Introduction
Respiratory failure (RF) is a state in which the
respiratory system fails by its gas exchange functions. Usually, RF
is defined by an arterial oxygen tension (PaO2) of
<8.0 kPa (60 mmHg) and an arterial carbon dioxide tension
(PaCO2) of >6.0 kPa (45 mmHg). These cut-off values
of respiratory failure serve as a general guide and are coupled
with the history and clinical assessment of RF patients (1). The respiratory system includes the lung
and the pump that ventilates the lungs (2). Failure of the lung, which is caused by
all types of lung diseases leads to hypoxemia with type I
respiratory failure. Failure of the pump leads to hypercapnia or
type II respiratory failure (3).
In the RF research, one of the main aims is to
identify biomarkers for a better understanding of RF pathogenesis
and improving diagnosis. Aberrant functions of the lymphocytic
regulatory pathway were widely associated with the pathological
mechanism of certain diseases and peripheral blood mononuclear
cells (PBMCs) were used as feasible samples in these studies
(4). In the past several years, the
developments in proteomics research of numerous rheumatic diseases
have been reported (4–6). Isobaric tags for relative and absolute
quantification (iTRAQ) technology has increasingly been used in
biomarker research for numerous diseases (7–10). However,
to the best of our knowledge, there is no previous study of iTRAQ
technology applied to PBMCs of RF. If the differentially expressed
proteins in the PBMCs of RF patients could be identified using
proteomic analysis, then these proteins could serve as a basis for
the development of proteomics research of RF.
iTRAQ reagents comprise three parts: A peptide
reactive group, a reporter group and a molecular mass balance.
Different protein samples are noted with a corresponding iTRAQ mass
group (i.e., 113, 114 and 115 Da) which could be quantified. The
proteomics workflow comprises two parts: 2-dimensional orthogonal
resolution of peptides by strong cation exchange (SCX) and
high-performance liquid chromatography (HPLC). Subsequently, the
fractions were analyzed through tandem mass spectrometry (MS/MS).
The sequence information (from peptide fragments) and relative
quantification (from reporter group ions) are provided from the
resultant mass spectra. In the present study, the total proteins in
the PBMCs of RF patients were analyzed through iTRAQ technology.
Further research of the molecular mechanism of the proteins can
better clarify the pathogenesis and identify novel biomarkers of
RF.
Materials and methods
Main reagents
Triton X-100 and a Strata-X 33u Polymeric Reversed
Phase column were separately purchased from Amersham Biosciences
(Waukesha, WI, USA) and Phenomenex (Los Angeles, CA, USA). The
Bicinchoninic Acid (BCA) Protein Assay Reagent kit and
triethylammonium bicarbonate buffer were respectively acquired from
Pierce (Thermo Fisher Scientific, Inc., Rockford, IL, USA) and
Sigma-Aldrich (Supelco, Bellefonte, Pennsylvania, USA). ZipTip
Pipette tips and Milli-Q water were obtained from Millipore
(Billerica, MA, USA). The iTRAQ Reagent-8Plex Multiplex kit and
Trypsin Gold, mass spectrometry grade were respectively acquired
from Applied Biosystems (Carlsbad, CA, USA) and Promega (Madison,
WI, USA). All the other reagents were obtained from commercial
sources.
Patients and healthy controls
The samples included 5 patients and 5 healthy
controls. The participants were from Shenzhen People's Hospital
(Shenzhen, China), between April and October 2013. The 5 patients,
who were diagnosed as type IIRF, included 4 women and 1 man with an
average age of 35.22 years; range, 28–45 years. The age and gender
of the 5 healthy controls were matched with the 5 patients. The
diagnosis of RF was confirmed by pathological diagnosis and
clinical evidence.
The healthy controls were confirmed to have no
clinical evidence of RF. All the subjects provided informed
consents. The study was approved by the regional ethics committee
and abided by the Helsinki Declaration.
PBMC isolation, protein extraction and
quantitation
One 5-ml fasting venous blood sample from each
participant was collected into the corresponding heparinize
vacutainers. According to the manufacturer's protocol (Cedarlane
Laboratories, Burlington, ON, Canada), PBMCs were isolated with
lymphocyte-H medium. The concentration of total protein for PBMCs,
which was extracted, was measured with the BCA protein assay kit.
The protein samples were kept at −80°C for further analysis.
iTRAQ labeling and SCX
fractionation
Firstly, a ratio of protein:trypsin = 30:1 was used
to generate the Trypsin Gold. Subsequently, the Trypsin Gold was
used to digest the equal amounts (100 µg) of protein in the PBMCs
of the samples at 37°C for 16 h. Following the digestion, peptides
were dried through vacuum centrifugation. Following the
manufacturer's protocol, peptides were reconstituted in 0.5 M TEAB
and processed for 8-plex iTRAQ reagent. In brief, samples were
thawed and reconstituted in one unit of iTRAQ reagent in 24 µl
isopropanol. Samples were labeled with the iTRAQ tags (samples 114
and 115). The peptides labeled with the isobaric tags were
incubated with them at room temperature for 2 h. Subsequently the
labeled peptide mixtures were pooled and dried by vacuum
centrifugation.
SCX chromatography was performed through an LC-20AB
HPLC Pump system (Shimadzu, Kyoto, Japan). The iTRAQ-labeled
peptide mixtures were reconstituted with 4 ml of buffer A [25 mM
NaH2PO4 in 25% acetonitrile (ACN) (pH 2.7)].
Subsequently, the iTRAQ-labeled peptide mixtures were loaded onto a
4.6×250 mm Ultremex SCX column containing 5-µm particles
(Phenomenex). After the loading process, the peptides were eluted
at a flow rate of 1 ml/min with a gradient of 100% buffer A for 10
min. Secondly, the peptides were eluted at a flow rate of 1 ml/min
with a gradient of 5–60% buffer B [25 mM
NaH2PO4, 1 M KCl in 25% ACN (pH 2.7)] for 27
min. Thirdly, the peptides were eluted at a flow rate of 1 ml/min
with a gradient of 60–100% buffer B for 1 min. Before equilibrating
with buffer A for 10 min prior to the next injection, the system
was maintained at 100% buffer B for 1 min. The elution was analyzed
through measuring the absorbance at 214 nm, and collecting the
fractions every 1 min. Following this, the eluted peptides were
pooled into 20 fractions and the eluted peptides were desalted with
a Strata X C18 column (Phenomenex) and vacuum-dried.
LC-ESI-MS/MS analysis based on
Q-Exactive
Each fraction was resuspended in buffer A [2% ACN,
0.1% formic acid (FA)] and centrifuged at 20,000 × g for 10 min.
The final concentration of the peptide was ~0.5 µg/µl. In total, 10
µl supernatant was loaded on an LC-20AD nano-HPLC (Shimadzu)
through the autosampler onto a 2-cm C18 trap column. The peptides
were eluted onto a 10-cm analytical C18 column (inner diameter 75
µm) packed in-house. The samples were loaded at 8 µl/min for 4 min.
The 44 min gradient was run at 300 nl/min from 2 to 35% B (98% ACN,
0.1% FA), followed by a 2-min linear gradient to 80%, and
maintained at 80% B for 4 min, prior to reverting to 5% for 1
min.
The peptides were subjected to nanoelectrospray
ionization through MS/MS in an Q-Exactive (Thermo Fisher
Scientific, San Jose, CA, USA), which was online to the HPLC and
detected the intact peptides in the orbitrap at a resolution of
70,000. The peptides for MS/MS were selected through high-energy
collision dissociation. A data-dependent procedure was used, which
alternated between one MS scan through 15 MS/MS scans. The
data-dependent procedure was applied to the 15 most abundant
precursor ions above a threshold ion count of 20,000 in the MS
survey scan with a following dynamic exclusion duration of 15 sec.
The operating electrospray voltage was 1.6 kV. Automatic gain
control (AGC) was used to optimize the spectra generated through
the orbitrap. The AGC target for a full MS was 3e6. The m/z scan
range was 350–2,000 Da for the MS scans.
Data analysis
MASCOT version 2.3.02 (Matrix Science, Ltd., London,
UK) was used to analyze the identification and quantification of
the proteins. The peptide sequences were searched in the
nonredundant NCBI database. The search criteria were set to permit
a maximum of 1 missed cleavage. Certain peptide modifications were
permitted: For example, Gln->pyro-Glu, iTRAQ 8plex, Phospho. The
values supplied with the Applied Biosystems reagents were used to
carry out automatic isotope correction through both software
packages. Subsequently, Gene Ontology (GO) (http://www.geneontol ogy.org) was used to elucidate
the molecular function, biological process and cellular component
associated with each individual protein.
Results
Identification of proteins
A default significance threshold of <0.05 for
individual variation was used as the cut-off. There were 19,711
iTRAQ-labeled unique peptides that mapped to a total of 4,795
proteins identified and quantified from PBMCs (Table I). The overlap or commonality of up-
and downregulated proteins were subtracted, and 403 proteins were
upregulated and 421 proteins were downregulated (Table II). Of these, a difference of a
multiple of ≥0.5 points identified 604, a difference of upregulated
>18 points included 7 proteins (Table
III) and a difference of downregulated <0.13 points included
5 proteins (Table IV).
 | Table I.Identification results. |
Table I.
Identification results.
| Source | Total spectra | Spectra | Unique spectra | Peptide | Unique peptide | Protein |
|---|
| Homosapien | 342,324 | 81,690 | 67,339 | 21,788 | 19,771 | 4,795 |
| Table II.Differential proteins. |
Table II.
Differential proteins.
| Participants | Upregulation
proteins | Downregulation
proteins | Total differentially
expressed proteins |
|---|
| Healthy versus
respiratory failure | 403 | 421 | 824 |
| Table III.Proteins identified as upregulated
from the isobaric tags for relative and absolute quantification
experiment, and an indication of the molecular function and
biological processes of these proteins. |
Table III.
Proteins identified as upregulated
from the isobaric tags for relative and absolute quantification
experiment, and an indication of the molecular function and
biological processes of these proteins.
| No. | Accession | Protein name | Molecular
function | Biological
process | Max multiple |
|---|
| 1 | gi|58257696 | KIAA1520 protein | TAP2 binding | Positive regulation
of T-cell mediated cytotoxicity | 34.504 |
| 2 | gi|182443 | 5 fibrinogen type B
(AA at 202) | Protein binding,
bridging | platelet
activation | 27.155 |
| 3 | gi|193244949 | β-globin | Peroxidase
activity | Hydrogen peroxide
catabolic process | 26.911 |
| 4 | gi|62088878 | Protein 4.1
variant | Calmodulin
binding | Positive regulation
of protein binding | 20.145 |
| 5 | gi|229959 | β-globin
(fragment) | Peroxidase
activity | Hydrogen peroxide
catabolic process | 19.487 |
| 6 | gi|284521122 | A-γ globin Osilo
variant | Oxygen transporter
activity | Oxygen transport | 19.257 |
| 7 | gi|28332 | cDNA FLJ35730 fis,
clone TESTI2003131, highly similar to α-1-antichymotrypsin | Serine-type
endopeptidase inhibitor activity | Negative regulation
of endopeptidase activity | 18.890 |
| Table IV.Proteins identified as downregulated
from the isobaric tags for relative and absolute quantification
experiment, and an indication of the molecular function and
biological processes of these proteins. |
Table IV.
Proteins identified as downregulated
from the isobaric tags for relative and absolute quantification
experiment, and an indication of the molecular function and
biological processes of these proteins.
| No. | Accession | Protein name | Molecular
function | Biological
process | Max multiple |
|---|
| 1 | gi|809369 | Chain A, crystal
structure of recombinant human platelet factor 4 | Heparin binding | Negative regulation
of angiogenesis | 0.091 |
| 2 | gi|431896311 | Myosin regulatory
light polypeptide 9 | Calcium ion
binding | Regulation of cell
shape | 0.092 |
| 3 | gi|406717976 | MHC class II
antigen | Glutamate receptor
binding | T-cell
costimulation | 0.101 |
| 4 | gi|206581665 | Chain A, human Ll-37
structure | Protein binding | Protein localization
to microtubule | 0.124 |
| 5 | gi|122939159 | Protein-arginine
deiminase type-2 | Protein-arginine
deiminase activity | Peptidyl-citrulline
biosynthetic process from peptidyl-arginine | 0.128 |
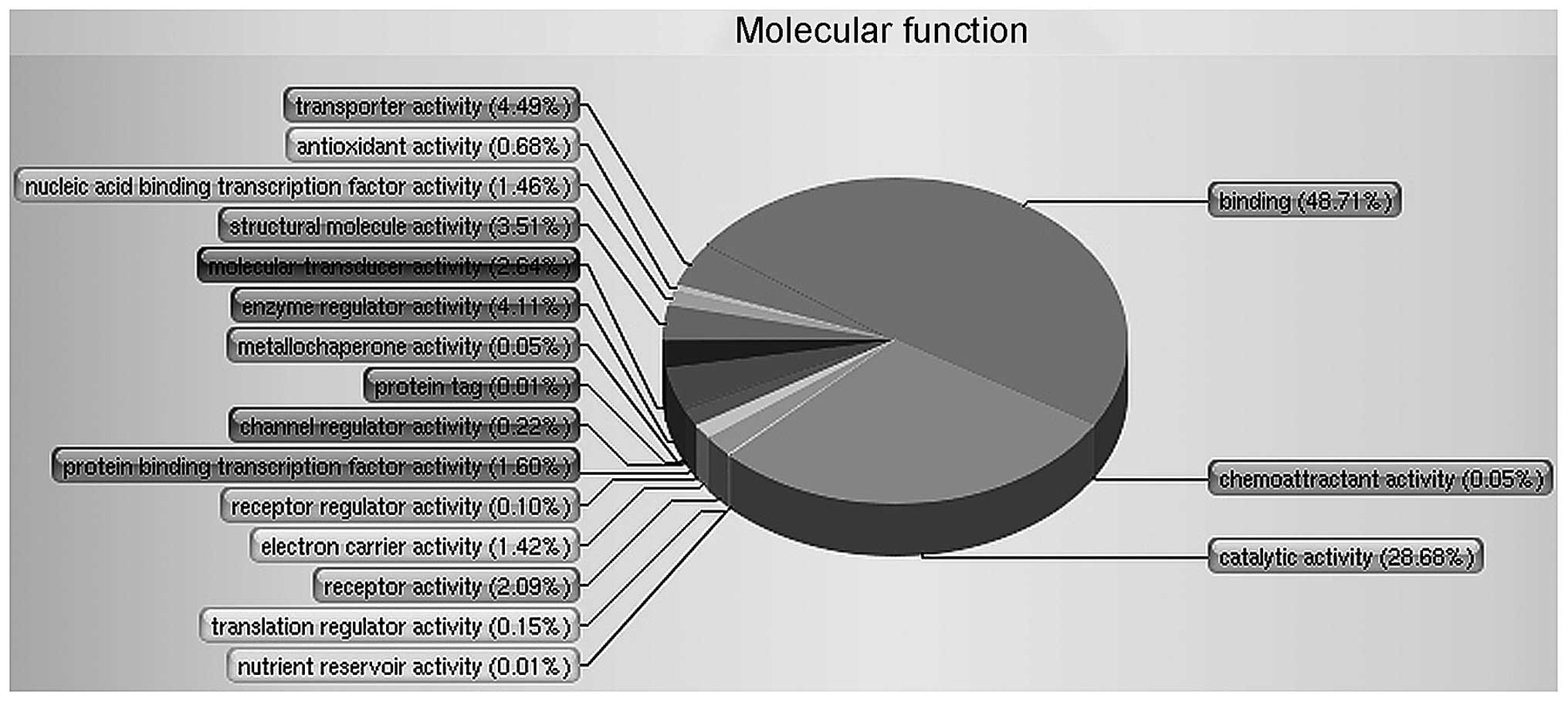
The functional distribution of these proteins is
shown in Fig. 1. The GO was used to
perform an analysis of 4,795 proteins and to divide the proteins
into respective classes based on their molecular function. It found
that two of the major groups involved binding (48.71%) and
catalytic activity (28.68%) that were apparently changed in RF
patients versus healthy controls.
Discussion
In the past few years, protein quantification has
become a critical component of modern MS-based proteomic research
(11,12). In addition, numerous quantification
strategies have been improved. Almost all of them depend on the
incorporation of steady isotopes for subsequent mass spectrometric
sorting and relative quantification (13,14).
Through measuring the peak intensities of reporter
ions, which are released from iTRAQ-tagged peptides, iTRAQ can
compare the protein abundance. iTRAQ could be a potential tool for
quantitative proteomic study. In the present study, the proteomics
of PBMCs were analyzed in RF patients and healthy controls
quantitatively through iTRAQ technology. As a result, 824 proteins,
which are involved with different biological functions and cellular
locations, were identified, and a proteome database was built for
the RF proteome, which to the best of our knowledge has not been
reported previously.
The up- and downregulated proteins for the RF and
healthy controls are shown in Tables I
and II. Among them, there was a
significant difference of 4 proteins [upregulated KIAA1520 protein,
γ fibrinogen type B (AA at 202) and downregulated chain A, crystal
structure of recombinant human platelet factor 4 and myosin
regulatory light polypeptide 9] in the present iTRAQ study. This
provided additional certification that the iTRAQ technique could
quantify relative changes in the proteins of PBMC accurately. In
addition, the iTRAQ technique has been used for detecting
pathological stages or prognosis in certain diseases (12–14). While
iTRAQ-based biomarker profiles from tissue have been used for
analyzing the pathological stages or prognosis of certain diseases
(15,16), the analysis of PBMC in diagnosing the
pathological result or progression of RF is only beginning to be
explored.
Nagase et al (17) reported the entire sequences of 100 cDNA
clones of KIAA1444 to KIAA1543 human genes from cDNA libraries.
They found that open reading frames (ORFs) in 10 clones (KIAA1513,
KIAA1515, KIAA1520-KIAA1522, KIAA1524, KIAA1525, KIAA1529, KIAA1531
and KIAA1538) carried single or multiple deletions; however,
certain ORFs in 23 clones (KIAA1509-KIAA1512, KIAA1514-KIAA1517,
KIAA1519-KIAA1521, KIAA1523, KIAA1524, KIAA1526-KIAA1528, KIAA1530
and KIAA1532-KIAA1537) carried single or multiple insertions. The
study also reported that 48 gene products have functions correlated
with nucleic acid management, cell structure/motility or cell
signaling/communication.
The γA and γB were the two forms of the γ chain of
human fibrinogen. The differences between them are only in their
carboxyl termini. The protein sequence of γ-fibrinogen in rats and
humans is generally highly conserved (18). The unique γB sequence, which is coded
by human fibrinogen, contained 1 basic residue and 7 acidic
(19). Song et al (20) found that the levels of plasma
viscosity, blood viscosity, hematocrit, fibrinogen and D-dimer were
significantly higher in acute exacerbations of chronic obstructive
pulmonary disease (AECOPD) patients. The levels of fibrinogen and
D-dimer had significantly positively associated with the
PaCO2 and negatively associated with the PaO) in AECOPD
patients combined with RF.
Myosin regulatory light polypeptide 9 is a type of
myosin regulatory subunit. It exhibited a critical role in
regulating the activities of the smooth muscle and non-muscle cell
contractile. In addition, it was involved with cell locomotion,
receptor capping and cytokinesis. In lipopolysaccharide-induced
lung inflammatory injury, which is the chief cause of the acute
respiratory distress syndrome, non-muscle myosin light-chain kinase
mediates increased lung vascular endothelial permeability (21).
The functions of certain other novel candidates,
such as the chain A, crystal structure of human platelet factor 4
(downregulated in RF), remain to be elucidated. The novel
candidates would be more worthy for further investigation.
In the present study, every candidate protein was
not discussed in detail. The aim of this preparatory investigation
was centered on illustrating the primary comparative protein
profiles of RF patients and healthy controls using iTRAQ
technology. Furthermore, future studies require the collection of
more patient samples to identify the beneficial biomarker
candidates of the pathogenesis in RF. In conclusion, the present
study showed the potential application of iTRAQ-based quantitative
proteomics for the identification of protein changes and potential
biomarker candidates in certain diseases.
Acknowledgements
The authors would like to thank the patients and
volunteers who participated in the present study. The study was
supported by a grant from the Science and Technology Plan of
Shenzhen (no. JCYJ20130401093116730).
References
|
1
|
Roussos C and Koutsoukou A: Respiratory
failure. Eur Respir J Suppl. 47:3s–14s. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roussos C and Macklem PT: The respiratory
muscles. N Engl J Med. 307:786–797. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burt CC and Arrowsmith JE: Respiratory
failure. Surgery. 27:475–479. 2009.
|
|
4
|
Chang X, Cui Y, Zong M, Zhao Y, Yan X,
Chen Y and Han J: Identification of proteins with increased
expression in rheumatoid arthritis synovial tissues. J Rheumatol.
36:872–880. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gibson DS, Blelock S, Curry J, Finnegan S,
Healy A, Scaife C, McAllister C, Pennington S, Dunn M and Rooney M:
Comparative analysis of synovial fluid and plasma proteomes in
juvenile arthritis - proteomic patterns of joint inflammation in
early stage disease. J Proteomics. 72:656–676. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai Y, Hu C, Huang Y, Huang H, Liu J and
Lv T: A proteomic study of peripheral blood mononuclear cells in
systemic lupus erythematosus. Lupus. 17:799–804. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
DeSouza LV, Grigull J, Ghanny S, Dubé V,
Romaschin AD, Colgan TJ and Siu KW: Endometrial carcinoma biomarker
discovery and verification using differentially tagged clinical
samples with multidimensional liquid chromatography and tandem mass
spectrometry. Mol Cell Proteomics. 6:1170–1182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al Badaai Y, DiFalco MR, Tewfik MA and
Samaha M: Quantitative proteomics of nasal mucus in chronic
sinusitis with nasal polyposis. J Otolaryngol Head Neck Surg.
38:381–389. 2009.PubMed/NCBI
|
|
9
|
Zhou L, Beuerman RW, Chan CM, Zhao SZ, Li
XR, Yang H, Tong L, Liu S, Stern ME and Tan D: Identification of
tear fluid biomarkers in dry eye syndrome using iTRAQ quantitative
proteomics. J Proteome Res. 8:4889–4905. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hergenroeder G, Redell JB, Moore AN,
Dubinsky WP, Funk RT, Crommett J, Clifton GL, Levine R, Valadka A
and Dash PK: Identification of serum biomarkers in brain-injured
adults: Potential for predicting elevated intracranial pressure. J
Neurotrauma. 25:79–93. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bantscheff M, Schirle M, Sweetman G, Rick
J and Kuster B: Quantitative mass spectrometry in proteomics: A
critical review. Anal Bioanal Chem. 389:1017–1031. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li X, Hu B, Ding J and Chen H: Rapid
characterization of complex viscous samples at molecular levels by
neutral desorption extractive electrospray ionization mass
spectrometry. Nat Protoc. 6:1010–1025. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Couttas TA, Raftery MJ, Erce MA and
Wilkins MR: Monitoring cytoplasmic protein complexes with blue
native gel electrophoresis and stable isotope labelling with amino
acids in cell culture: Analysis of changes in the 20S proteasome.
Electrophoresis. 32:1819–1823. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Albaum SP, Hahne H, Otto A, Haußmann U,
Becher D, Poetsch A, Goesmann A and Nattkemper TW: A guide through
the computational analysis of isotope-labeled mass
spectrometry-based quantitative proteomics data: An application
study. Proteome Sci. 9:302011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hergenroeder G, Redell JB, Moore AN,
Dubinsky WP, Funk RT, Crommett J, Clifton GL, Levine R, Valadka A
and Dash PK: Identification of serum biomarkers in brain-injured
adults: Potential for predicting elevated intracranial pressure. J
Neurotrauma. 25:79–93. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matta A, DeSouza LV, Shukla NK, Gupta SD,
Ralhan R and Siu KW: Prognostic significance of head-and-neck
cancer biomarkers previously discovered and identified using
iTRAQ-labeling and multidimensional liquid chromatography-tandem
mass spectrometry. J Proteome Res. 7:2078–2087. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagase T, Kikuno R, Ishikawa K, Hirosawa M
and Ohara O: Prediction of the coding sequences of unidentified
human genes. XVII. The complete sequences of 100 new cDNA clones
from brain which code for large proteins in vitro. DNA Res.
7:143–150. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Crabtree GR and Kant JA: Organization of
the rat gamma-fibrinogen gene: Alternative mRNA splice patterns
produce the gamma A and gamma B (gamma ') chains of fibrinogen.
Cell. 31:159–166. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fornace AJ Jr, Cummings DE, Comeau CM,
Kant JA and Crabtree GR: Structure of the human gamma-fibrinogen
gene. Alternate mRNA splicing near the 3′ end of the gene produces
gamma A and gamma B forms of gamma-fibrinogen. J Biol Chem.
259:12826–12830. 1984.PubMed/NCBI
|
|
20
|
Song YJ, Zhou ZH, Liu YK, Rao SM and Huang
YJ: Prothrombotic state in senile patients with acute exacerbations
of chronic obstructive pulmonary disease combined with respiratory
failure. Exp Ther Med. 5:1184–1188. 2013.PubMed/NCBI
|
|
21
|
Xu J, Gao XP, Ramchandran R, Zhao YY,
Vogel SM and Malik AB: Nonmuscle myosin light-chain kinase mediates
neutrophil transmigration in sepsis-induced lung inflammation by
activating beta2 integrins. Nat Immunol. 9:880–886. 2008.
View Article : Google Scholar : PubMed/NCBI
|