindopril and time to reach the
Cmax (Tmax) were obtained by visual
inspection in the concentration-time curve. The area under the
perindopril concentration-time curve (AUC0-t) from time
zero to the time of the measured concentration (Clast)
was calculated using the linear trapezoidal rule (17). The AUC zero to infinity
(AUC0-inf) was obtained by adding AUC0-t and
the extrapolated area was determined by
Clast/Kel. The mean residence time to
infinity (MRTinf) was calculated by dividing the first
moment curve (AUMC0-inf) by AUC0-inf.
Statistical analyses
All the data are presented as average values ±
standard error of the mean (SEM). Data for body weights and
perindopril concentration were examined by testing the homogeneity
of variance, followed by analysis of variance (ANOVA) with the
group as a main effect. The day on which the body weights were
measured or the time collected for plasma samples was treated as
repeated measurements. When the data passed at the test of
homogeneity of variance, they were compared by independent t-test
for post hoc test, otherwise, the data were compared by
Mann-Whitney U test. All the pharmacokinetic parameters were
examined by Mann-Whitney U test as a non-parametric comparison due
to the small sample sizes, which have difficulties reaching a
normal distribution. For all analyses, P<0.05 was considered to
indicate a statistically significant difference.
Results
Single oral administration of perindopril
combination with CSORS within 5 min
Body weight changes
There were no differences in the body weights
between the combination and control treatment (F=0.02, P>0.10).
The weight changes were 26.0±1.5 and 27.6±1.0 g in the combination
and control groups, respectively.
Perindopril concentration
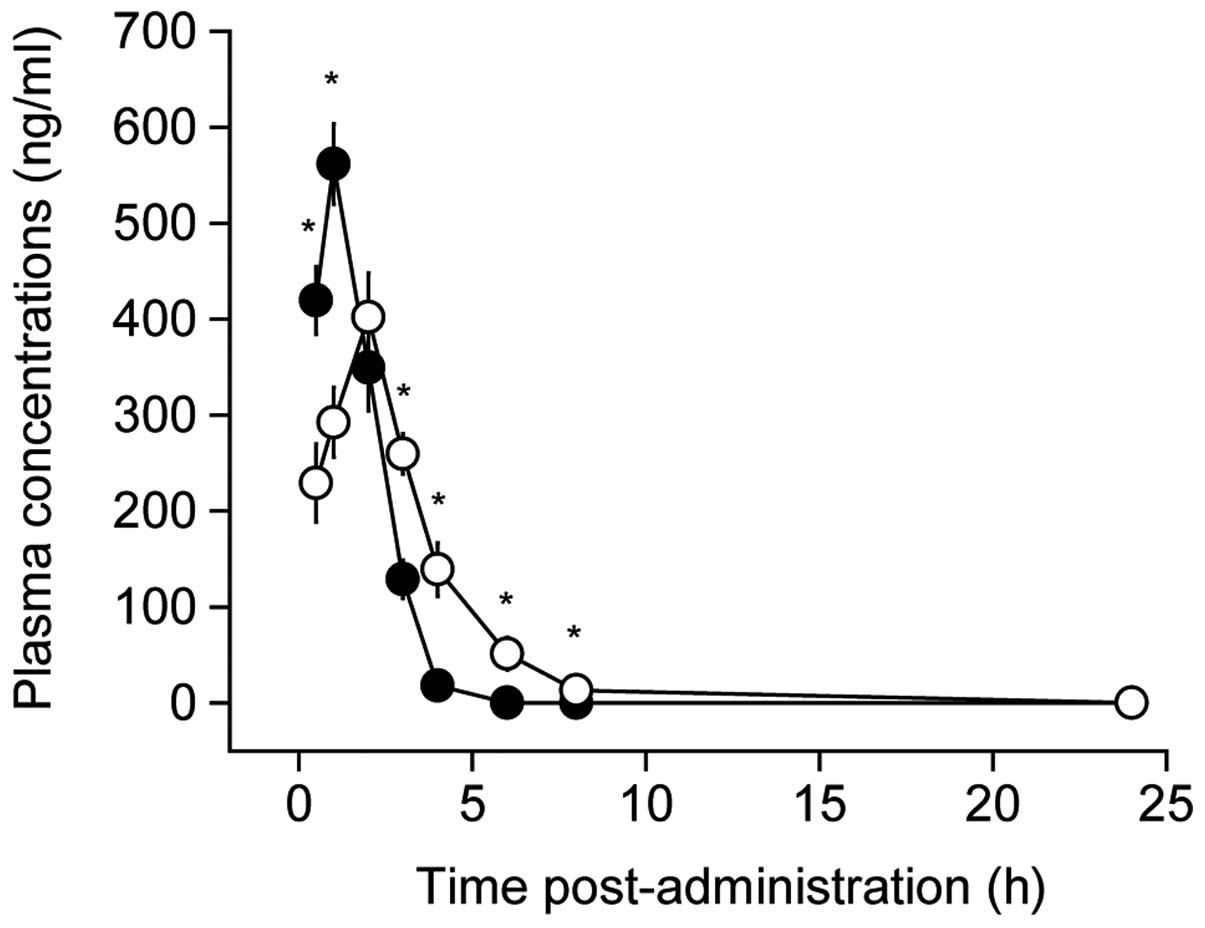
Perindopril was detected until 8 h
post-administration in the combination treatment, whereas it was
detected until 4 h in the control treatment (Fig. 1). The kinetics of perindopril
concentration were examined by ANOVA with the group as a main
effect and the collected time was treated as a repeated
measurement. Overall, there were significant main effects for time
(F=143.8, P<0.01), indicating time-dependent perindopril
concentration. Although no main effects for group were found
(F=0.3, P>0.10), there were significant interactions between
time and group (F=24.0, P<0.01). Post hoc test revealed that the
combination treatment reduced the perindopril concentration by 55%
at 0.5 h post-administration and 52% at 1 h, and increased by 202,
771, 613 and 231% at 3, 4, 6 and 8 h, respectively, compared to the
control (P<0.01) (Fig. 1). This
indicates altered perindopril pharmacodynamics by CSORS.
Perindopril pharmacokinetics
Although it was not significant (P=0.06), for group
analysis the Cmax showed a 28% reduction in the
combination (401.9±41.9 ng/ml) compared to the control treatment
(561.5±46.0 ng/ml) (Fig. 2A).
However, there were significant main effects for group for
Tmax (P<0.01) and t1/2 (P<0.01)
(Fig. 2B and C). Tmax
for the combination group was increased by 200% compared to the
control and t1/2 was reduced by 60%. Tmax was
2.0±0.0 vs. 1.0±0.0 h in the combination versus the control
treatment and t1/2 was 0.59±0.03 h vs. 1.47±0.15 h,
respectively. AUC0-t of perindopril was not
significantly increased in the combination (1,319.6±160.4 ng h/ml)
compared to the control group (1,117.9±82.5 ng h/ml) (P>0.10)
(Fig. 2D). No differences were
detected in AUC0-inf between the groups (P>0.10)
(Fig. 2E). However,
MRTinf was significantly increased by 78% in the
combination (2.7±0.1 h) compared to the control treatment (1.5±0.0
h) (P<0.01) (Fig. 2F). These
results indicate delayed absorption and excretion of perindopril by
combination with CSORS within 5 min.
Repeated oral administration of
perindopril combination with CSORS for a week at a 2-h
interval
Body weight changes
No evident differences were found in the gross
aspects of behavior and weight changes (Table II). ANOVA revealed no main effects
for the group (F=0.001, P>0.10) and no interactions between
group and measured days (F=0.2, P>0.10).
| Table IIBody weight changes following
repeated administration of perindopril combination with
Chungsinoryungsan for a week at a 2-h interval. |
Table II
Body weight changes following
repeated administration of perindopril combination with
Chungsinoryungsan for a week at a 2-h interval.
| Body weight
(g) |
|---|
|
|
|---|
| Perindopril
combination | Distilled
water | CSORS |
|---|
| Initial
co-administration [A] | 227.4±2.8 | 227.8±6.0 |
| Last
co-administration [B] | 248.2±3.6 | 248.0±4.6 |
| Changes
[B]-[A] | 20.8±2.7 | 20.2±2.3 |
Perindopril concentration
Following the initial and last co-administration,
the perindopril was detected up until 4 h post-administration in
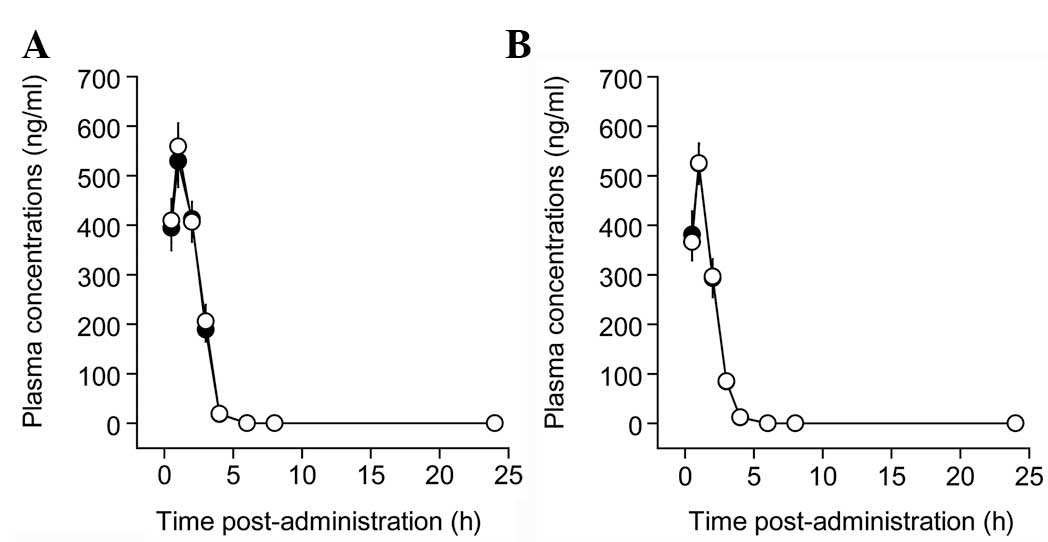
the combination and control groups (Fig. 3). The time-concentration graph was
similar between the combination and control groups. Following the
initial co-administration (Fig.
3A), there were significant main effects for time (F=185.3,
P<0.01), but no main effects for group (F=0.1, P>0.10) and no
interaction between time and group (F=0.1, P>0.10). Following
the last co-administration of the repeated administration (Fig. 3B), there were significant main
effects for time (F=205.3, P<0.01), but no main effects for
group (F=0.07, P>0.10) and no interaction between time and group
(F=0.04, P>0.10). These indicate limited interaction between
perindopril and CSORS by co-administration at a 2-h interval.
Perindopril pharmacokinetics
The perindopril combination with CSORS at a 2-h
interval showed no differences in Tmax, Cmax,
t1/2, AUC0-t, AUC0-inf and
MRTinf compared to the control following the initial and
last co-administration of the repeated administration for a week
(Fig. 4). Mann-Whitney U test
revealed no main effects for group for any of the parameters
assessed (P>0.10).
Discussion
The effects of CSORS administration on
pharmacokinetics of perindopril were examined in the present study.
When perindopril was co-administered with CSORS within 5 min, the
perindopril plasma concentration was different from the normal
pharmacokinetics of the control (Fig.
1). The pharmacokinetic parameters showed reduced
t1/2 and increased Tmax and MRTinf
in the combination compared to the control group. This indicates a
drug-drug interaction between perindopril and CSORS (Fig. 2). Perindopril was hypothesized to
possibly have a limited interaction with CSORS co-administration at
an interval gap that was more than perindopril MRTinf of
the control treatment (1.51±0.09 h). When perindopril was
co-administered with CSORS at a 2-h interval, the perindopril
concentration and pharmacokinetic parameters were not different
between the combination and control groups following the initial
and last administration of a weekly repeated dosing (Figs. 3 and 4). These results provide detailed
information for the drug regimen of perindopril combination with
CSORS.
Perindopril has been shown to have various drug-drug
interactions with diuretics (18,19),
gentamicin (20) and lithium
(21,22). However, there have been limited
studies regarding the interactions between perindopril and natural
herbal products, except for digoxin (23,24).
In the present study, single oral administration of perindopril
combination with CSORS within 5 min markedly delayed the absorption
of perindopril and its excretion, whereas the co-administration of
the combination at a 2-h interval showed no interaction between
perindopril and CSORS even by a weekly repeated dosing. Perindopril
is well-absorbed in the gastrointestinal tract with a high
bioavailability of 75% via the oral route (25), however, it is extensively
metabolized to six metabolites, including perindoprilat, an active
metabolite, in the liver (26,27).
The maximal concentration of plasma perindoprilat is reached 2–6 h
after oral administration of perindopril and 70% of perindoprilat
is cleared by the kidneys. Food does not influence the rate or
extent of perindopril absorption but reduces conversion to
perindoprilat by ~35% (28). The
present study results showed a Tmax of 1 h in the
control group, which had a similarity with that of humans (Figs 2B and 4B) (26).
However, perindopril combination with CSORS within 5 min resulted
in 2 h of Tmax. Although the exact mechanism regarding
how CSORS interacted with perindopril is unclear, it may be due to
partial interruption of perindopril absorption by coexistence with
CSORS or delayed conversion of perindopril to perindoprilat.
In the present study, CSORS had no interaction with
perindopril in a weekly repeated co-administration at 2-h
intervals, which indicates the suitable dosing regimen for the
combination therapy. However, there are numerous clinical factors
that alter perindopril pharmacokinetics. Since the active
metabolites of perindopril are hydrolyzed in the liver and
primarily excreted into the urine, the elimination kinetics can be
altered in hepatic impairment (26,29),
renal failure (30) or chronic
heart failure (31). Ageing is also
associated with the alteration in enhanced conversion to
perindoprilat and the reduced renal clearance (32). Therefore, perindopril combination
therapy requires further clinical studies for the pharmacokinetics
in specific disease conditions. To the best of our knowledge, this
is the first study to monitor the use of CSORS in combination with
antihypertensive drugs. The results showed CSORS co-administration
has limited interaction with perindopril at an interval that was
more than mean residence time of perindopril. These results provide
detailed information for a drug dosing regimen of perindopril with
CSORS in human clinical studies of novel combination therapy.
Acknowledgements
The present study was supported in part by grant of
Korea of Health and Welfare, Republic of Korea (grant no.
20-11-0-090-091-3000-3033-320).
References
|
1
|
Kearney PM, Whelton M, Reynolds K, Whelton
PK and He J: Worldwide prevalence of hypertension: a systematic
review. J Hypertens. 22:11–19. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nesbitt SD: Antihypertensive combination
therapy: optimizing blood pressure control and cardiovascular risk
reduction. J Clin Hypertens (Greenwich). 9(Suppl 4): 26–32. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salahuddin A, Mushtaq M and Materson BJ:
Combination therapy for hypertension 2013: an update. J Am Soc
Hypertens. 7:401–407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakao N, Yoshimura A, Morita H, Takada M,
Kayano T and Ideura T: Combination treatment of angiotensin-II
receptor blocker and angiotensin-converting-enzyme inhibitor in
non-diabetic renal disease (COOPERATE): a randomised controlled
trial. Lancet. 361:117–124. 2003. View Article : Google Scholar
|
|
5
|
Todd PA and Fitton A: Perindopril. A
review of its pharmacological properties and therapeutic use in
cardiovascular disorders. Drugs. 42:90–114. 1991.PubMed/NCBI
|
|
6
|
Cleland JG, Tendera M, Adamus J, et al:
The perindopril in elderly people with chronic heart failure
(PEP-CHF) study. Eur Heart J. 27:2338–2345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waeber B, de la Sierra A and Ruilope LM:
The ADVANCE trial: clarifying the role of perindopril/indapamide
fixed-dose combination in the reduction of cardiovascular and renal
events in patients with diabetes mellitus. Am J Cardiovasc Drugs.
9:283–291. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Santa Cruz Biotechnology. Material safety
data sheet of perindopril (sc-205799). http://pdf.analysis1.org/perindopril-santa-cruz-biotechnology-w1778uri.
Accessed on December 6, 2010
|
|
9
|
Clark LT: Safety profile of perindopril.
Am J Cardiol. 88:36i–40i. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ji HF, Li XJ and Zhang HY: Natural
products and drug discovery. Can thousands of years of ancient
medical knowledge lead us to new and powerful drug combinations in
the fight against cancer and dementia? EMBO Rep. 10:194–200.
2009.PubMed/NCBI
|
|
11
|
Scheid V, Bensky B, Ellis A and Barolet R:
Chinese Herbal Medicine: Formulas and Strategies. 2nd edition.
Eastland Press; Seattle, WA: pp. 724–728. 2009
|
|
12
|
Liu IM, Tzeng TF, Liou SS and Chang CJ:
The amelioration of streptozotocin diabetes-induced renal damage by
Wu-Ling-San (Hoelen Five Herb Formula), a traditional Chinese
prescription. J Ethnopharmacol. 124:211–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He L, Rong X, Jiang JM, Liu PQ and Li Y:
Amelioration of anti-cancer agent adriamycin-induced nephrotic
syndrome in rats by Wulingsan (Gorei-San), a blended traditional
Chinese herbal medicine. Food Chem Toxicol. 46:1452–1460. 2008.
View Article : Google Scholar
|
|
14
|
Ding XQ, Pan Y, Wang X, Ma YX and Kong LD:
Wuling san ameliorates urate under-excretion and renal dysfunction
in hyperuricemic mice. Chin J Nat Med. 11:214–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu YP: Chinese Materia Medica: Chemistry,
Pharmacology, and Applications. Harwood Academic; Amsterdam: pp.
311–344. 1998
|
|
16
|
DeVane CL: Pharmacokinetics (2nd edition,
revised and expanded), M. Gibaldi and D. Perrier (vol. 15 of Drugs
and the Pharmaceutical sciences), Marcel Dekker, New York, 1982.
Biopharmaceutics and Drug Disposition. 4:201. 1983. View Article : Google Scholar
|
|
17
|
Chiou WL: Critical evaluation of the
potential error in pharmacokinetic studies of using the linear
trapezoidal rule method for the calculation of the area under the
plasma level - time curve. J Pharmacokinet Biopharm. 6:539–546.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brown CL, Backhouse CI, Grippat JC and
Santoni JP: The effect of perindopril and hydrochlorothiazide alone
and in combination on blood pressure and on the renin-angiotensin
system in hypertensive subjects. Eur J Clin Pharmacol. 39:327–332.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scalbert E, Abdon D, Devissaguet M and
Juggi JS: Interaction between an angiotensin converting enzyme
inhibitor, perindopril, and a thiazide diuretic in the
spontaneously hypertensive rat. Can J Cardiol. 8:381–386.
1992.PubMed/NCBI
|
|
20
|
Morin JP, Thomas N, Toutain H, Borghi H
and Fillastre JP: Treatment with an angiotensin converting enzyme
inhibitor may increase the nephrotoxicity of gentamicin in rats.
Pathol Biol (Paris). 37:652–656. 1989.(In French).
|
|
21
|
Vipond AJ, Bakewell S, Telford R and
Nicholls AJ: Lithium toxicity. Anaesthesia. 51:1156–1158. 1996.
View Article : Google Scholar
|
|
22
|
Christensen S, Shalmi M, Hansen AK and
Marcussen N: Effects of perindopril and hydrochlorothiazide on the
long-term progression of lithium-induced chronic renal failure in
rats. Pharmacol Toxicol. 80:132–141. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vandenburg MJ, Stephens JD, Resplandy G,
Dews IM, Robinson J and Desche P: Digoxin pharmacokinetics and
perindopril in heart failure patients. J Clin Pharmacol.
33:146–149. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnston D and Duffin D: Drug-patient
interactions and their relevance in the treatment of heart failure.
Am J Cardiol. 70:109C–112C. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Setiawati E, Deniati SH, Yunaidi DA, et
al: Bioequivalence study of two perindopril erbumine tablet
formulations in healthy volunteers. Arzneimittelforschung.
61:234–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Devissaguet JP, Ammoury N, Devissaguet M
and Perret L: Pharmacokinetics of perindopril and its metabolites
in healthy volunteers. Fundam Clin Pharmacol. 4:175–189. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Georgakakou S, Kazanis M and Panderi I:
Hydrophilic interaction liquid chromatography/positive ion
electrospray ionization mass spectrometry method for the
quantification of perindopril and its main metabolite in human
plasma. Anal Bioanal Chem. 397:2161–2170. 2010. View Article : Google Scholar
|
|
28
|
Lecocq B, Funck-Brentano C, Lecocq V, et
al: Influence of food on the pharmacokinetics of perindopril and
the time course of angiotensin-converting enzyme inhibition in
serum. Clin Pharmacol Ther. 47:397–402. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grislain L, Mocquard MT, Dabe JF, et al:
Interspecies comparison of the metabolic pathways of perindopril, a
new angiotensin-converting enzyme (ACE) inhibitor. Xenobiotica.
20:787–800. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Begg EJ, Robson RA, Bailey RR, Lynn KL,
Frank GJ and Olson SC: The pharmacokinetics and pharmacodynamics of
quinapril and quinaprilat in renal impairment. Br J Clin Pharmacol.
30:213–220. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Resplandy G and Genissel P:
Pharmacokinetics of perindopril in high-risk populations. J
Cardiovasc Pharmacol. 18(Suppl 7): S9–S18. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parker E, Aarons L, Rowland M and
Resplandy G: The pharmacokinetics of perindoprilat in normal
volunteers and patients: influence of age and disease state. Eur J
Pharm Sci. 26:104–113. 2005. View Article : Google Scholar : PubMed/NCBI
|