Introduction
Hyperuricemia has been linked to cardiovascular
disease (CVD) recent since it is regarded as an independent risk
factors in the contribution of CVD including vascular disease,
hypertension, metabolic syndrome and renal disease (1). Generally, uric acid (UA) levels >7
mg/dl in males and >6 mg/dl in females are considered as
hyperuricemia in a clinical aspect (2). Risk factors for hyperuricemia include
alcohol consumption, intake of high fat diet and refined
carbohydrate, and the medicines of diuretics and angiotensin
converting enzyme antagonists (1,2). Population
epidemiological studies support the notion that there is a causal
link between hyperuricemia and CVD. For example, Rotterdam
(3) and a NHANES I study (4) found that there is an association between
high levels of UA and myocardial infarctions and cardiovascular
death even after adjustment for related factors such as age,
dyslipidemia and obesity. Multiple center studies also suggested
that hyperuricemia prompts hypertension and coronary heart disease,
as well as vascular diseases such as cerebrovascular disease,
preeclampsia, vascular dementia and kidney disease (5,6). Some of the
potential pathophysiological mechanisms have been investigated to
explain the association between UA and vascular injury. One study
suggested that high dose UA significantly increased angiotensin II
and oxidative stress in cultured endothelial cells (ECs) and this
further prompted EC senescence and apoptosis (7). To this point, increase inflammation and
reduction of nitric oxide (NO) are the characteristics of
endothelium dysfunction (8). However,
few studies have been conducted to investigate the role of UA on
vascular relaxation, NO production, inflammation and its signaling
pathway on ECs. In the current study, the authors hypothesized that
UA-induced impairment of vascular relaxation is associated with a
reduction of NO and expression of inflammatory cytokines through
the nuclear factor (NF)-κB pathway.
Materials and methods
Study population
The study investigated 21 patients with
hyperuricemia and 16 control subjects at The First Clinical
Hospital of Hubei University of Science and Technology and Xianning
Central Hospital in China, between February 2014 and March 2016.
Blood samples were collected at 8:00 a.m. while subjects were
regularly fasting. The study was approved by the institutional
review board on human investigation at The First Clinical Hospital
of Hubei University of Science and Technology and Xianning Central
Hospital (Xianning, China). Informed consent was obtained from all
individuals in the study.
Brachial flow-mediated dilation (FMD)
and nitroglycerine-mediated dilation (NMD)
The diameter of the right brachial arteries in all
subjects were measured on B-mode ultrasound with 10 megahertz
linear-array transducer (Beijing ADSS Development Co., Ltd.,
Beijing, China) by one investigator at rest and sublingual spray
nitroglycerin (350 µg), respectively. A pressure cuff was placed
and automatically inflated on the forearm in order to initial
forearm ischemia, the pressure cuff was then released after 5 min
so that blood flow in the forearm could be increased. The maximum
diameters were recorded after cuff release (9,10). The
percentage increase in FMD and NMD were calculated as the increased
diameters in brachial arteries compared with the corresponding
baseline at rest (9,10).
EC culture
Primary human umbilical vein ECs were purchased from
(cat. no. 211–500, Shanghai Biotechnology Co., Ltd, Shanghai,
China) and cultured at cell incubator with 37°C in 5%
CO2 atmosphere. The sub-cultured strains of ECs were
used in experiments between passages 3–5. Prior to the experiments,
ECs were serum-starved in Dulbecco's modified Eagle's medium for 24
h. UA and NF-κB inhibitor II were purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany).
Measurement of NO production
Nitrite, a stable metabolite of NO, is converted
from nitrate by nitrate reductase and is an indicator of released
NO in the media of cultured ECs (11).
The NO productions in cultured ECs were measured using a
nitrate/nitrite assay kit (cat. no. 23479; Sigma-Aldrich; Merck
KGaA) following the protocol instructions (12).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was isolated from the cultured human
umbilical vein ECs using RNAzol (Sigma-Aldrich; Merck KGaA). After
the first strand, cDNA was synthesized by using Improm II reverse
transcription kit (Sigma-Aldrich; Merck KGaA), qPCR was done using
8 µl cDNA, 10 μl SYBR-Green PCR Master Mix and forward and reverse
primers (10 pM/µl) using a qPCR system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The following primer sequences were used:
interleukin (IL)-6 forward, 5′-ACAGTGTGGAATCATATATGA-3′ and
reverse, 5′-GCGTCGTTATATGTTGAACGC-3′; IL-8 forward,
5′-CGTGGTTATGCTACAAATGT-3′ and reverse, 5′-ACGGGCAACCCGTGGAGAGC-3′;
Tumor necrosis factor (TNF)-α forward, 5′-GCAGCCGTTAACGTAAACCT-3′
and reverse, 5′-TGTGTTAGGGCCTGTGGCAC-3′; 18S forward,
5′-GACGGTTGCCATCAAGTTCGA-3′ and reverse, 5′-TGCTGAGCATCGCTGGGAA-3′.
The PCR cycling conditions were 2 min at 95°C for initial
denaturation, 40 cycles of 45 sec at 95°C, 30 sec at 58°C and 45
sec at 72°C. The data were normalized to 18S ribosomal RNA that is
a housekeeping gene.
Western blot analysis
Human umbilical vein ECs were collected and lysed
with RIPA protein lysis buffer (cat. no. 20–188, Sigma-Aldrich;
Merck KGaA) and the protein concentration was investigated by Bio
Rad protein assay (Bio Rad Laboratories, Inc.). Proteins were
resolved by 10% SDS-PAGE and transferred to nitrocellulose
membranes (Sigma-Aldrich; Merck KGaA). Membranes were incubated
with antibody targeting NF-κB (cat. no. SAB4502609; 1:1,000
dilution; Sigma-Aldrich; Merck KGaA) and horseradish peroxidase
(HRP) conjugated secondary antibody (cat. no. A9542; 1:5000
dilution; Sigma-Aldrich; Merck KGaA) at room temperature for 1
hour. The HRP activity was examined by an enhanced
chemiluminescence kit (Bio Rad Laboratories, Inc.). As a loading
control, nuclear protein lamin B was probed in all the protein
expression.
Activation of transcription factor
NF-κB
Human umbilical vein ECs were transfected with 5 µg
of pNF-κB-Luc (Clontech Laboratories, Inc., Mountainview, CA, USA).
Transfection of human umbilical vein ECs was performed using the
nucleofection device and solutions to deliver electrical stimuli to
the ECs (Lonza Group, Basel, Switzerland) (13). Following 24 h transfection, 10 mg/dl UA
was used to treat the cells for 3 h and luciferase activity was
examined by using a Dual Luciferase Reporter Assay System and a
luminometer (Promega Corporation, Madison, WI, USA) as per the
manufacturer's protocol (14).
Statistical analysis
Results are calculated as the mean ± standard error
of the mean. Data were analyzed by using SAS software (version,
9.4; SAS Institute Inc., Cary, NC, USA). Differences in data were
determined using one-way analysis of variance or paired Student's t
tests and P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical characteristics of the
subjects
There was no significant difference in age, sex,
body mass index, blood pressure, plasma levels of creatinine,
glucose, triglyceride and low/high-density lipoprotein between
health control subjects and hyperuricemia patients (Table I). However, the plasma UA level is
significantly higher in hyperuricemia patients than in control
group (Table I).
 | Table I.Clinical characteristic of the study
population. |
Table I.
Clinical characteristic of the study
population.
| Variable | Control subjects
(n=16) | Hyperuricemia
patients (n=21) |
|---|
| Age (years) | 65.2±7.28 | 68.5±6.6 |
| Males in population
(%) | 50% | 47.6% |
| Body mass index
(kg/m2) | 25.1±2.2 | 25.6.±1.7 |
| Systolic blood
pressure (mmHg) | 140±6 | 142±10 |
| Diastolic blood
pressure (mmHg) | 85±4 | 86±4 |
| Creatinine
(mg/dl) | 0.78±0.11 | 0.77±0.14 |
| Plasma uric acid
(mg/dl) | 4.51±1.09 |
16.78±4.69† |
| Plasma glucose
(mg/dl) | 98.65±11.35 | 100.47±9.69 |
| Triglyceride
(mg/dl) | 99.46±37.33 | 128.44±51.22 |
| Low-density
lipoprotein | 121.25±27.98 | 145.66±43.21 |
| High-density
lipoprotein (mg/dl) | 53.15±6.23 | 62.22±8.09 |
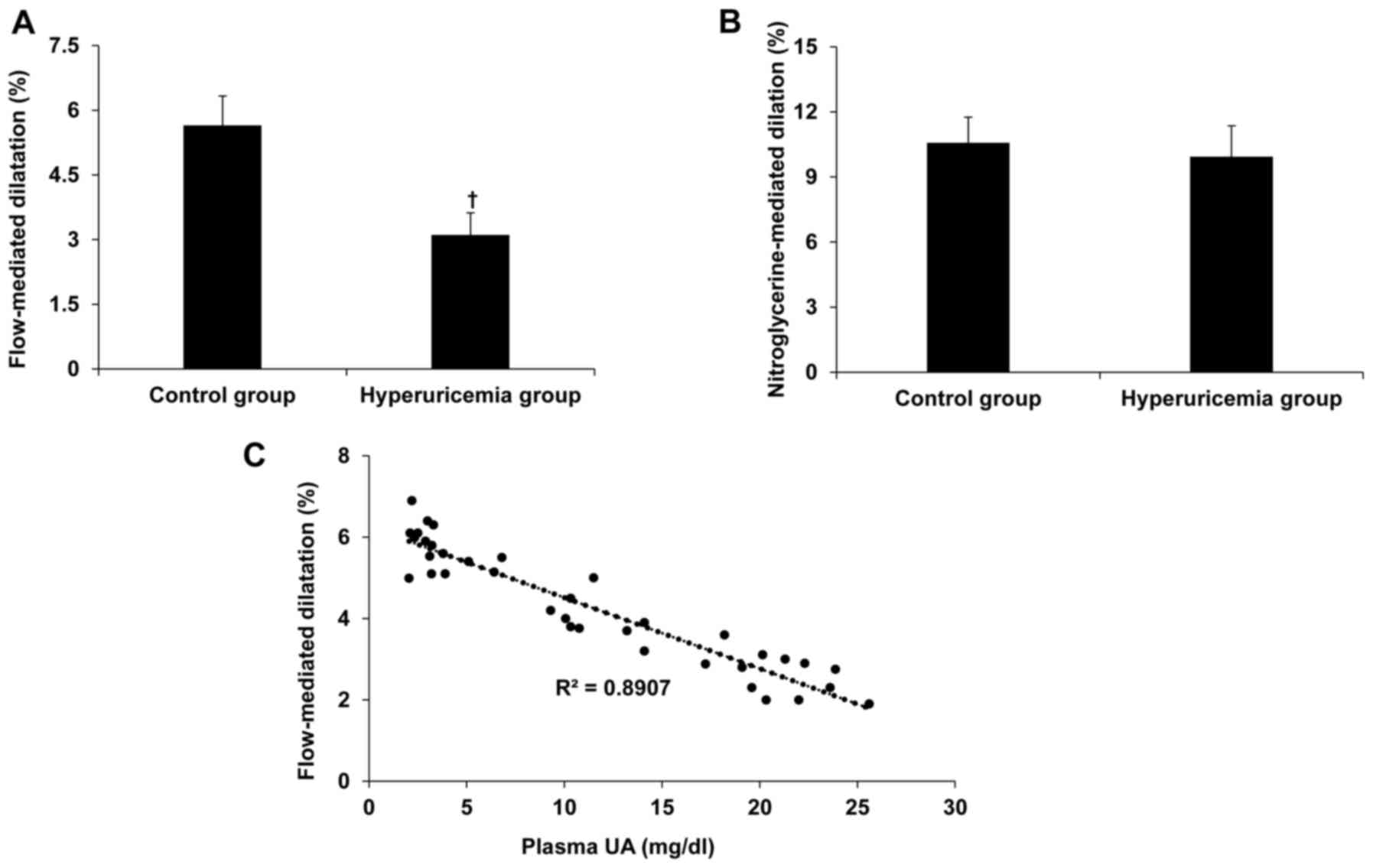
Hyperuricemia induces arterial
endothelium dysfunction
FMD and NMD are applied to access the
endothelium-dependent and -independent arterial dilation,
respectively and thus arterial endothelial function can be
evaluated using FMD and NMD (9,10). In
current study, the authors explored arterial endothelial function
using FMD in hyperuricemia patients and control subjects to
disclose whether an increase in serum UA is associated with
arterial endothelial dysfunction. Hyperuricemia inhibited brachial
FMD but not NMD (Figs. 1A and B).
Furthermore, there was a positive correlation between hyperuricemia
and impairment of FMD (R2=0.8907; Fig.
1C), suggesting that hyperuricemia impairs
endothelium-dependent arterial dilation.
UA reduced NO production in cultured
human umbilical vein ECs
The authors further investigated the effect of high
dose of UA on NO production in cultured human umbilical vein ECs
with various concentrations of UA treatments and different time
points. As presented in Fig. 2, 5
mg/dl UA (normal dose) did not alter NO production. However, 10 and
15 mg/dl UA (high dose) significantly inhibited NO expression from
12–48 h. Compared to the 5 mg/dl group, both 10 and 15 mg/dl UA
also significantly inhibited NO expression at 24 and 48 h (Fig. 2).
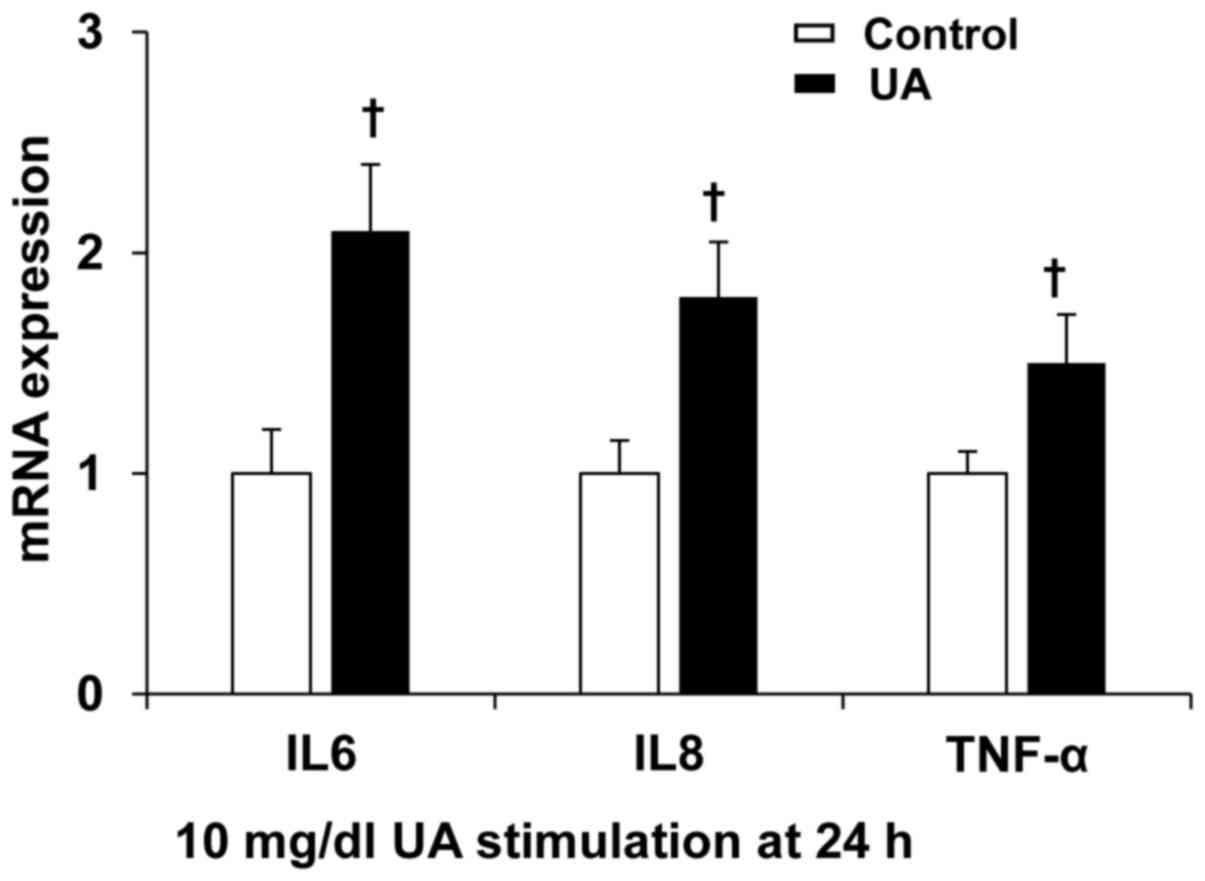
UA increased inflammation cytokine
expression in cultured human umbilical vein ECs
To further confirm the roles of UA on the expression
of inflammation cytokines, human umbilical vein ECs were incubated
with 10 mg/dl UA for 24 h. UA significantly induced IL-6, IL-8,
TNF-α expression (Fig. 3), suggesting
that UA induces endothelium inflammation.
UA increased activation of
transcription factor NF-κB in cultured human umbilical vein
ECs
In order to investigate the role of UA on the
activation of transcription factor NF-κB, human umbilical vein ECs
were co-transfected with pNF-κB-Luc and 10 mg/dl UA. UA
significantly induced activation of transcription factor NF-κB
luciferase activity (Fig. 4A).
Meanwhile, UA also induced NF-κB nuclear translocation at 5, 15 and
30 min in ECs (Fig. 4B), suggesting
that UA increased transcription factor NF-κB activity.
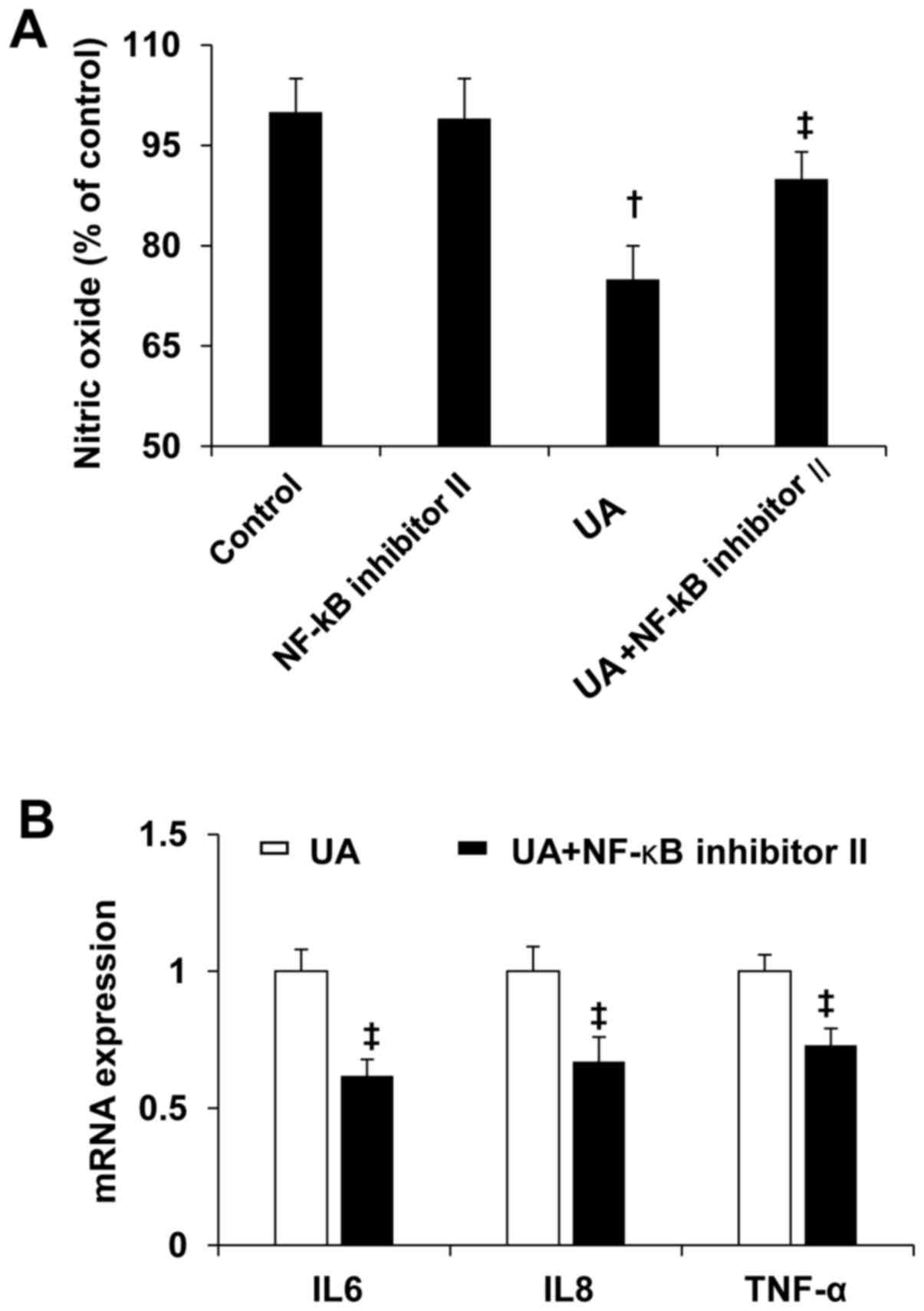
NF-κB antagonist prevented UA-induced
reduction of NO and increased inflammation cytokines
To determine the role of NF-κB in UA-induced EC
dysfunction, human umbilical vein ECs were treated by 10 mg/dl UA
with or without NF-κB inhibitor II (20 nmol/l) for 24 h. NF-κB
inhibitor II prevented UA-induced reduction of NO (Fig. 5A). In addition, NF-κB inhibitor II
prevented UA-induced increases in expression of IL6, IL8, and TNF-α
in human umbilical vein ECs.
Discussion
Hyperuricemia is an independent risk factor in the
pathogenesis of vascular disease, hypertension and metabolic
syndrome (15). The current study
further found that hyperuricemia inhibited endothelium-dependent
arterial dilation and brachial FMD. While UA significantly
inhibited NO expression with time course and dose dependent manner
in the cultured HUVECs, high dose UA also increased expression of
inflammation cytokine IL-6, IL-8 and TNF-α in vitro. These
abnormalities were associated with UA-induced activation of
transcription factor NF-κB. However, NF-κB inhibitor II, an
antagonist of NF-κB, prevented UA-induced reduction of NO and
increased inflammation cytokines. Thereby, these data implicated
that activated NF-κB plays an important role in UA-induced
endothelial inflammation and injury.
Hyperuricemia is a common clinical finding in
patients with obesity, dyslipidemia, hypertension and other CVDs
(16). Epidemiological studies have
confirmed the association of hyperuricemia and CVD. Recent data
from cross-sectional, interventional, and cohort studies have found
that hyperuricemia is an independent risk factor for hypertension
(17,18). To this point, endothelium injury and
dysfunction is regarded as the first step to develop the
hypertension, coronary heart disease and other CVDs (19). Thus, high dose UA has ability to
initial the endothelium injury to development of CVD by increases
in platelet aggregation and pro-inflammatory activity and
inhibition of EC NO production (20).
In the present study, FMD is used for assessing the vascular NO
response and endothelial function in patients with hyperuricemia
that have similar clinical characteristics with control individuals
in age, body mass index, blood pressure, plasma levels of
creatinine, glucose, triglyceride and low/high-density lipoprotein.
Interestingly, hyperuricemia inhibited brachial FMD but not NMD and
there is a positive correlation between hyperuricemia and
impairment of FMD. Therefore, hyperuricemia induces the impairment
of endothelium-dependent arterial dilation.
Inhibition of UA through targeting of xanthine
oxidase has been found to decrease vascular dysfunction and improve
cardiovascular function recent (21).
The current study further found that high dose UA directly impaired
NO generation in cultured human umbilical vein ECs, suggesting that
hyperuricemia impairs vascular function by a mechanism of
inhibition of NO production. In this regard, high dose may increase
the activity of nicotinamide adenine dinucleotide phosphate oxidase
that prompts oxidative stress and decreases the bioavailability of
NO (22). Further, hyperuricemia
impairs vascular function by increasing inflammation cytokine
expression in IL-6, IL-8 and TNF-α. Therefore, UA-induced
inflammatory immune response is as an important mechanism in UA's
vascular effects.
The nuclear factor transcription factor NF-κB
pathway is regarded as a proinflammatory signaling pathway since
activated NF-κB mediates the expression of proinflammatory
cytokines, chemokines, and adhesion molecules (23,24). In the
current study, UA increased NF-κB activity and its nuclear protein
translocation that further prompted the maladaptive immune
response. Inhibition of NF-κB reduced the UA-induced inflammation
cytokine expression of IL-6, IL-8 and TNF-α. Interestingly,
inhibition of NF-κB also inhibited UA-induced reduction of NO.
Indeed, NF-κB is a negative regulator of endothelial NO synthase
expression via upregulation of microRNA155 under inflammatory
conditions (25). Sustained activation
of NF-κB decreases NO bioavailability in response to shear and
endothelial inflammation (26).
Therefore, activated NF-κB mediates UA-induced reduction of NO and
increased inflammation response.
In conclusion, these data reveal that the NF-κB
pathway mediates hyperuricemia-induced endothelium impairment and
vascular dysfunction by a reduction of NO and expression of
inflammatory cytokines. The link between activated NF-κB, reduction
of NO, maladaptive immune responses and vascular dysfunction offers
possibilities for identification of UA-induced vascular dysfunction
and allows for development of novel therapeutic strategies for
protection of cardiovascular disease.
Acknowledgements
The present research was supported by Hubei
University of Science and Technology (grant. no. HK201608 to
H.Z.).
References
|
1
|
Mallat SG, Al Kattar S, Tanios BY and
Jurjus A: Hyperuricemia, Hypertension, and Chronic Kidney Disease:
An Emerging Association. Curr Hypertens Rep. 18:742016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Essex MN, Hopps M, Bienen EJ, Udall M,
Mardekian J and Makinson GT: Evaluation of the relationship between
serum uric acid levels and cardiovascular events in patients with
gout: A retrospective analysis using electronic medical record
data. J Clin Rheumatol. 23:160–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bos MJ, Koudstaal PJ, Hofman A, Witteman
JC and Breteler MM: Uric acid is a risk factor for myocardial
infarction and stroke: The Rotterdam study. Stroke. 37:1503–1507.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang J and Alderman MH: Serum uric acid
and cardiovascular mortality the NHANES I epidemiologic follow-up
study, 1971–1992. National Health and Nutrition Examination Survey.
JAMA. 283:2404–2410. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ford ES, Li C, Cook S and Choi HK: Serum
concentrations of uric acid and the metabolic syndrome among US
children and adolescents. Circulation. 115:2526–2532. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lehto S, Niskanen L, Rönnemaa T and Laakso
M: Serum uric acid is a strong predictor of stroke in patients with
non-insulin-dependent diabetes mellitus. Stroke. 29:635–639. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li P, Zhang L, Zhang M, Zhou C and Lin N:
Uric acid enhances PKC-dependent eNOS phosphorylation and mediates
cellular ER stress: A mechanism for uric acid-induced endothelial
dysfunction. Int J Mol Med. 37:989–997. 2016.PubMed/NCBI
|
|
8
|
Cai W, Duan XM, Liu Y, Yu J, Tang YL, Liu
ZL, Jiang S, Zhang CP, Liu JY and Xu JX: Uric Acid Induces
Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling
Pathway. BioMed Res Int. 2017:43919202017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong CK, Chen Y, Ho LM, Zhen Z, Siu CW,
Tse HF and Yiu KH: The effects of hyperuricaemia on flow-mediated
and nitroglycerin-mediated dilatation in high-risk patients. Nutr
Metab Cardiovasc Dis. 24:1012–1019. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Naka KK, Papathanassiou K, Bechlioulis A,
Kazakos N, Pappas K, Tigas S, Makriyiannis D, Tsatsoulis A and
Michalis LK: Determinants of vascular function in patients with
type 2 diabetes. Cardiovasc Diabetol. 11:1272012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Calvert JW: Cardioprotective effects of
nitrite during exercise. Cardiovasc Res. 89:499–506. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang R, Zhang Y, Xu L, Lin Y, Yang X, Bai
L, Chen Y, Zhao S, Fan J, Cheng X, et al: Protein Inhibitor of
Activated STAT3 Suppresses Oxidized LDL-induced Cell Responses
during Atherosclerosis in Apolipoprotein E-deficient Mice. Sci Rep.
6:367902016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slanina H, Schmutzler M, Christodoulides
M, Kim KS and Schubert-Unkmeir A: Effective plasmid DNA and small
interfering RNA delivery to diseased human brain microvascular
endothelial cells. J Mol Microbiol Biotechnol. 22:245–257. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang D, Atlasi SS, Patel KK, Zhuang Z and
Heaney AP: False responses of Renilla luciferase reporter control
to nuclear receptor TR4. Mol Cell Biochem. 430:139–147. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ding XH, Wang X, Cao R, Yang X, Xiao W,
Zhang Y, Bai Y, Wu H and Ye P: A higher baseline plasma uric acid
level is an independent predictor of arterial stiffness: A
community-based prospective study. Medicine (Baltimore).
96:e59572017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Volterrani M, Iellamo F, Sposato B and
Romeo F: Uric acid lowering therapy in cardiovascular diseases. Int
J Cardiol. 213:20–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang J, Sun Y, Niu K, Wan Z, Yao W, Gao
Y, Zhang W, Li Y, Zhao H and Wu X: Does elevated serum uric acid
level predict the hypertension incidence? A Chinese prospective
cohort study. Clin Exp Hypertens. 37:498–504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang TY, Fang CY, Chen JS, Po HL, Chou LP,
Chiang CY and Ueng KC: Association of Serum Uric Acid with
Cardiovascular Disease in Taiwanese Patients with Primary
Hypertension. Acta Cardiol Sin. 31:42–51. 2015.PubMed/NCBI
|
|
19
|
McCarthy CG, Goulopoulou S, Wenceslau CF,
Spitler K, Matsumoto T and Webb RC: Toll-like receptors and
damage-associated molecular patterns: Novel links between
inflammation and hypertension. Am J Physiol Heart Circ Physiol.
306:H184–H196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Verdoia M, Barbieri L, Schaffer A,
Cassetti E, Nardin M, Bellomo G, Aimaretti G, Marino P, Sinigaglia
F and De Luca G: Novara Atherosclerosis Study Group (NAS): Impact
of diabetes on uric acid and its relationship with the extent of
coronary artery disease and platelet aggregation: A single-centre
cohort study. Metabolism. 63:640–646. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Borghi C and Desideri G: Urate-lowering
drugs and prevention of cardiovascular disease: The emerging role
of xanthine oxidase inhibition. Hypertension. 67:496–498.
2016.PubMed/NCBI
|
|
22
|
Kaneko C, Ogura J, Sasaki S, Okamoto K,
Kobayashi M, Kuwayama K, Narumi K and Iseki K: Fructose suppresses
uric acid excretion to the intestinal lumen as a result of the
induction of oxidative stress by NADPH oxidase activation. Biochim
Biophys Acta. 1861:559–566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Spiga R, Marini MA, Mancuso E, Di Fatta C,
Fuoco A, Perticone F, Andreozzi F, Mannino GC and Sesti G: Uric
acid is associated with inflammatory biomarkers and induces
inflammation via activating the NF-κB Signaling Pathway in HepG2
Cells. Arterioscler Thromb Vasc Biol. 37:1241–1249. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo C, Lian X, Hong L, Zou J, Li Z, Zhu Y,
Huang T, Zhang Y, Hu Y, Yuan H, et al: High Uric Acid Activates the
ROS-AMPK Pathway, Impairs CD68 Expression and Inhibits
OxLDL-Induced Foam-Cell Formation in a Human Monocytic Cell Line,
THP-1. Cell Physiol Biochem. 40:538–548. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee KS, Kim J, Kwak SN, Lee KS, Lee DK, Ha
KS, Won MH, Jeoung D, Lee H, Kwon YG, et al: Functional role of
NF-κB in expression of human endothelial nitric oxide synthase.
Biochem Biophys Res Commun. 448:101–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grumbach IM, Chen W, Mertens SA and
Harrison DG: A negative feedback mechanism involving nitric oxide
and nuclear factor kappa-B modulates endothelial nitric oxide
synthase transcription. J Mol Cell Cardiol. 39:595–603. 2005.
View Article : Google Scholar : PubMed/NCBI
|