Introduction
Pulmonary lymphangioleiomyomatosis (PLAM) is a rare
hamartomatous proliferation of the smooth muscle involved in blood
vessels and lymphatics in the lung (1,2). It extends
into the pulmonary interstitium, leading to diffuse thin-walled
cystic lesions and pulmonary hemorrhage, and lymph node involvement
may result in chylous effusion (3).
PLAM is clinically characterized by progressive dyspnea, cough,
chest pain, hemoptysis and pneumothorax, leading to progressive
airflow obstruction and impaired lung diffusion function,
culminating in respiratory failure commonly in premenopausal women
(4,5).
Chest radiographs show diffuse interstitial infiltrates and
thin-walled cystic lesions. Pulmonary function tests usually reveal
obstructive ventilation dysfunction and airflow limitation with
impaired lung diffusion function. PLAM is considered to occur alone
or is associated with tuberous sclerosis (6). Thus far, the pathogenesis of PLAM has not
been clearly clarified and its treatment remains limited.
PLAM was first reported in China in 1986, and since
then ~100 Chinese cases have been reported. However, case reports
of PLAM are rare in Northwestern China. A recent retrospective
study in China showed that PLAM commonly occurs in women (99.2%);
dyspnea was the most common clinical manifestation (94.6%); the
most common extrapulmonary manifestation was retroperitoneal lymph
node involvement (52.1%); the most common chest imaging
manifestation was multiple different sized thin-walled cystic
shadows in whole lungs (93.8%); the most common lung biopsy method
was bronchoscopic biopsy (41.0%); in the patients with pulmonary
dysfunction, obstructive ventilation dysfunction was 65.1%, and
diffusion dysfunction was 38.4%. However, there is no effective
medical therapy, and lung transplant may be the only possible
treatment for the cure of patients with PLAM (7).
Regardless of the extent of the disease, it is
notable that the patients are usually female, and in general are in
the reproductive years. The development of PLAM is possibly
associated with hormone secretion. In the present study, a woman
with PLAM, renal cyst, oophoritic cyst and uterine myoma, who had a
hysterectomy for treating uterine myoma 12 years previously is
reported. This is a rare case report in Northwestern China.
Case report
A 46-year-old female in Northwestern China presented
with exertional dyspnea, which occurred 1 month prior and
progressed gradually. The patient did not report any fever, chills,
cough, chest pain or hemoptysis. Due to worsening of symptoms, the
patient visited the Department of Respiratory and Critical Care
Medicine (The First Affiliated Hospital of Xi'an Jiaotong
University, Xi'an, Shaanxi, China). The patient had a history of
hysterectomy for treating uterine myoma 12 years previously and had
no history of smoking or alcohol consumption. Physical examination
appeared normal. The laboratory findings including routine tests of
blood, urine and feces, liver and kidney function tests, and tumor
markers detection were normal. Blood gas analysis showed slight
hypoxemia, and the pulmonary function test showed a normal
ventilation function with a reduced diffusing capacity of the lung
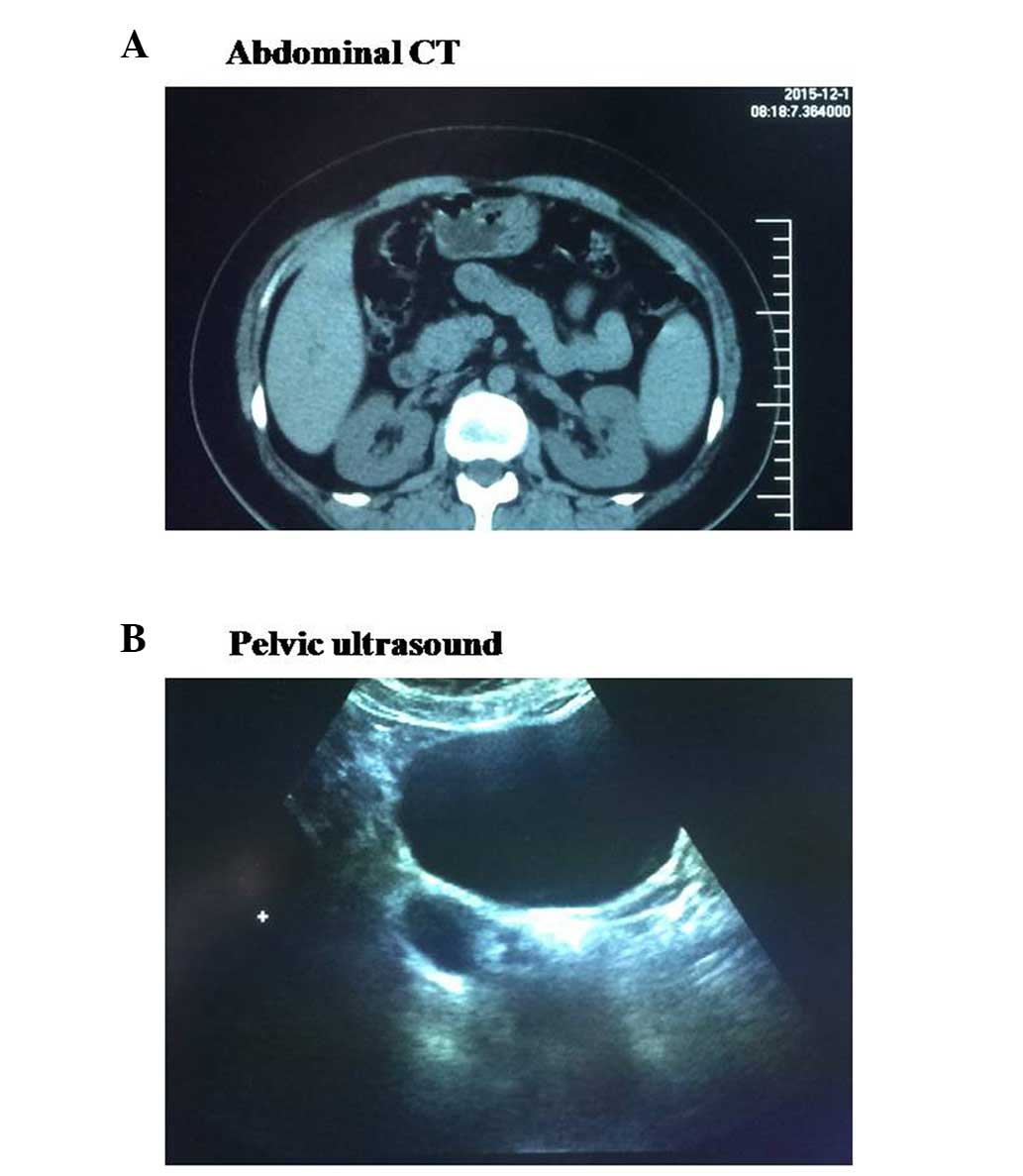
for carbon monoxide (68%). Additionally, abdominal computed
tomography (CT) showed a right renal cyst, and pelvic ultrasound
exhibited a right oophoritic cyst (Fig.
1).
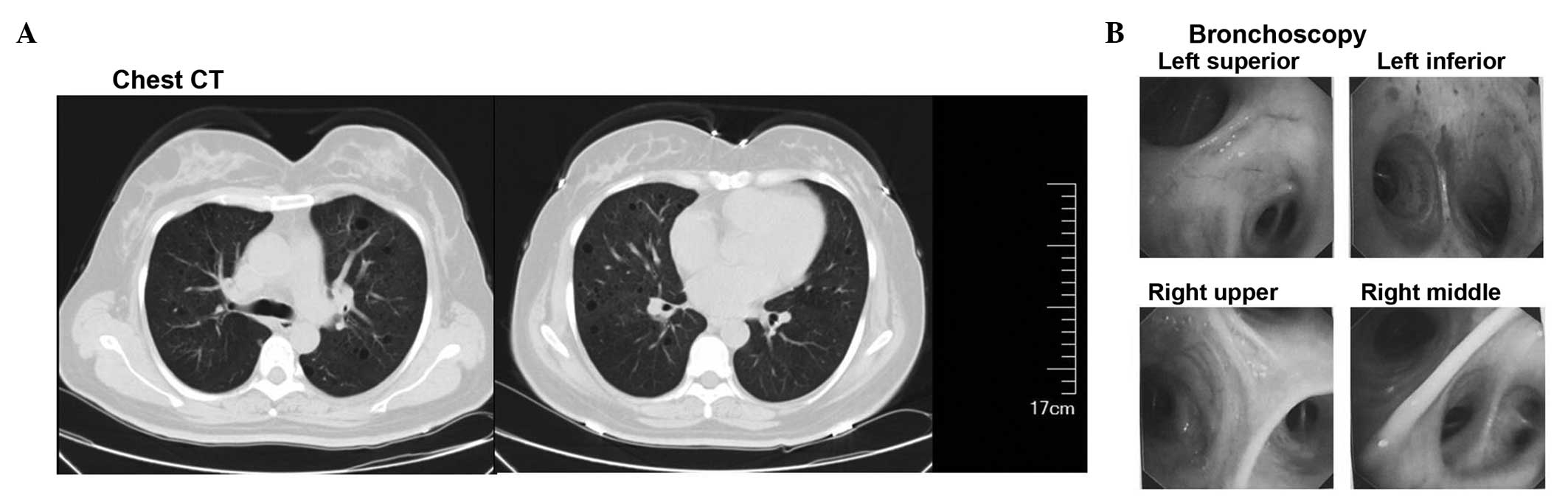
Chest CT exhibited bilateral, diffuse and
thin-walled cystic changes (Fig. 2A).
Based on the chest radiographs, lung biopsy was required for a
precise diagnosis. Firstly, bronchoscopy was performed and no
abnormality was detected in bronchial lumen (Fig. 2B). The patient was not diagnosed by the
bronchoscopic biopsy, as the size of the specimen was too small.
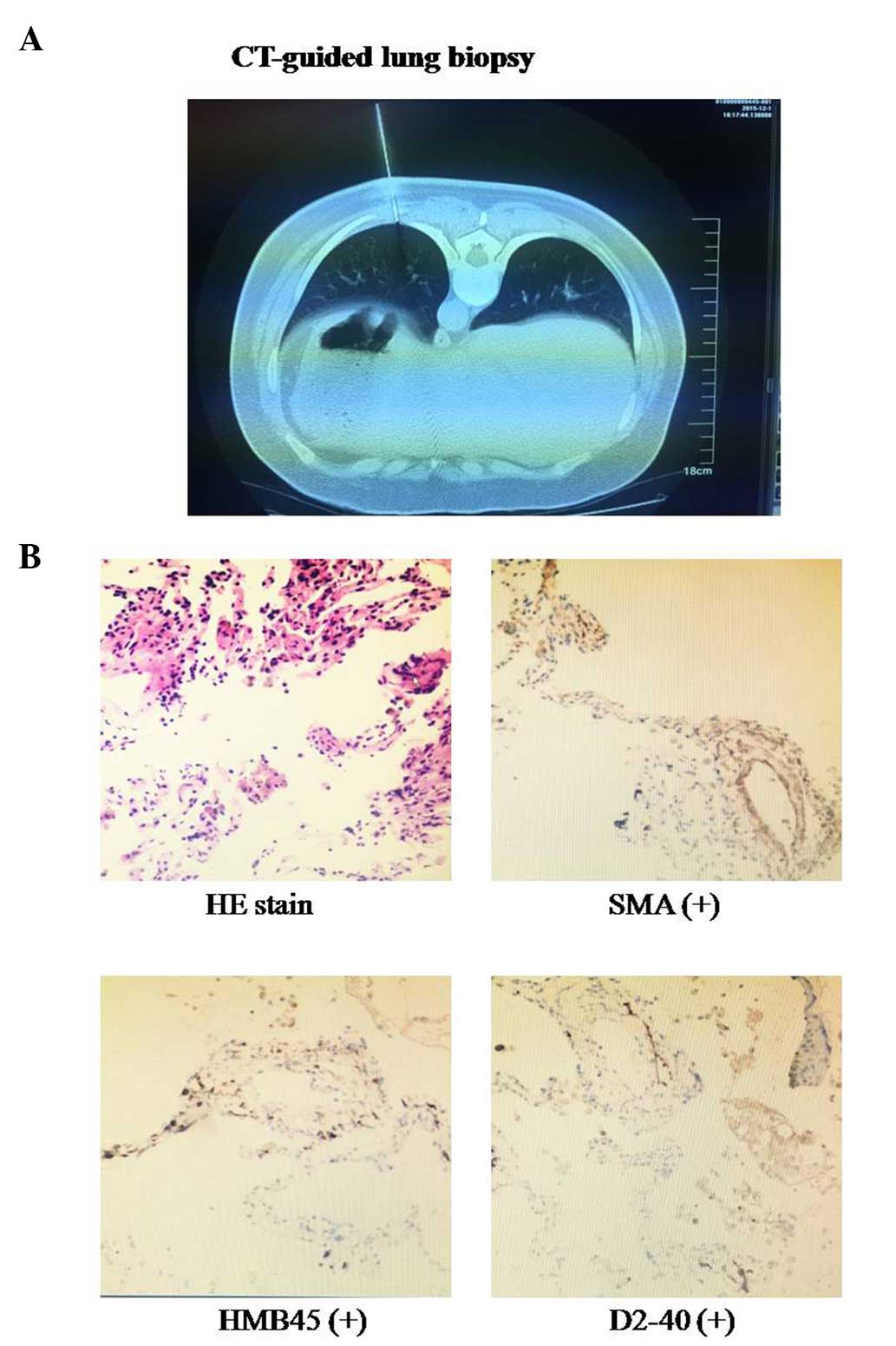
Subsequently, CT-guided percutaneous needle-aspiration biopsy of
the lung was performed for the patient (Fig. 3A). Initial microscopic examination of
the lung biopsy was interpreted as fibrosis and chronic
inflammation. Due to the CT findings, the pathology was
questionable and the diagnosis required further assessment.
Reexamination of the biopsy specimen exhibited hyperplasia of
smooth muscle and a proliferation of spindle cells around small
blood vessels and respiratory bronchioles (Fig. 3B). Histology and immunohistochemistry
(IHC) demonstrated that characteristic smooth muscle cells were
positive for smooth muscle actin (SMA), human melanoma black (HMB)
45 and D2-40 (a specific lymphatic endothelial marker) (Fig. 3B). These findings established the
diagnosis of PLAM. Sirolimus was administered for the patient,
which improved the dyspnea and pulmonary function temporarily.
However, the patient with PLAM is in an early stage in the process
of the disease and requires long-term follow-up, as there is
currently no effective medical therapy for PLAM. The present study
was approved by the local ethics review committees, and the patient
provided informed consent.
Discussion
PLAM is characterized by the progressive
proliferation of smooth muscle in alveolar septa, pulmonary vessels
and lymphatics frequently occurring in females between the ages of
17 and 50 years (8). The chest
radiographs usually show that bilateral multiple pulmonary cysts.
When pulmonary vessels are involved and occluded, pulmonary
hemorrhage may occur in the interstitium; when the lymphatics and
the thoracic duct was occluded, chylous effusion may occur. Similar
results have been found in patients with tuberous sclerosis
(9). PLAM is considered to be
associated with tuberous sclerosis. However, in contrast to
tuberous sclerosis, there is no family history, mental retardation
or adenoma sebaceum in PLAM.
The most frequent clinical manifestation worldwide
is dyspnea, followed by cough, pneumothorax, chest pain, hemoptysis
and chylothorax. The progressive proliferation of smooth muscle may
lead to obstruction of bronchioles and pulmonary vessels, so
pulmonary function tests show obstructive ventilation dysfunction
and diffusion dysfunction, culminating in hypoxemia and respiratory
failure. The treatment of PLAM is limited, and the prognosis
remains poor. The majority of patients with PLAM may succumb to
respiratory failure within 10 years (10).
In the present case, chest CT showed that the cysts
were bilateral, diffuse and thin-walled. According to statistical
analysis, the most common chest imaging manifestation was also
multiple different sized thin-walled cystic shadows in the whole
lungs. Although cysts frequently exist in various types of
interstitial lung diseases, the chest radiographs of PLAM in CT are
different from other interstitial lung diseases. For example,
idiopathic pulmonary fibrosis is characterized by reticular
abnormalities and honeycombing in the basilar and subpleural areas,
while PLAM is more diffuse in the whole lungs. Furthermore, the
cysts in idiopathic pulmonary fibrosis are surrounded by abnormal
parenchyma, while the cysts in PLAM are surrounded by normal
tissues (11). Thus, the chest CT
findings are helpful in the diagnosis of PLAM.
Currently, bronchoscopic biopsy was the most common
lung biopsy method. However, sufficient specimens for the diagnosis
of PLAM by bronchoscopy were not obtained, as no abnormality was
identified in the bronchial lumen. This disease is rare in
Northwestern China, and therefore the diagnosis procedure is
unfamiliar. However, CT-guided lung biopsy showed that
characteristic smooth muscle cells were positive for SMA, HMB45 and
D2-40 by histology and IHC techniques, resulting in a diagnosis of
PLAM. Therefore, the present study summarized the experience in
clinical diagnosis and treatment of PLAM in order to improve the
diagnostic accuracy rating and reduce an ignored diagnosis.
The case of PLAM in a female complicated with renal
cyst, oophoritic cyst and uterine myoma that was removed by a
hysterectomy 12 years previously was reported in the present study.
A recent study showed that the development of PLAM is associated
with hormone secretion, particularly estrogen (12). Whether renal cyst, oophoritic cyst and
uterine myoma are involved in PLAM remains to be elucidated.
Therefore, the patient requires long-term follow-up for observation
on the process of the disease.
The results of the recent sirolimus phase III trial
showed that sirolimus treatment significantly slowed the decline in
the pulmonary function, and improved dyspnea and the quality of
life (13,14). In the present case, sirolimus treatment
efficiently stabilized pulmonary function and alleviated the
symptoms during the observation period. Recently, certain results
have demonstrated that abnormality of the tuberous sclerosis
complex gene 1/2 induced LAM cell proliferation via the activation
of mechanistic target of rapamycin (mTOR) (15). Thus, sirolimus, as an mTOR inhibitor,
could stabilize the pulmonary function and improve dyspnea. We
believe that the continuous administration of sirolimus is
necessary for stabilizing the pulmonary function and improving the
quality of life, and the safety and efficacy of sirolimus should be
further studied in the future.
Acknowledgements
The authors appreciate the technical support and
materials from the Department of Pathology (The First Affiliated
Hospital of Xi'an Jiaotong University). The present study was
supported by the programs from the National Natural Science
Foundation of China (general program nos. 81270094 and
81500016).
References
|
1
|
Carrington CB, Cugell DW, Gaensler EA,
Marks A, Redding RA, Schaaf JT and Tomasian A:
Lymphangioleiomyomatosis. Physiologic-pathologic-radiologic
correlations. Am Rev Respir Dis. 116:977–995. 1977.PubMed/NCBI
|
|
2
|
Bernstein SM, Newell JD Jr, Adamczyk D,
Mortenson RL, King TE Jr and Lynch DA: How common are renal
angiomyolipomas in patients with pulmonary lymphangiomyomatosis? Am
J Respir Crit Care Med. 152:2138–2143. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Corrin B, Liebow AA and Friedman PJ:
Pulmonary lymphangiomyomatosis. A review. Am J Pathol. 79:348–382.
1975.PubMed/NCBI
|
|
4
|
Hayashida M, Seyama K, Inoue Y, Fujimoto K
and Kubo K: Respiratory Failure Research Group of the Japanese
Ministry of Health, Labor, and Welfare: The epidemiology of
lymphangioleiomyomatosis in Japan: A nationwide cross-sectional
study of presenting features and prognostic factors. Respirology.
12:523–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hohman DW, Noghrehkar D and Ratnayake S:
Lymphangioleiomyomatosis: A review. Eur J Intern Med. 19:319–324.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valensi QJ: Pulmonary lymphangiomyoma, a
probable forme frust of tuberous sclerosis. A case report and
survey of the literature. Am Rev Respir Dis. 108:1411–1415.
1973.PubMed/NCBI
|
|
7
|
Zhang L, Liang Y, Zhong X and Liu J:
Literature review of clinical and pathological features of
pulmonary lymphangioleiomyomatosis for 130 cases in China in the
last thirty years. Chin Gen Pract. 3:329–334. 2015.
|
|
8
|
Silverstein EF, Ellis K, Wolff M and
Jaretzki A III: Pulmonary lymphangiomyomatosis. Am J Roentgenol
Radium Ther Nucl Med. 120:832–850. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sato T, Seyama K, Fujii H, Maruyama H,
Setoguchi Y, Iwakami S, Fukuchi Y and Hino O: Mutation analysis of
the TSC1 and TSC2 genes in Japanese patients with pulmonary
lymphangioleiomyomatosis. J Hum Genet. 47:20–28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taylor JR, Ryu J, Colby TV and Raffin TA:
Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J
Med. 323:1254–1260. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kitaichi M, Nishimura K, Itoh H and Izumi
T: Pulmonary lymphangioleiomyomatosis: A report of 46 patients
including a clinicopathologic study of prognostic factors. Am J
Respir Crit Care Med. 151:527–533. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Numata T, Araya J, Mikami J, Hara H,
Harada T, Takahashi H, Nakayama K and Kuwano K: A case of pulmonary
lymphangioleiomyomatosis complicated with uterine and
retroperitoneal tumors. Respir Med Case Rep. 15:71–76.
2015.PubMed/NCBI
|
|
13
|
McCormack FX, Inoue Y, Moss J, Singer LG,
Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM,
et al: National Institutes of Health Rare Lung Diseases Consortium;
MILES Trial Group: Efficacy and safety of sirolimus in
lymphangioleiomyomatosis. N Engl J Med. 364:1595–1606. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carsillo T, Astrinidis A and Henske EP:
Mutations in the tuberous sclerosis complex gene TSC2 are a cause
of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci
USA. 97:6085–6090. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Q and Guan KL: Expanding mTOR
signaling. Cell Res. 17:666–681. 2007. View Article : Google Scholar : PubMed/NCBI
|