Introduction
Atherosclerosis is an autoimmune condition
characterized by the development of complex atherosclerotic
plaques, leading to hardening and narrowing of the arterial lumen.
Chronic exposure to cardiovascular risk factors, such as
hyperlipidemia, hypertension, smoking, male gender and diabetes,
can increase the rate and severity of atherosclerosis. Among the
different risk factors, increased plasma low-density lipoprotein
(LDL) level has been identified as the most significant, which
alone is sufficient to produce atherosclerosis in monogenetic
hyperlipidemia disorders, such as familial hypercholesterolemia
(1). In individuals with normal LDL
levels, other factors are responsible for the development and
progression of atherosclerosis (2,3). However,
these risk factors are rather insignificant in individuals with low
LDL levels, who are unlikely to develop atherosclerosis
irrespective of the presence of additional risk factors (4).
Excess LDL in plasma accumulates in the
sub-endothelial space of the arterial wall, undergoing oxidation to
become oxidized LDL (oxLDL). This in turn, triggers an inflammatory
response, thereby inducing the expression of a number of different
molecules, such as vascular cell adhesion molecule-1, E-selectin
and P-selectin in the endothelium (5).
This response provides the necessary conditions for chemotaxis,
where blood cells are recruited into the injured arterial wall
(6). Monocytes are the most prominent
cell type involved (7). After entry
into the tunica intima, monocytes undergo differentiation into
macrophages, which take up oxLDL to become foam cells (8). Foam cells function as antigen-presenting
cells, and activate circulating monocytes and T-cells (9). They also secrete mediators to further
perpetuate inflammation, and stimulate the migration of smooth
muscle cells from the tunica media into sub-endothelial space
(10). Mediated by platelet-derived
growth factor, the smooth muscle cells exhibit abnormally high
proliferation rates and secrete extracellular matrix proteins that
contribute to fibrous cap formation (11). The fibrous cap protects the core of the
plaque from circulating blood cells, especially platelets
responsible for the thrombosis associated with rupture plaques.
This maladaptive non-resolving inflammation is the driving force of
atherosclerotic plaque development (12). SMCs from different regions of the
microvasculature have different developmental origins (13), which can contribute to site-specific
atherosclerotic responses (14).
During plaque evolution, macrophages proliferate,
undergo apoptosis and efferocytosis (15,16).
Apoptotic cells may be removed, leading to lesion size reduction,
or may accumulate and be subjected to secondary necrosis, producing
the necrotic core characteristic of advance plaques. Accumulation
of apoptotic bodies may enhance the plaque instability by
triggering inflammation. While foam cells are the most abundant
leukocytes within the atherosclerotic lesions, other cell types,
including neutrophils, mast cells, T-lymphocytes and B-lymphocytes
are also involved in atherogenesis (17,18).
Although these cells contribute little in mass to the lesions, they
can secrete different signalling proteins that regulate other cells
or components within the plaques (19–21).
Plaque rupture is responsible for the adverse
clinical consequences of ischaemia in cerebrovascular accidents,
myocardial infarction and heart failure, producing significant
morbidity and mortality in affected patients. In advanced stages of
atherosclerosis, rupture of vulnerable plaques exposes their
thrombogenic compounds, producing luminal thrombosis.
Destabilization of plaques into a vulnerable state is in part
mediated by macrophage-derived proteases, such as metalloproteases;
however, the precise mechanisms remain incompletely characterized
(22).
Animal models of atherosclerosis
In 1908, Ignatowski provided the first experimental
demonstrations that atherosclerosis can be induced in laboratory
animals. He fed rabbits a protein-rich diet (mainly meat, milk, and
egg yolk), which led to the formation of atherosclerotic lesions in
the aortic wall. Since then, a number of species, such as rabbits,
mice, rats, guinea pigs, hamsters, birds, dogs and non-human
primates, have been developed. Despite differences between the
animal models, a common finding is the necessary condition of
hypercholesterolaemia in plaque development. Animal models have
been extensively used for the study of human cardiovascular
diseases (23–41). In the present review, we review rabbit,
porcine and non-primate models of atherosclerosis, together with
their advantages and disadvantages (Table
I).
 | Table I.Models of atherosclerosis with their
advantages and disadvantages. |
Table I.
Models of atherosclerosis with their
advantages and disadvantages.
| Animal experimental
models |
|---|
| Rabbits |
|
Advantages |
|
Similar lipid
metabolism with human |
|
Similar morpology
of lesion development |
|
Low cost for
maintenance |
|
High
availability |
|
Larger artery
allow clinical evaluation: Ultrasound and MRI can be applied to
determine plaque composition and its vulnerability |
|
Low cost of
maintenance due to its small size |
|
Disadvantages |
|
Not always
responsive to dietary cholesterol |
|
Different
cardiovascular physiology with human: HDL as the predominant plasma
lipoprotein, absence of Apo AII, low hepatic lipase activity |
|
Low hepatic lipase
acitivity leads to hepatotoxicity following prolonged cholesterol
feeding |
|
Plaque lesion
dissimilar with human: foam cells with more fatty streak and
macrophage rich, advanced lesion (e.g., Fibrosis and haemorrhage
and ulceration) are not seen |
|
Different
predilection site: Atherosclerotic plaque preferentially deposited
in aorta, iliac arteries |
| Porcine |
|
Advantages |
|
Similar
haemodynamics and pathogenesis to humans: Lesion location,
morphology and content |
|
Similar heart size
and cardiovascular anatomy |
|
Similar lipid
metabolism, except for Apo II deficiency in porcine |
|
Highly defined
genotypes for genetic manipulation |
|
Minipig version
offer option with lower cost |
|
Unlike mouse and
rabbit, it can spontaneously develop atherosclerosis with an
accelrated rate when fed with atherogenic diet |
|
Easier to carry
out imaging, e.g., Ultrasound, CT and MRI compared to smaller
species |
|
Disadvantages |
|
Toxic diet needed
for induction of atherosclerosis |
|
Large in size,
which limits its practical use |
| Non-human
primates |
|
Advantages |
|
Similar cardiac
anatomy: Same predilection site for atherosclerosis |
|
Similar cardiac
physiology: Comparable lipid metabolism and advanced
atherosclerotic lesion found |
|
Closest
phylogenetic relationship with human |
|
Highest
resemblance to human atherosclerotic clinical condition |
|
Susceptible to
spontaneous atherosclerosis |
|
Similar omnivorous
diet |
|
Disadvantages |
|
High cost for
purchase and maintenance |
|
Long lifespan and
hence long period of time needed for induction of
atherosclerosis |
|
Significant
ethical concern |
|
Large size with
difficulty of management |
|
Low
availability |
Rabbit
Rabbit is the first animal model developed for
atherosclerosis research, leading to the identification of the
crucial role of elevated plasma cholesterol in atherogenesis. It
served as the mainstay of pre-clinical model until genetically
modified mouse models became widely available.
New Zealand White (NZW) strain
The most common strain is the NZW, in which the
roles of lipoproteins of differing sizes on atherosclerosis were
examined. For example, atherosclerosis was unexpectedly inhibited
in an alloxan-induced diabetes rabbit model, explicably by the
accumulation of large triglyceride-rich lipoprotein (>75 mm
diameter), to which the vascular wall has limited permeability
(42,43). However, NZW rabbits show high
biological variability with respect to individual responsiveness to
dietary cholesterol and the lesion morphology varies significantly
depending on the cholesterol content of the diet (44). This strain is not prone to
atherosclerotic risk due to its low plasma cholesterol level of 50
mg/dl when exposed to standard diet. The induction of vascular
lesion in NZW rabbits generally requires feeding of a high
cholesterol diet (from 0.2 to 2% cholesterol) which increases
plasma cholesterol level by ≤8-fold and leads to the formation of
foam cells-enriched fatty streaks in several vascular regions,
especially the aortic arch and the thoracic aorta (45). For complex atherosclerotic plaques with
lipid core surrounded by smooth muscle cells to develop, a long
period of cholesterol feeding, from six months to several years, is
required. The disadvantage of this diet is its hepatic toxicity,
which increases mortality.
Genetically modified rabbits
Due to the noxious side effects of the high-fat
diet, genetically modified rabbits have been developed to produce
spontaneous atherosclerotic lesions. For example, Watanabe
hereditary hypercholesterolemic rabbit (WHHL), a LDLR-deficient
model, was used by Buja et al, who identified LDL as the
lipoprotein underlying human familial hypercholesterolemia
(46). The advantage of this WHHL
model is that the morphology of lesions and lipid metabolism are
largely similar to those observed in humans. When WHHL rabbit is
fed with 1% cholesterol for 12 months, the atherosclerotic plaques
resemble those seen in homozygous familial hypercholesterolemia
patients (46,47), with areas of necrosis, cholesterol
clefts and calcification (48), with
foam cells originating from smooth muscle cells. Furthermore, WHHL
rabbits share the same gender predisposition patterns, with males
being more prone to coronary atherosclerosis (49).
Advantages and disadvantages of rabbit
models
Rabbits share the same advantages with mice with
their small size and hence ease in maintenance, high availability
and low economical cost. They are frequently preferred because of
similar lipoprotein metabolism to humans. With the expression of
CETP, the predominant lipoprotein in rabbit is LDL (45). Rabbits are sensitive to dietary
cholesterol overload, demonstrating subsequent hyperlipidemia
without the need of the toxic high cholesterol diet. Since rabbits
are larger than mice, catheter-based procedure and non-invasive
imaging can be used for experimental interrogation. Some important
differences are that the frequent sites of atherosclerotic lesions
in rabbits are the aortic arch and descending thoracic aorta,
whereas those in humans are the coronary arteries and the abdominal
aorta (45). Nevertheless, application
of an ameroid constrictor to induce arterial stenosis in rabbit
coronary arteries led to intimal proliferation together with
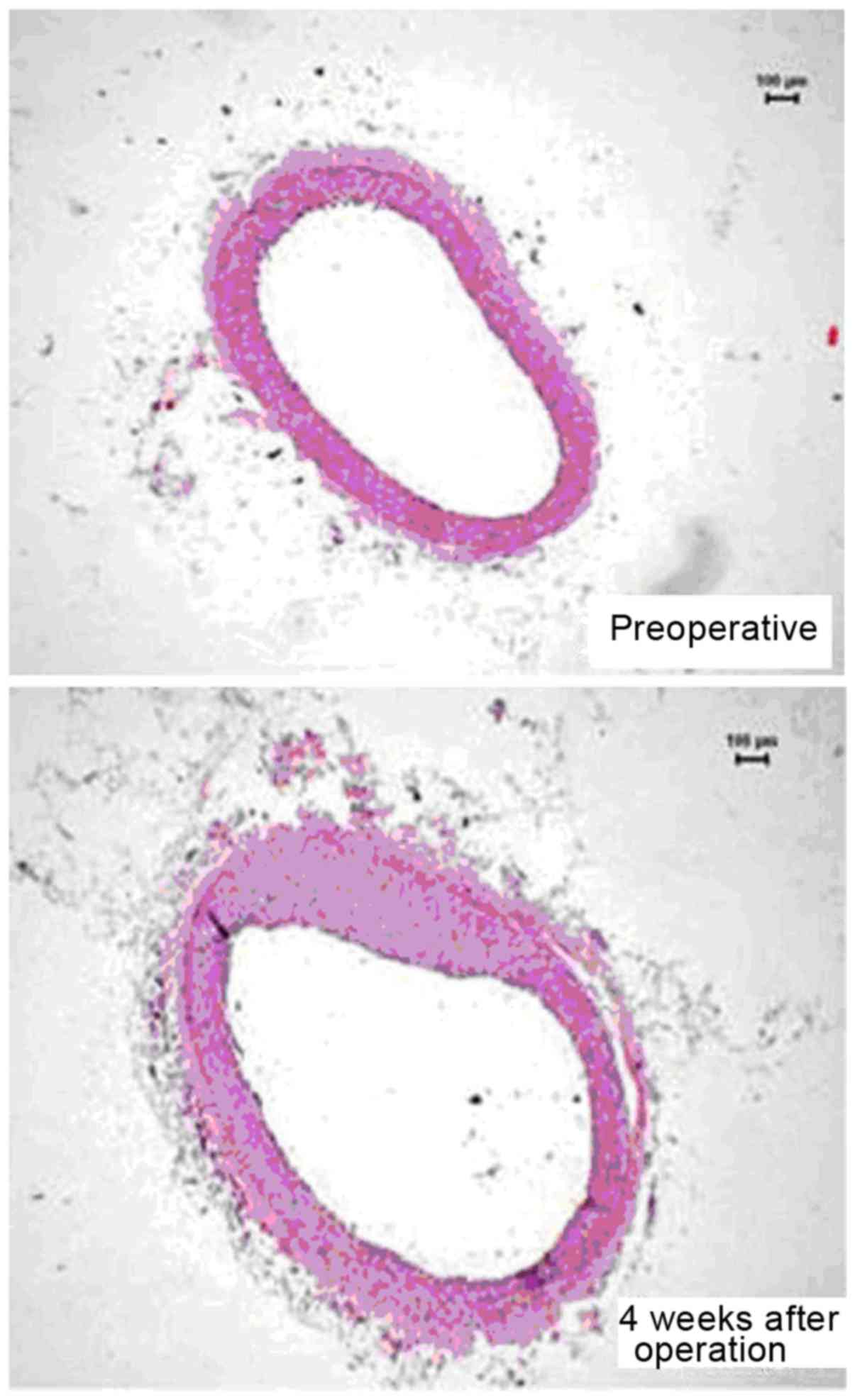
eccentric narrowing 4 weeks later (Fig.
1) (30).
Porcine
The different porcine models can broadly be divided
into wild-type or genetically modified models.
Wild-type pig models
Rapacz pig is a wild-type model with a natural
mutation in ApoB and LDLR genes, which were produced
by selective breeding of pigs with high cholesterol by Davis et
al (50). Within 2–4 years on a
normal diet, these pigs developed increased hypercholesterolemia,
with LDL as the main circulating lipoprotein, associated with the
development of coronary atherosclerosis. A mini-pig model bearing
the same gene mutation but with lower cost, the
hypercholesterolemia Bretoncelles Meishan (FBM) pigs, was
subsequently made available (51). The
diabetic hypercholesterolemic wild-type pig can provide a humanoid
model for investigations. An example is the type I diabetes model
produced by intravenous streptozotocin injection, which has been
used to destroy >80% of pancreatic β-cells in Yorkshire pigs
(52). Its combination with a high
cholesterol diet used to induce hypercholesterolemia increased
atherosclerotic risk by 2-fold, compared with hypercholesterolemia
alone. Diabetic Yorkshire pigs demonstrated an accelerated rate of
atherosclerotic lesion development in the aorta, coronary and
femoral arteries (53). The lesions
developed exhibit human-like features of advanced plaques,
including necrotic cores, fibrous caps, calcification, medial
thinning and intraplaque haemorrhages.
Genetically modified porcine
models
In genetic mini-pig models, atherosclerosis can be
induced without the use of cholic acid, thereby avoiding its toxic
inflammatory side effects (54). An
example of a model system is the Yucatan mini-pig with
liver-specific expression of D374Y, a gain-of-function mutation in
human protein convertase PCSK9, which consistently downregulated
LDL receptor levels and increased LDL concentrations following a
high-fat diet. This led to a severe form of autosomal dominant
hypercholesterolemia (55,56), successfully inducing atherosclerosis in
thoracic, abdominal, ilio-femoral and coronary arteries at 1 year
of age. This model is applicable for validating equipment designed
for human use, such as clinical scanners and intravascular devices.
Another transgenic model is the LDLR knockout Yucatan mini-pigs,
which demonstrated similar hypercholesterolemia and progression of
atherosclerotic development. A high-fat diet led to the development
of atherosclerotic lesions at 6–11 months (57).
Advantages and disadvantages of
porcine models
The use of porcine models has the advantage of
bearing close resemblance of cardiac anatomy and physiology to the
human counterpart. LDL, as in humans, is the major circulating
lipoprotein in plasma, except for apoliproprotein II deficiency in
pigs. Another advantage is the highly defined genotypes, which
enable the development of porcine models with multiple genetic
alterations. The emergence of site-specific nucleases, including
the clustered regularly interspaced short palindromic repeats
(CRISPR)/Cas9 system, was a breakthrough that allowed multiple
genes to be targeted at the same time by the expression of multiple
sgRNAs together with the Cas9 nuclease. The CRISPR/Cas9 system
allows pronuclear injection protocols with a high success rate
(58), and only one animal cloning
round is needed, reducing the time needed for breeding and the cost
of production.
As with humans, pigs are susceptible to diet-induced
hypercholesterolemia, but they require high dietary cholesterol,
typically combined with cholic acid to block the conversion from
cholesterol to bile (59,60). The atherosclerotic lesions usually do
not progress beyond the foam cell stage and the duration of
atheroma formation is longer than that observed in mice. Combining
the use of an atherogenic diet with artificial vascular injury is
one of the methods to accelerate atherosclerosis development in
pigs. First, normal pigs are fed with a high cholesterol diet and
percutaneous intramural injection of cholesteryl esters and human
oxLDL (61,62). Two weeks later, vascular injury is
produced by methods such as guidewire-induced injury, endovascular
balloon inflation and partial vessel ligation (63). This method produces rapid
atherosclerotic plaque development, thereby reducing the duration
and cost of the experimental studies. The histopathology of
atherosclerotic lesions are similar to humans, including their
location and content (57). This model
therefore provides a platform for the investigation of the disease
complications, including plaque rupture, ischemic reperfusion
injury, arterial thrombosis and restenosis after angioplasty
(64–66), and explorations for therapeutic
interventions such as drug-eluting stents (66).
The use of the porcine model as an in vivo
validation of imaging tools is valuable, in contrast to the post
mortem specimen and ex vivo model, which failed to produce
satisfactory validation. Post-mortem specimens cannot imitate the
dynamic cell components in atherosclerotic plaques, whereas ex
vivo models do not demonstrate the pulsatile flow normally
observed in elastic arteries, moving coronary arteries on a beating
heart, irregular heart rhythms and the moving tissues surrounding
the vessels. Initial validation is crucial for the development of
intravascular imaging technique to guide therapy in symptomatic
patients (67,68). With the authentic human-like dimension
and morphology of coronary arteries, pigs provide an ideal platform
for the insertion of intravascular imaging tools, such as
intravascular ultrasound (69) and
near-infrared spectroscopy (70).
Real-time imaging of tissues and cells in evolving atherosclerotic
plaques is also possible.
Due to lack of sensitive and reliable biomarkers for
monitoring disease progression, imaging tools are important in
monitoring plaque evolution and the efficacy of therapeutic
treatment. For non-invasive imaging with PET-CT, MRI or CT, high
resolution imaging of large arteries in smaller animals is possible
using dedicated and modified scan protocols. Porcine models are
useful to establish the relationship between plaque size and system
resolution, with similar extents of motion and artefacts compared
to humans. The protocol of scan parameter can be used subsequently
without the need of modification.
Another valuable use of the porcine model is drug
development. In view of the close phylogenetic relationship and
relevant atherosclerotic pathology observed in porcine systems
(71,72), drug testing is predictive for efficacy
of drugs in humans compared to the mouse model. It can be used to
guide the decision on endpoint of drug efficacy in clinical trials.
Currently, there is no standard imaging endpoint that is capable of
detecting all beneficial effects of pharmacological intervention
(73,74). Some drugs target lipid content
reduction, which is potentially measurable using near-infrared
spectroscopy (70). By contrast, other
drugs that aim to reduce inflammation can be assessed by 18
fluorodeoxyglucose PET-CT techniques (75). Porcine models offer a test platform
where both pathological examinations of atherosclerosis and
evaluation with clinical imaging end point can be performed
concurrently (71).
Nonetheless, the large size of pigs limits their
widespread use. Recently, genetically engineered mini-pigs
(76,77)
in which hyperlipidemia and consequently atherosclerosis were
successfully induced, became available; they are cheaper to
maintain compared to full-sized pigs. A close examination of its
pathophysiological mechanisms revealed similarities with human
atherosclerosis, as in the full-sized pigs, that are not observed
in mouse models.
Non-human primate models
Non-human primate models bear closest similarities
to humans compared to other species, in terms of phylogeny with 98%
genetic material being identical.
Rhesus and cynomolgus monkeys
Complex atherosclerotic lesions in coronary arteries
of Rhesus monkeys were successfully induced using a high fat, high
cholesterol diet (57). The lesions
demonstrated intimal thickening and increased density of vasa
vasorum in the tunica media (78),
which are features also observed in humans. The identification of
regression of coronary atherosclerosis upon reversion to a low-fat
diet was first established using this model system (79). This was associated with a lower
cholesterol content within the lesions and a decrease in the number
of foam cells as well as their lipid content. Cynomolgus monkeys
have been used because of their higher sensitivity to a high-fat
diet. When these animals were fed with 12.5% coconut oil and 12.5%
of butter fat (50,80), their plasma cholesterol level was twice
as high as those of the Rhesus monkeys, associated with a higher
number of lipid-loaded monocytes in the blood and skin xanthomata,
as well as faster disease progression.
Advantages and disadvantages of
primate models
The major advantage of using primates is that they
have very similar cardiac anatomy and physiology compared to
humans. Abnormal cardiovascular physiology in terms of lesion
morphology, plaque vulnerability and development of spontaneous
luminal thrombosis are observed in both species (51,80–82). Primates bear similar susceptibility to
atherosclerosis, with younger-aged animals being reasonably
resistant to development of atherosclerosis, but have an increased
risk of becoming susceptible with increasing age (83). Gender difference in the susceptibility
of atherosclerosis have been demonstrated in these primate models,
with a male preponderance to development of atherosclerosis
following the introduction of a high-fat diet (84). High cholesterol diet greatly
accelerated the development of atherosclerosis and frequently
induced fatal MI in these primates (85). Conversely, disease regression upon low
fat feeding was also evident, in keeping with clinical findings
(86,87). Finally, associations between
psychosocial factors and atherosclerosis have similarly been
established for these primate models (88). Taken together, these factors lead to a
greater applicability of experimental data on the clinical
scenario.
Despite bearing close resemblances to humans,
primate models are less popular than the other types of model due
to its difficulty in maintainance due to their large size, high
cost, limited availability and the special facilities required for
their accommodation. Secondly, a considerate length of time is
needed to induce significant atherosclerosis. Thirdly, the ethical
concern of experimenting with human-like primates limits their
widespread use. Nevertheless, non-human primates are ideal for the
development of reliable biomarker tools for risk stratification and
monitoring of the effects of pharmacological interventions on
disease progression.
Concluding remarks
Animal models have been extensively used to study
the pathophysiology of cardiovascular disorders (89–106). There
is no one single ideal animal model for all the diseases (107,108). The
general criteria for an appropriate animal model includes the size,
docility, ease of breeding and housing, known genetic profile,
analogies with humans and the cost associated. A smaller animal
model, such as mouse and rabbit, generally provide valuable
information on etiology and pathophysiology of atherosclerosis.
Understanding of the risk factors and the natural history of
atherosclerosis offer insights on disease prevention. On the other
hand, larger animal models, such as porcine and non-human primates,
are more reliable homologies with human disease. The advanced
lesions developed share similar histological features with humans,
from initial content of fatty streak to final advanced lesion of
ulceration and thrombus formation. Their use is therefore more
valuable for the development of disease management, such as
analysing the utility imaging methods and assessing the efficacy of
pharmacological intervention.
With the advancement in genetic technology, the
development of mini-pigs is a favourable trade-off between
human-like physiology compared to non-human primate; and ease of
handling compared with small animal, with high resemblance to human
cardiac anatomy, physiology, lipid metabolism and atherosclerotic
pathophysiology. IT is expected to act as an important in
vivo model, for developing sensitive biomarkers and validated
imaging tools to predict plaque rupture, as the most important
clinical event that cost life in atherosclerosis.
Acknowledgements
GT was supported by a BBSRC Doctoral Training Award
and thanks the Croucher Foundation of Hong Kong for the generous
support of his clinical assistant professorship. YC is supported by
the ESRC.
References
|
1
|
Nordestgaard BG, Chapman MJ, Humphries SE,
Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ,
Defesche JC, et al: European Atherosclerosis Society Consensus
Panel: Familial hypercholesterolaemia is underdiagnosed and
undertreated in the general population: guidance for clinicians to
prevent coronary heart disease: consensus statement of the European
Atherosclerosis Society. Eur Heart J. 34:3478–3490a. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lim SS, Vos T, Flaxman AD, Danaei G,
Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee
M, et al: A comparative risk assessment of burden of disease and
injury attributable to 67 risk factors and risk factor clusters in
21 regions, 1990–2010: A systematic analysis for the Global Burden
of Disease Study 2010. Lancet. 380:2224–2260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yusuf S, Hawken S, Ounpuu S, Dans T,
Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, et al:
INTERHEART Study Investigators: Effect of potentially modifiable
risk factors associated with myocardial infarction in 52 countries
(the INTERHEART study): Case-control study. Lancet. 364:937–952.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Steinberg D, Glass CK and Witztum JL:
Evidence mandating earlier and more aggressive treatment of
hypercholesterolemia. Circulation. 118:672–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma S, Tian XY, Zhang Y, Mu C, Shen H,
Bismuth J, Pownall HJ, Huang Y and Wong WT: E-selectin-targeting
delivery of microRNAs by microparticles ameliorates endothelial
inflammation and atherosclerosis. Sci Rep. 6:229102016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee WH, Kim SH, Jeong EM, Choi YH, Kim DI,
Lee BB, Cho YS, Kwon BS and Park JE: A novel chemokine,
Leukotactin-1, induces chemotaxis, pro-atherogenic cytokines, and
tissue factor expression in atherosclerosis. Atherosclerosis.
161:255–260. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tabas I: 2016 Russell Ross Memorial
Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the
Progression of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2016
Dec 15;pii: ATVBAHA.116.308036. PMID: 27979856. PubMed/NCBI
|
|
8
|
Di Pietro N, Formoso G and Pandolfi A:
Physiology and pathophysiology of oxLDL uptake by vascular wall
cells in atherosclerosis. Vascul Pharmacol. 84:1–7. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Randolph GJ, Jakubzick C and Qu C: Antigen
presentation by monocytes and monocyte-derived cells. Curr Opin
Immunol. 20:52–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gerthoffer WT: Mechanisms of vascular
smooth muscle cell migration. Circ Res. 100:607–621. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao Y, Biswas SK, McNulty PH, Kozak M,
Jun JY and Segar L: PDGF-induced vascular smooth muscle cell
proliferation is associated with dysregulation of insulin receptor
substrates. Am J Physiol Cell Physiol. 300:C1375–C1385. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shah PK: Mechanisms of plaque
vulnerability and rupture. J Am Coll Cardiol. 41:15S–22S. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Majesky MW: Developmental basis of
vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol.
27:1248–1258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
VanderLaan PA, Reardon CA and Getz GS:
Site specificity of atherosclerosis: Site-selective responses to
atherosclerotic modulators. Arterioscler Thromb Vasc Biol.
24:12–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ley K, Miller YI and Hedrick CC: Monocyte
and macrophage dynamics during atherogenesis. Arterioscler Thromb
Vasc Biol. 31:1506–1516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baetta R and Corsini A: Role of
polymorphonuclear neutrophils in atherosclerosis: Current state and
future perspectives. Atherosclerosis. 210:1–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weber C, Zernecke A and Libby P: The
multifaceted contributions of leukocyte subsets to atherosclerosis:
Lessons from mouse models. Nat Rev Immunol. 8:802–815. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lahoute C, Herbin O, Mallat Z and Tedgui
A: Adaptive immunity in atherosclerosis: Mechanisms and future
therapeutic targets. Nat Rev Cardiol. 8:348–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Getz GS, Vanderlaan PA and Reardon CA:
Natural killer T cells in lipoprotein metabolism and
atherosclerosis. Thromb Haemost. 106:814–819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Butcher M and Galkina E: Current views on
the functions of interleukin-17A-producing cells in
atherosclerosis. Thromb Haemost. 106:787–795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Newby AC: Metalloproteinase expression in
monocytes and macrophages and its relationship to atherosclerotic
plaque instability. Arterioscler Thromb Vasc Biol. 28:2108–2114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tse G, Wong ST, Tse V, Lee YT, Lin HY and
Yeo JM: Cardiac dynamics: alternans and arrhythmogenesis. J
Arrhythm. 32:411–417. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tse G, Wong ST, Tse V and Yeo JM:
Depolarization vs. repolarization: What is the mechanism of
ventricular arrhythmogenesis underlying sodium channel
haploinsufficiency in mouse hearts? Acta Physiol (Oxf). Apr
16–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tse G: (Tpeak - Tend)/QRS and (Tpeak -
Tend)/(QT × QRS): Novel markers for predicting arrhythmic risk in
the Brugada syndrome. Europace. Oct 5–2016.(Epub ahead of print).
View Article : Google Scholar
|
|
26
|
Tse G, Wong ST, Tse V and Yeo JM:
Determination of action potential wavelength restitution in
Scn5a/−mouse hearts modelling human Brugada syndrome. J Physiol.
(In press).
|
|
27
|
Tse G: Both transmural dispersion of
repolarization and transmural dispersion of refractoriness are poor
predictors of arrhythmogenicity: A role for the index of Cardiac
Electrophysiological Balance (QT/QRS)? J Geriatr Cardiol. (In
press).
|
|
28
|
Tse G: Novel conduction-repolarization
indices for the stratification of arrhythmic risk. J Geriatr
Cardiol. (In press).
|
|
29
|
Tse G, Wong ST, Tse V and Yeo JM:
Variability in local action potential durations, dispersion of
repolarization and wavelength restitution in aged wild-type and
Scn5a/−mouse hearts modelling human Brugada syndrome. J Geriatr
Cardiol. (In press).
|
|
30
|
Hu Z, Chen Z, Wang Y, Jiang J, Tse G, Xu
W, Ge J and Sun B: Effects of granulocyte colony-stimulating factor
on rabbit carotid and swine heart models of chronic obliterative
arterial disease. Mol Med Rep. (In press).
|
|
31
|
Tse G, Yeo JM, Tse V, Kwan J and Sun B:
Gap junction inhibition by heptanol increases ventricular
arrhythmogenicity by reducing conduction velocity without affecting
repolarization properties or myocardial refractoriness in
Langendorff-perfused mouse hearts. Mol Med Rep. 14:4069–4074.
2016.PubMed/NCBI
|
|
32
|
Tse G, Tse V and Yeo JM: Ventricular
anti-arrhythmic effects of heptanol in hypokalaemic,
Langendorff-perfused mouse hearts. Biomed Rep. 4:313–324.
2016.PubMed/NCBI
|
|
33
|
Tse G, Tse V, Yeo JM and Sun B: Atrial
anti-arrhythmic effects of heptanol in Langendorff-perfused mouse
hearts. PLoS One. 11:e01488582016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tse G, Wong ST, Tse V and Yeo JM:
Restitution analysis of alternans using dynamic pacing and its
comparison with S1S2 restitution in heptanol-treated, hypokalaemic
Langendorff-perfused mouse hearts. Biomed Rep. 4:673–680.
2016.PubMed/NCBI
|
|
35
|
Tse G, Wong ST, Tse V and Yeo JM:
Monophasic action potential recordings: Which is the recording
electrode? J Basic Clin Physiol Pharmacol. 27:457–462. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tse G, Lai ET, Yeo JM, Tse V and Wong SH:
Mechanisms of electrical activation and conduction in the
gastrointestinal system: Lessons from cardiac electrophysiology.
Front Physiol. 7:1822016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tse G, Lai ET, Tse V and Yeo JM: Molecular
and electrophysiological mechanisms underlying cardiac
arrhythmogenesis in diabetes mellitus. J Diabetes Res.
2016:28487592016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tse G, Lai ET, Yeo JM and Yan BP:
Electrophysiological mechanisms of Bayés syndrome: Insights from
clinical and mouse studies. Front Physiol. 7:1882016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tse G, Sun B, Wong ST, Tse V and Yeo JM:
Anti-arrhythmic effects of hypercalcemia in hyperkalemic,
Langendorff-perfused mouse hearts. Biomed Rep. 5:301–310.
2016.PubMed/NCBI
|
|
40
|
Chen Z, Sun B, Tse G, Jiang J and Xu W:
Reversibility of both sinus node dysfunction and reduced HCN4 mRNA
expression level in an atrial tachycardia pacing model of
tachycardia-bradycardia syndrome in rabbit hearts. Int J Clin Exp
Pathol. 9:8526–8531. 2016.
|
|
41
|
Tse G, Yeo JM, Chan YW, Lai ET and Yan BP:
What is the arrhythmic substrate in viral myocarditis? Insights
from clinical and animal studies. Front Physiol. 7:3082016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nordestgaard BG and Zilversmit DB: Large
lipoproteins are excluded from the arterial wall in diabetic
cholesterol-fed rabbits. J Lipid Res. 29:1491–1500. 1988.PubMed/NCBI
|
|
43
|
Kritchevsky D: Herman Award Lecture, 1992:
Lipid nutrition - a personal perspective. Am J Clin Nutr.
56:730–734. 1992.PubMed/NCBI
|
|
44
|
Spagnoli LG, Orlandi A, Mauriello A,
Santeusanio G, de Angelis C, Lucreziotti R and Ramacci MT: Aging
and atherosclerosis in the rabbit. 1. Distribution, prevalence and
morphology of atherosclerotic lesions. Atherosclerosis. 89:11–24.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bocan TM, Mueller SB, Mazur MJ, Uhlendorf
PD, Brown EQ and Kieft KA: The relationship between the degree of
dietary-induced hypercholesterolemia in the rabbit and
atherosclerotic lesion formation. Atherosclerosis. 102:9–22. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shiomi M and Ito T: The Watanabe heritable
hyperlipidemic (WHHL) rabbit, its characteristics and history of
development: A tribute to the late Dr. Yoshio Watanabe.
Atherosclerosis. 207:1–7. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Buja LM, Kita T, Goldstein JL, Watanabe Y
and Brown MS: Cellular pathology of progressive atherosclerosis in
the WHHL rabbit. An animal model of familial hypercholesterolemia.
Arteriosclerosis. 3:87–101. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Atkinson JB, Hoover RL, Berry KK and Swift
LL: Cholesterol-fed heterozygous Watanabe heritable hyperlipidemic
rabbits: A new model for atherosclerosis. Atherosclerosis.
78:123–136. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shiomi M, Ito T, Shiraishi M and Watanabe
Y: Inheritability of atherosclerosis and the role of lipoproteins
as risk factors in the development of atherosclerosis in WHHL
rabbits: Risk factors related to coronary atherosclerosis are
different from those related to aortic atherosclerosis.
Atherosclerosis. 96:43–52. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Davis HR, Vesselinovitch D and Wissler RW:
Reticuloendothelial system response to hyperlipidemia in rhesus and
cynomolgus monkeys. J Leukoc Biol. 36:63–80. 1984.PubMed/NCBI
|
|
51
|
Mott GE, Jackson EM, McMahan CA and McGill
HC Jr: Dietary cholesterol and type of fat differentially affect
cholesterol metabolism and atherosclerosis in baboons. J Nutr.
122:1397–1406. 1992.PubMed/NCBI
|
|
52
|
Gerrity RG: The role of the monocyte in
atherogenesis: I. Transition of blood-borne monocytes into foam
cells in fatty lesions. Am J Pathol. 103:181–190. 1981.PubMed/NCBI
|
|
53
|
Gerrity RG, Natarajan R, Nadler JL and
Kimsey T: Diabetes-induced accelerated atherosclerosis in swine.
Diabetes. 50:1654–1665. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Civelek M, Manduchi E, Riley RJ, Stoeckert
CJ Jr and Davies PF: Coronary artery endothelial transcriptome in
vivo: Identification of endoplasmic reticulum stress and enhanced
reactive oxygen species by gene connectivity network analysis. Circ
Cardiovasc Genet. 4:243–252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakashima Y, Plump AS, Raines EW, Breslow
JL and Ross R: ApoE-deficient mice develop lesions of all phases of
atherosclerosis throughout the arterial tree. Arterioscler Thromb.
14:133–140. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Getz GS and Reardon CA: Apoprotein E as a
lipid transport and signaling protein in the blood, liver, and
artery wall. J Lipid Res. 50:S156–S161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Williams JK, Armstrong ML and Heistad DD:
Vasa vasorum in atherosclerotic coronary arteries: Responses to
vasoactive stimuli and regression of atherosclerosis. Circ Res.
62:515–523. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hai T, Teng F, Guo R, Li W and Zhou Q:
One-step generation of knockout pigs by zygote injection of
CRISPR/Cas system. Cell Res. 24:372–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Getz GS and Reardon CA: Animal models of
atherosclerosis. Arterioscler Thromb Vasc Biol. 32:1104–1115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Al-Mashhadi RH, Sørensen CB, Kragh PM,
Christoffersen C, Mortensen MB, Tolbod LP, Thim T, Du Y, Li J, Liu
Y, et al: Familial hypercholesterolemia and atherosclerosis in
cloned minipigs created by DNA transposition of a human PCSK9
gain-of-function mutant. Sci Transl Med. 5:166ra12013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Granada JF, Moreno PR, Burke AP, Schulz
DG, Raizner AE and Kaluza GL: Endovascular needle injection of
cholesteryl linoleate into the arterial wall produces complex
vascular lesions identifiable by intravascular ultrasound: Early
development in a porcine model of vulnerable plaque. Coron Artery
Dis. 16:217–224. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Granada JF, Wallace-Bradley D, Win HK,
Alviar CL, Builes A, Lev EI, Barrios R, Schulz DG, Raizner AE and
Kaluza GL: In vivo plaque characterization using intravascular
ultrasound-virtual histology in a porcine model of complex coronary
lesions. Arterioscler Thromb Vasc Biol. 27:387–393. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Thim T, Hagensen MK, Drouet L, Bal Dit
Sollier C, Bonneau M, Granada JF, Nielsen LB, Paaske WP, Bøtker HE
and Falk E: Familial hypercholesterolaemic downsized pig with
human-like coronary atherosclerosis: A model for preclinical
studies. EuroIntervention. 6:261–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dansky HM, Charlton SA, Harper MM and
Smith JD: T and B lymphocytes play a minor role in atherosclerotic
plaque formation in the apolipoprotein E-deficient mouse. Proc Natl
Acad Sci USA. 94:4642–4646. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nakashima Y, Raines EW, Plump AS, Breslow
JL and Ross R: Upregulation of VCAM-1 and ICAM-1 at
atherosclerosis-prone sites on the endothelium in the
ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. 18:842–851.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Iiyama K, Hajra L, Iiyama M, Li H,
DiChiara M, Medoff BD and Cybulsky MI: Patterns of vascular cell
adhesion molecule-1 and intercellular adhesion molecule-1
expression in rabbit and mouse atherosclerotic lesions and at sites
predisposed to lesion formation. Circ Res. 85:199–207. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fazio S, Babaev VR, Murray AB, Hasty AH,
Carter KJ, Gleaves LA, Atkinson JB and Linton MF: Increased
atherosclerosis in mice reconstituted with apolipoprotein E null
macrophages. Proc Natl Acad Sci USA. 94:4647–4652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Van Eck M, Herijgers N, Vidgeon-Hart M,
Pearce NJ, Hoogerbrugge PM, Groot PH and Van Berkel TJ: Accelerated
atherosclerosis in C57Bl/6 mice transplanted with ApoE-deficient
bone marrow. Atherosclerosis. 150:71–80. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mazzone T and Reardon C: Expression of
heterologous human apolipoprotein E by J774 macrophages enhances
cholesterol efflux to HDL3. J Lipid Res. 35:1345–1353.
1994.PubMed/NCBI
|
|
70
|
Strong JP and McGill HC Jr: Diet and
experimental atherosclerosis in baboons. Am J Pathol. 50:669–690.
1967.PubMed/NCBI
|
|
71
|
Kaplan JR, Manuck SB, Clarkson TB, Lusso
FM and Taub DM: Social status, environment, and atherosclerosis in
cynomolgus monkeys. Arteriosclerosis. 2:359–368. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Thorngate FE, Rudel LL, Walzem RL and
Williams DL: Low levels of extrahepatic nonmacrophage ApoE inhibit
atherosclerosis without correcting hypercholesterolemia in
ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 20:1939–1945.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Linton MF and Fazio S: Macrophages,
lipoprotein metabolism, and atherosclerosis: Insights from murine
bone marrow transplantation studies. Curr Opin Lipidol. 10:97–105.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang X, Peterson L, Thieringer R, Deignan
JL, Wang X, Zhu J, Wang S, Zhong H, Stepaniants S, Beaulaurier J,
et al: Identification and validation of genes affecting aortic
lesions in mice. J Clin Invest. 120:2414–2422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Barcat D, Amadio A, Palos-Pinto A, Daret
D, Benlian P, Darmon M and Bérard AM: Combined
hyperlipidemia/hyperalphalipoproteinemia associated with premature
spontaneous atherosclerosis in mice lacking hepatic lipase and low
density lipoprotein receptor. Atherosclerosis. 188:347–355. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Davis BT, Wang XJ, Rohret JA, Struzynski
JT, Merricks EP, Bellinger DA, Rohret FA, Nichols TC and Rogers CS:
Targeted disruption of LDLR causes hypercholesterolemia and
atherosclerosis in Yucatan miniature pigs. PLoS One. 9:e934572014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Agarwala A, Billheimer J and Rader DJ:
Mighty minipig in fight against cardiovascular disease. Sci Transl
Med. 5:166fs12013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Heistad DD and Armstrong ML: Blood flow
through vasa vasorum of coronary arteries in atherosclerotic
monkeys. Arteriosclerosis. 6:326–331. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Armstrong ML and Megan MB: Lipid depletion
in atheromatous coronary arteries in rhesus monkeys after
regression diets. Circ Res. 30:675–680. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Davis HR, Vesselinovitch D and Wissler RW:
Histochemical detection and quantification of macrophages in rhesus
and cynomolgus monkey atherosclerotic lesions. J Histochem
Cytochem. 32:1319–1327. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wolfe MS, Sawyer JK, Morgan TM, Bullock BC
and Rudel LL: Dietary polyunsaturated fat decreases coronary artery
atherosclerosis in a pediatric-aged population of African green
monkeys. Arterioscler Thromb. 14:587–597. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Vesselinovitch D, Getz GS, Hughes RH and
Wissler RW: Atherosclerosis in the rhesus monkey fed three food
fats. Atherosclerosis. 20:303–321. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bullock BC, Clarkson TB, Lehner ND,
Lofland HB Jr and St Clair RW: Atherosclerosis in Cebus albifrons
monkeys. Clinical and pathologic studies. Exp Mol Pathol. 10:39–62.
1969. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Clarkson TB, Koritnik DR, Weingand KW and
Miller LC: Nonhuman primate models of atherosclerosis: Potential
for the study of diabetes mellitus and hyperinsulinemia.
Metabolism. 34:(Suppl 1). 51–59. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Taylor CB, Cox GF, Hall-Taylor BJ and
Nelson LG: Atherosclerosis in areas of vascular injury in monkeys
with mild hypercholesterolemia. Circulation. 10:6131954.
|
|
86
|
Clarkson TB, Lehner NDM, Wagner WD, St
Clair RW, Bond MG and Bullock BC: A study of atherosclerosis
regression in Macaca mulatta. I. Design of experiment and lesion
induction. Exp Mol Pathol. 30:360–385. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Clarkson TB, Bond MG, Bullock BC,
McLaughlin KJ and Sawyer JK: A study of atherosclerosis regression
in Macaca mulatta. V. Changes in abdominal aorta and carotid and
coronary arteries from animals with atherosclerosis induced for 38
months and then regressed for 24 or 48 months at plasma cholesterol
concentrations of 300 or 200 mg/dl. Exp Mol Pathol. 41:96–118.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kaplan JR and Manuck SB: Status, stress,
and atherosclerosis: The role of environment and individual
behavior. Ann N Y Acad Sci. 896:145–161. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Choy L, Yeo JM, Tse V, Chan SP and Tse G:
Cardiac disease and arrhythmogenesis: Mechanistic insights from
mouse models. Int J Cardiol Heart Vasc. 12:1–10. 2016.PubMed/NCBI
|
|
90
|
Tse G and Yan BP: Novel arrhythmic risk
markers incorporating QRS dispersion: QRSd × (Tpeak - Tend)/QRS and
QRSd × (Tpeak - Tend)/(QT × QRS). Ann Noninvasive Electrocardiol.
Aug 18–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tse G and Yan BP: Electrophysiological
mechanisms of long and short QT syndromes: insights from mouse
models. IJC Heart & Vasculature. 2016.
|
|
92
|
Tse G, Lai ETH, Lee APW, Yan BP and Wong
SH: Electrophysiological mechanisms of gastrointestinal
arrhythmogenesis: Lessons from the heart. Front Physiol. 7:2302016.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Tse G and Yan BP: Traditional and novel
electrocardiographic conduction and repolarization markers of
sudden cardiac death. Europace. Oct 4–2016.(Epub ahead of print).
View Article : Google Scholar
|
|
94
|
Tse G, Yan BP, Chan YW, Tian XY and Huang
Y: Reactive oxygen species, endoplasmic reticulum stress and
mitochondrial dysfunction: The link with cardiac arrhythmogenesis.
Front Physiol. 7:3132016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tse G, Wong ST, Tse V and Yeo JM:
Schrödinger's cat in cardiac electrophysiology: Quinidine both
increases and decreases left ventricular endocardial action
potential durations in Langendorff-perfused mouse hearts. Acta
Physiol (Oxf). 2016.PubMed/NCBI
|
|
96
|
Sun B, Chen Z, Gu J, et al: Tight junction
proteins and gap junction proteins play important roles in high fat
dietary atherosclerosis pathogenesis. Int J Clin Exp Pathol.
9:7969–7976. 2016.
|
|
97
|
Chen-Izu Y, Shaw RM, Pitt GS,
Yarov-Yarovoy V, Sack JT, Abriel H, Aldrich RW, Belardinelli L,
Cannell MB, Catterall WA, et al: Na+ channel function,
regulation, structure, trafficking and sequestration. J Physiol.
593:1347–1360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tse G, Ali A, Prasad SK, Vassiliou V and
Raphael CE: Atypical case of post-partum cardiomyopathy: an overlap
syndrome with arrhythmogenic right ventricular cardiomyopathy? BJR
Case Rep. 1:201501822015.
|
|
99
|
Tse G, Ali A, Alpendurada F, Prasad S,
Raphael CE and Vassiliou V: Tuberculous constrictive pericarditis.
Res Cardiovasc Med. 4:e296142015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tse G and Yeo JM: Conduction abnormalities
and ventricular arrhythmogenesis: The roles of sodium channels and
gap junctions. Int J Cardiol Heart Vasc. 9:75–82. 2015.PubMed/NCBI
|
|
101
|
Tse G: Mechanisms of cardiac arrhythmias.
J Arrhythm. 32:75–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Tse G, Hothi SS, Grace AA and Huang CL:
Ventricular arrhythmogenesis following slowed conduction in
heptanol-treated, Langendorff-perfused mouse hearts. J Physiol Sci.
62:79–92. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yeo JM, Tse V, Kung J, Lin HY, Lee YT,
Kwan J, Yan BP and Tse G: Isolated heart models for studying
cardiac electrophysiology: a historical perspective and recent
advances. J Basic Clin Physiol Pharmacol. (In press).
|
|
104
|
Tse G, Liu T, Li KH, Laxton V, Wong A,
Chan YW, Keung W, Chan C and Li RA: 2016.Molecular and
electrophysiological mechanisms of tachycardia-bradycardia
syndrome. Int J Mol Med. (In press).
|
|
105
|
Fu H, Li G, Liu C, Li J, Cheng L, Yang W,
Tse G, Zhao J and Liu T: Probucol prevents atrial ion channel
remodeling in an alloxan-induced diabetes rabbit model. Oncotarget.
Nov 14–2016.(Epub ahead of print). View Article : Google Scholar
|
|
106
|
Tse G, Liu T, Li KH, Laxton V, Chan YW,
Keung W, Li RA and Yan BP: Electrophysiological mechanisms of
Brugada syndrome: insights from pre-clinical and clinical studies.
Front Physiol. 7:4672016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wong P, Laxton V, Srivastava S, Chan YW
and Tse G: The role of gap junctions in inflammatory and neoplastic
disorders. Int J Mol Med. (In press).
|
|
108
|
Wong P, Tan T, Chan C, Laxton V, Chan YWF,
Liu T, Wong WT and Tse G: The role of connexins in wound healing
and erpair: Novel therapeutic approaches. Front. Physiol.
7:5962016.
|