1. Introduction
Definition of DNase I-hypersensitive
sites (DHSs)
DNase I is an endonuclease with little DNA-sequence
specificity (1). In the early 1960s,
DNase I was used to probe how the nucleosome was organized
(2). Weintraub and Groudine (3) found that active chromatin was
prioritized for decomposition by this enzyme. These specific
regions are termed DHSs, which are distinct markers of active
chromatin that co-position with various cis-regulatory
elements (CREs), including the expression regulation sequences of
enhancers and promoters, negative regulation sequence of
insulators, silencers, and certain locus control regions. DHSs
usually disperse around transcriptionally active genes and are
accessible to regulatory proteins. Therefore, by contrast, certain
regions within DHSs are resistant to degradation as they are
protected by these gene regulatory proteins, including
transcription factors.
Generation and erasure of DHSs
The underlying molecular mechanisms of gene
regulation remain to be fully elucidated. The first step of gene
regulation is that the cells adopt an ‘open’ structure in response
to external stimulation at a group of specific sequences located in
certain chromatin regions. These exposed sequences are bound by
site-specific transcriptional regulatory elements, leading to
chromatin structure remodeling marked by significant accessibility
to the nucleases (4). DHSs in the
open chromatin serve important roles in chromatin rearrangement
(5,6),
and are stimulated by the different state of histone acetylation
(7,8)
and the binding capacity of the chromatin remodeling multiprotein
complex (9). DHSs act as binding
anchors of activator proteins and mediating cofactors, and interact
with the preinitiation complex at promoters (2,10-14),
regulating gene expression.
DHSs can be eliminated by site-specific factors. It
was reported that DHS at a CCCTC-binding factor (CTCF)-dependent
silencer can be eliminated due to the eviction of CTCF and
remodeling of a nucleosome caused by inducible non-coding RNA
transcription in the chicken lysozyme (15). DHSs are dynamic and fine-tuned by
different classes of remodeling enzymes. For example, TFE3 combines
with ACF to stimulate the occurrence of an active DHS site in the
IgH intronic enhancer, whereas PU.1 has been demonstrated to
recruit Mi2β and subsequently erase this DHS (16).
Characteristic features of DHSs
In mammalian cells, >3% of the genome is found to
be DNase I-hypersensitive (17). To
date, 290,000,000 DHSs have been recognized. Each tissue/cell type
is represented by multiple distinguished DHS profiling derived from
different individuals. DHSs are considered to be the one of the
most useful discriminative features between cell types (18) and have several distinctive
characters.
First, DHSs are typically characterized by high
sensitivity to DNase I, particularly when the related gene is
actively transcribed. Regions with high transcriptional activity
are reported to be even more sensitive than those with no
transcriptional activity. That is, there exists two states of DHSs,
open and closed, in which the accessibility features of chromatin
is increased or decreased, respectively, associated with gene
expression (19).
Second, DHSs are short sequences of ~200 base pairs
with low methylation, and the majority are no more than several
hundred base pairs long. These low-methylation regions are
co-positioned at or close to the transcription starting sites
(20,21), which affect gene transcription
according to the degree of methylation.
Third, DHSs are representative markers of regulatory
DNA and overlap with multiple CREs, including promoters, enhancers
and active transcription sites (22).
In addition, DHSs have underpinned the identification of other
CREs, including insulators, silencers and locus control regions
(10).
Fourth, although each tissue/cell exhibits
distinguished DHS signatures, there exists specific core regions in
DHSs that can be identified by sequence-specific DNA-binding
proteins. The core regions are conserved in different cell types
across species and are enriched with binding sites of HMG14 and
HMG17 proteins (23).
2. High-throughput sequencing methods for
DHS identification
The techniques used for DHS identification do not
vary substantially, all of which are novel techniques based on
high-throughput sequencing. The differences are compared in
Table I.
 | Table IComparison of different techniques to
identify DHSs. |
Table I
Comparison of different techniques to
identify DHSs.
| Technique | Cell
requirement | Data
interpretation | Novel DHS
identification | Customized
DHSs | Complex
process | DNase I
digestion | Resolution |
|---|
| DNase-seq | Large number of
cells | Difficult | Yes | No | Yes | Yes | High |
| scDNase-seq | One or ~100
cells | Difficult | No | No | Yes | Yes | Low |
| liDNase-seq | <30 cells | Difficult | Yes | No | Reduced | Yes | High |
| ImmunoSEQ | Large number of
cells | Easy | Customized | Yes | Simpler | No | |
| | | | DHS region | | | | |
DNase-sequencing (DNase-seq)
Decades ago, Southern blot hybridization was the
major method used to identify DHSs by characterizing digested DNAs
following the titration of DNase I (24). However, the low-throughput nature of
this strategy profoundly restricted its further application.
Improvements and the wide application of the massively parallel
sequencing technique has allowed high-resolution genome-scale
mapping of various DHSs, which lays a foundation for assembling
comprehensive catalogs of regulatory sequences (25,26). The
first method used for identifying thousands of DHSs simultaneously
was introduced by Crawford et al (27). Firstly, the nuclei are cleaved by
DNase I, and the two ends are then digested blunt using T4 DNA
polymerase. The genome is then cleaved by adding BamHI and
BglII. The digested blunt or sticky fragments are ligated
into the pBluescript SK(+) plasmid and sequenced. This method
enriches the sequence within the genome that relates to active
chromatin and identifies DHSs on a genome-wide scale. Two years
later, these high-throughput strategies were renewed by attaching a
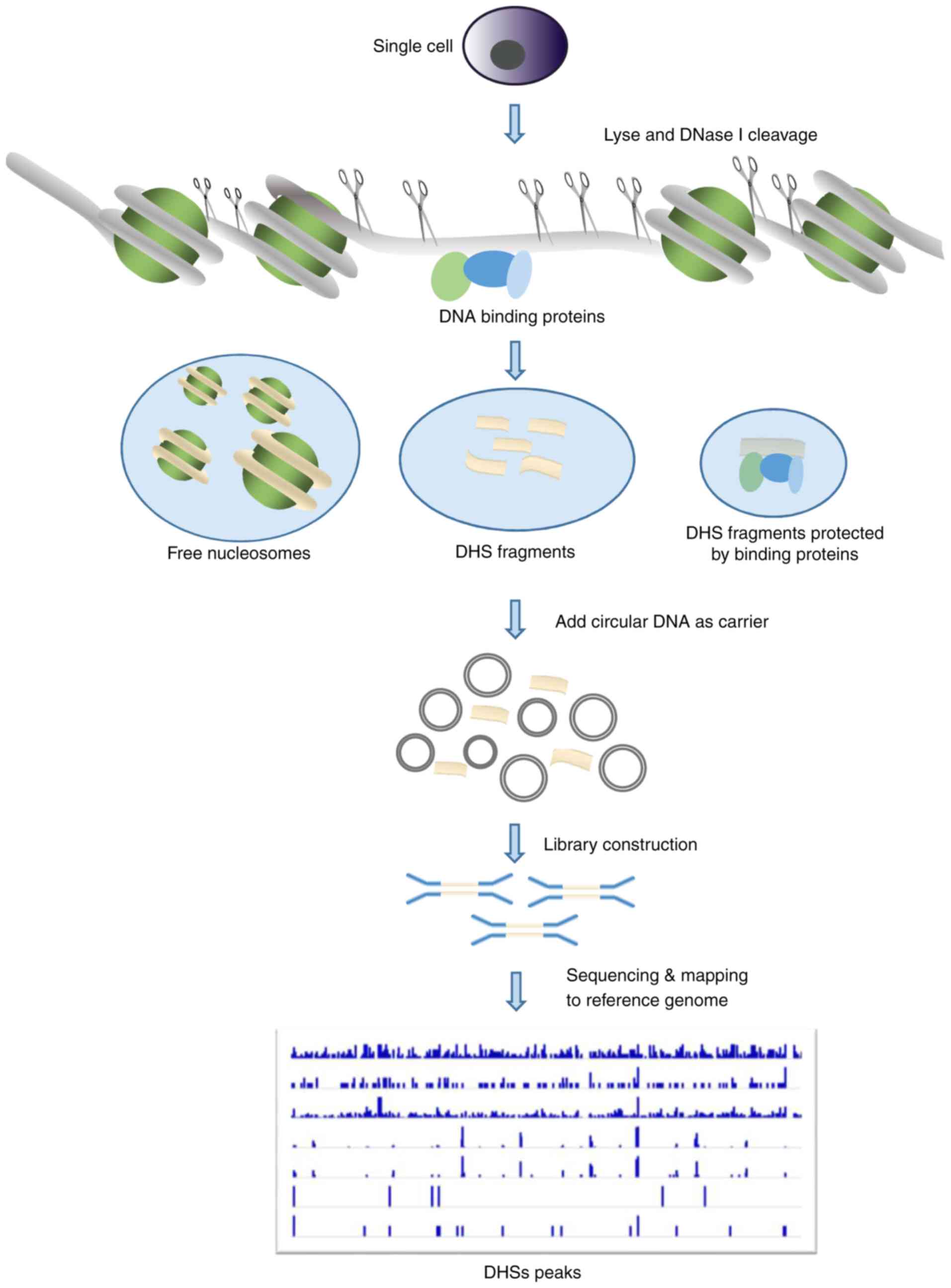
biotinylated linker to the DNase-digested ends (Fig. 1). The linker tags are used to extract
short joint DNA sequences, which can be identified by DNase-based
high-throughput next-generation sequencing (DNase-seq) (28) or DNase-based microarrays (DNase-chip)
(26). Similar strategies providing
accurate mapping of DHSs further assist in revealing a large
category of CREs in all types of mammalian tissues and cells
(29,30).
Morin et al (31) simulated the whole exon sequencing
paradigm, and developed a customized capture panel for known DHSs
(‘immune sequences’), specific for DHS detection in immune cells
and genetic variation in immune-related diseases.
Single-cell DNase-seq
(scDNase-seq)
Despite the robustness of DNase-seq technology,
millions of cells are required, which limits its application in
rare cases with limited cells, such as in certain cells from
patients. In addition, traditional DNase-seq suffers from low
sensitivity as a result of DNA loss during the multiple
purification steps. Therefore, scDNase-seq, also known as Pico-seq,
was developed to minimize DNA loss, and has been applied in the
analysis of chromatin accessibility using single cells (32,33). To
prevent loss of the small quantity of DNase I-hypersensitive DNA
released by DNase I digestion of single cells, a large amount of
circular plasmid DNA is added as carrier DNA in the subsequent
steps of library preparation (Fig.
2). The previous application of scDNase-seq to tumor cells,
NIH3T3 cells and pools of normal cells has shown that DHS patterns
at the single-cell level are highly reproducible among individual
cells (33). This method enables the
generation of a genome-wide DHS map for rare samples, which is more
valuable for clinical application.
Low-input DNase-seq (liDNase-seq)
Although scDNase-seq can identify DHSs from a small
quantity of starting material, the annotation of sequencing results
requires a pre-known DHS database, which is a challenge in the
identification of de novo DHSs sites. Lu et al
(34) introduced liDNase-seq by
modifying the scDNase-seq method to achieve de novo
genome-wide DHS identification using no more than 30 cells. The
major technical improvements, including reducing the complexity of
the purification process prior to the adaptor ligation reaction and
the first amplification reaction, and modifying the size selection
step by using SPRI affinity beads in place of gel purification. The
process for generating DHS maps is similar to that of the ENCODE
project (www.encodeproject.org and http://genome.ucsc.edu/ENCODE) and allows the
identification of de novo DHSs at much higher resolution
than in previous methods.
ImmunoSEQ technique
Morin et al (31) developed an ImmunoSEQ technique for the
detection of known DHSs. Using this technique, whole-exome
sequencing or customized DHS region sequencing can be efficiently
performed. The analysis focuses on the variation of non-coding
regions of immune-related diseases.
3. Functions of DHSs
DHSs are involved in gene expression
regulation
DHSs are essential features of all defined types of
active CREs and are often co-positioned with them (35-37).
DHSs are directly involved in chromatin modeling and structural
reestablishment, the recognition of regulatory proteins and
regulating the initiation of transcription. DHSs are associated
with nearby gene expression changes through the binding of certain
regulatory proteins to their specific sequence at promoters or
other CREs regions, and are thus involved in cell fate decisions,
individual variation and development (22). Frank et al (19) detected thousands of CREs at which the
accessibility of chromatin increased or decreased. These changes
coincided with the transcription level of adjacent genes, which is
important in the regulation of global gene expression, and most
likely infers activation or deactivation of enhancer elements.
Huang and Liew (38) identified DHSs
in the 4-kb upstream locus of the cardiac myosin heavy chain-α
(MHC-α) gene in the hamster and revealed a conserved GATA-motif
site that interacts specifically with GATA-binding factors at
different stages of cardiomyocyte development, which provided
evidence for the role of GATA factors in the gene expression of
cardiac MHC-α.
Open DHSs often mark increases in local
transcription levels, which supports the observation that open DHSs
are enhancers. Similarly, closed DHSs may represent reduced
enhancer activity (19). This
influence is more apparent when genes are associated with two or
more directional matched DHS changes. The findings identified in
genome-wide association studies (GWASs) show that genetic
variations frequently lie in non-coding regions of the genome that
contain CREs, which suggests that gene expression change underlies
the development of several complex traits.
DHSs exhibit distinguished profiles
and contribute to define CREs
Cells in a specific stage and stature possess a
fixed set of CREs that are accessible to trans-acting factors, and
thus underlie a complex controlling network of chromatin (35,39). Each
cell type has a specific set of regulatory sequences and the
cumulative span of those sequence consists of >80% of the
non-coding region of the genome. Studies on DHSs help to disclose
delicate gene regulation mechanisms and enable extensive annotation
of the genome. The genome-wide mapping of DHSs provides a novel
platform for the promising investigation of a specific molecular
biological problems affected by the regulation of a given gene or a
group of genes (17).
In addition, DHSs form a complicated, spatially- and
temporally-specific network. Certain DHSs identified in one cell
type by DNase-seq may not occur in the other cell types. Pan et
al (40) reported that 12 DHSs in
chromatin related to the Msx2 gene varied in different cell types
in the chicken, when they examined anterior and posterior limb
mesenchymal cells, calvarial osteoblasts and fibroblasts in
embryos. Most of the DHSs were not detected as active in any of the
four typed of cells, and only the DHS in the basal promoter region
was present in all four tissues. One DHS was active and unique in
the cells with Msx2 transcripts, and a secondary DHS was unique in
non-expressing cells. The anterior and posterior limb mesenchyme
cells had a distinct group of DHSs, which were more complex than
those detected in calvarial osteoblast cells, which suggested that
a complicated DHS pattern may be involved in the different
regulation models of the Msx2 gene in these two tissues, and is
involved in cell fate decision by interaction with cell-specific
transcription factors to guide the transcription program of cell
fate decision and development. DHSs of a certain gene may also
change in response to different transcription activity. Grünweller
et al (41) examined the
5'-end of the vigilin gene in chickens using the DNase-seq
technique and reporter gene analysis method. They identified two
candidate DHSs. One DHS was active and unique under high
transcriptional activity of the vigilin gene promoter in the
chicken cells, which was termed DHS1, and a secondary DHS was only
found under low transcriptional activity, which was termed DHS2.
The activity of the promoter of the vigilin gene was enhanced over
10 times by upstream sequences of the transcription start site
(TSS). Identifying DHSs and comparing their features differs among
various cell types or within a similar cell type, but culture in
different circumstances is essential for revealing gene expression
patterns under different conditions. This can effectively
complement current understanding and may have potential clinical
applications for disease treatment. The exploitation of variable
and plastic patterns of active DHSs offers potential for the
identification of certain cell or tissue states, which may have
potential to be applied to clinical diagnoses and predictions or
the evaluation of therapeutic effects.
Studies investigating DHSs facilitate the
identification of novel CREs, as DHSs are more promising indicators
for the identification of chromatin accessibility, which have been
widely used to map functional regulation elements. DHSs overlie
CREs with parallel degrees of nuclease sensitivity and cover the
main sequence of regulatory factor (42). DHSs usually contain CREs related to
transcriptional activation on the reporter locus, such as
enhancers, but can also contain transcription inhibition, such as
silencers (17). A DHS map reveals
the state and pattern of the presence of CREs, in addition to the
variable and plastic states of chromatin in various cell types
(25,29,33,43). Liu
et al (44) identified 17,472
specific DHSs and transcription factor binding sites in two cell
lines, the hESC H1 cell line and trophoblast (TB)-treated cell
line, and constructed a transcription factor network for placental
development. The specific DHSs in the TB-treated cells were found
in the ‘blood vessel’ and ‘trophectoderm’, including members of the
transcription factor motif family: Leucine zipper,
helix-loop-helix, GATA and ETS. The model of a TB system induced by
bone morphogenetic protein 4 (BMP4) was demonstrated to be
important in investigating the mechanism of trophoblast development
and revealed novel candidate genes involved in the regulation of
human placental development. These findings indicate that DHSs
enable the precise delineation of genomic CREs. Further
investigations on DHSs are expected to reveal more novel regulatory
elements.
Identifying sequence variations of the DHSs of
phylogenetic trees instead of the coding region of genes may assist
in disclosing the changes and evolution of certain phenotypes. Dong
et al (45) analyzed and
evaluated the accelerated evolution of orthologous sequences at
DHSs from the human genome and primate genomes using systematic
biology methods, and constructed a comparison map between the DHSs
and ancient repeat elements (AREs). Their analysis identified the
local AREs of all DHSs and demonstrated that they were neutrally
evolving. Therefore, it is noteworthy that ~0.44% of DHSs in the
human genome are undergoing accelerated evolution (termed
ace-DHSs). Further analysis of ace-DHSs is warranted for
investigating the evolution of human-specific phenotypes. These DHS
analyses are important in basic studies and may be of potential
value in translational medicine and personalized medicine.
4. Available data of DHSs
The ENCODE project (www.encodeproject.org and http://genome.ucsc.edu/ENCODE) aims to evolve
comprehensive schemes to list all human DHSs in order to map and
catalog genome-wide CREs. DHSs mark transcriptionally active sites
of chromatin, which may be the origin of cell selectivity.
The ENCODE research institutes have performed
genome-wide mapping of DHSs in >100 human cell and tissue types,
and almost 3,000,000 DHSs have been identified, including 71 normal
differentiated primary cells, 16 immortalized primary cells, 30
malignancy-derived cell lines and eight multipotent and pluripotent
progenitor cells. The 20-50-bp reads from the DNase-seq experiments
enabled unique mapping to 86.9% of the genomic sequence, allowing
the interrogation of a large fraction of transposon sequences. The
DHS profiles of 125 different human cell types were obtained, and
of these, 34% were specific to individual cell types and only a
minority were detected in all cell types (3,692). The open state of
DHSs varied >100 times, but the constitution pattern was
consistent in distinct cell types. It was demonstrated that ~5% of
DHSs were detected in the TSS region, while the remaining 95%
represented distal DHSs dispersed uniformly in intronic and
intergenic regions. These data provide additional information for
disclosing the mechanism of transcription.
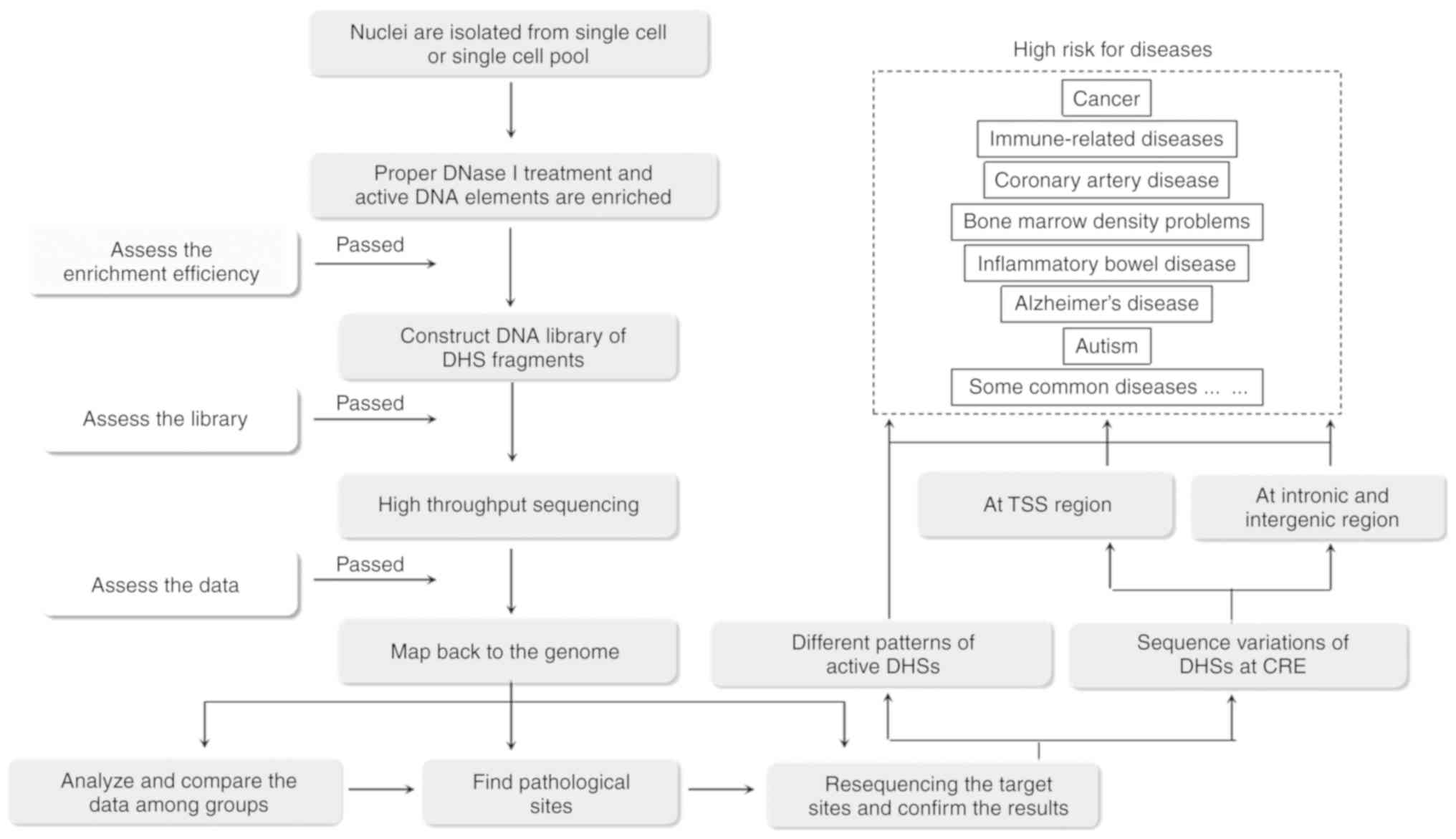
5. DHSs and diseases
DHSs are associated with multiple diseases and have
been suggested to serve distinct roles in the etiology of cancer,
immune-related diseases, inflammatory bowel disease, Alzheimer's
disease, bone marrow density problems, coronary artery disease,
autism, and certain common diseases and complex traits (Fig. 3) (46).
Recent evidence demonstrates the potential value of cell-specific
and disease-related DHSs in personalized medicine. Evidence showing
high overlap between human diseases and CREs has been
well-documented, which confirm that ‘critical’ cell types may
function as causal factors for certain diseases or help to maintain
certain phenotypic traits (46).
The accessibility or inaccessibility of the state of
DHSs is reported to be associated with diseases. An increasing
number of novel DHSs have been found to be associated with
diseases. Specific cell and tissue types have been identified as
being associated with different diseases. For example, specific
immune cell types are involved in immune-related diseases
(inflammatory bowel disease), and specific tissue types are
involved in diseases affecting specific organs (coronary artery
disease), with other associations including adrenal glands in
coronary artery disease, immune systems in Alzheimer's disease and
kidneys with bone marrow density (46).
Thousands of tumor-specific DHSs located at promoter
and enhancer regions have been detected, which have been shown to
be involved in the occurrence and development of cancer.
Function-related mutations of the DHS region are
closely correlated with transcription initiation activity and thus
result in the occurrence of certain diseases (Fig. 4). There are >100 studies on DHSs
that assessed various cell or tissue types by ENCODE Alliance (124
different cell types) and NIH roadmap epigenomics group (342
different adult fetal tissue samples), which demonstrate that an
overlap exists between mutations at non-coding DNA regulatory
sequences and diseases and traits.
GWASs have identified numerous single nucleotide
polymorphisms (SNPs) at DHSs, which are associated with various
types of quantitative traits and complex disorders. Local mutation
density is variable throughout the genome (47). A study on 1,161 human cancer genomes
revealed that the density of point mutations at the center of the
DHS in the gene promoter region of somatic cells was increased
(48). Numerous tissue types,
including brain, pancreatic and liver tissue, have also
demonstrated the enrichment of SNPs associated with DHSs in major
depressive disorder (49). A 14-kb
Down syndrome cell adhesion molecule deletion sequence, containing
12 CNS DHSs, was found in an autistic family, with regulatory
potential affecting the biology of the central nervous system
(50). De novo mutations, rich
in DHSs and proximal genes, have been significantly predicted to
result in the loss of transcription binding factors. For example,
deletion of lysine-specific demethylase 5B binding was found at the
promoter of the candidate autism risk gene, EFR3A (51).
A mutation at the SNP (chr18:52417839 G>C) site
was reported to be correlated with follicular thyroid cancer, which
had an influence on the binding of tumor suppressor protein p53 and
subsequently resulted in the decreased gene expression of
thioredoxin-like 1 (TXNL1) (33). The highest prevalence of mutations was
found in the hypothetical driving factor DHS chr5:1325957-1328153,
located in an intron of the Cleft lip and palate transmembrane
1-like (CLPTM1L) gene and 30 kb upstream of telomerase reverse
transcriptase (TERT), results in the overexpression of six adjacent
genes and four of these genes [TERT, CLPTM1L, thyroid hormone
receptor interactor 13 (TRIP13), lysophosphatidylcholine
acyltransferase 1 (LPCAT1)] are known to be associated with cancer
(52-54).
Recently, a statistical method has been developed to
identify distal regulatory elements with hypothetical driving
mutations in breast cancer, to identify DHSs in non-coding genomic
sequences associated with significant mutations in breast cancer
and abnormal expression of adjacent genes, which may be important
in the development of cancer (55).
The mutation of chr5:1325957-1328153 at the DHS region in breast
cancer was reported to be associated with the overexpression of
oncogene tripartite motif containing 27 (TRIM27). In addition,
abnormal activity was found with the mutation of
chr6:28948439-28951450 at the DHS region. Using data from The
Cancer Genome Atlas (TCGA) and breast cancer International Alliance
(metabonomics) molecular taxonomy, Guo et al (56) found two hypothetical functional
variants, rs62331150 and rs73838678, located at the DHS site and
transcription factor binding region. Among them, rs62331150 was
associated with the expression of tet methylcytosine dioxygenase 2
(TET2) in normal breast tissue and tumor tissue. Two new SNP
(rs12309362 and rs9970827) were found to be significantly
associated with reducing the risk of hepatocellular carcinoma (HCC)
by measuring the mutations at the peak of DHSs in 1,538 patients
with HCC and 1,465 normal controls (57).
A study on endometriosis, by re-sequencing 1.29 mb
of the 9p21 region, revealed that the mutation of rs17761446 at the
DHS was associated with endometriosis, the protective G allele at
this site had a strong interaction with the ANRIL promoter. Further
chromatin immunoprecipitation analysis confirmed that the
protective G allele also had a preferential binding capacity with
transcription factor 7-like 2, EP300 and may be involved in the
development of endometriosis (58).
Studies on prostate cancer and breast cancer cells
have revealed that different DHS patterns of androgen receptor (AR)
and estrogen receptor 1 (ESR1) were of high predictive value for
hormone receptor binding and may be involved in the development of
these types of cancer. The quantitative measurement of DHS changes
can predict the binding sites of perturbation-inducible
transcription factors (59).
Following activation of the transcription of AR in
LNCaP cells, 244 upregulated and 486 downregulated accessible DHS
regions were detected to be the candidate sites for further
investigation, which may be associated with prostate cancer. CTCF
and the ELK1-ETS transcription factor are potential upstream
regulator elements, which are rich in open promoter regions of
downregulated genes. The inhibitor of DNA-binding 1 HLH protein
(ID1) is the only transcription factor that is significantly
upregulated, exhibiting basal sequence enrichment in the promoter
region of the upregulated gene. Therefore, CTCF, ELK1 and ID1 may
be potential targets for the treatment of prostate cancer (60). Increasing evidence shows that changes
in the expression of BMP4 are involved in the pathogenesis of
cancer, which is associated with cancer metastasis and progression,
including rectal, hepatocellular and ovarian cancer (61). In order to determine the
characteristics of BMP4 transcription mediators in breast cancer,
RNA-Seq and DNase-seq were analyzed in T-47D and MDA-MB-231 breast
cancer cells treated with BMP4. It was confirmed that MBD2,
core-binding factor-β and hypoxia-inducible factor 1α were
downstream regulators of the BMP4 signal, which enhanced cell
migration and decreased cell growth (62).
In tumor therapy, particularly in acute myeloid
leukemia (AML), intratumoral heterogeneity caused by clonal
evolution has been found, which may have an influence on the effect
of treatment. In order to solve this problem, the chromatin
accessibility of subclones of AML was compared directly using
unsupervised clustering analysis. Marked differences in the
chromatin landscape and transcriptional regulation among the
subclones were detected and confirmed. The data indicated that the
common DHSs of individual AML subclones dominated in the clustering
analysis over the subclone-specific DHSs, most likely due to the
impact of shared founder mutations at the DHSs in each AML
subclone. Clone-specific DHSs, runt-related transcription factor
and ETS motifs are expressed in abundance in the two clones of
DHSs, although GATA motifs are particularly abundant in FLT3-WT
clones (63). It may be a potential
strategy to use DHSs analysis to improve the treatment effect,
particularly for those types of cancer with features of
intraturmoral heterogeneity.
Genetic variation at DHSs has been reported to be
correlated with carcinogenesis (64).
By analyzing 1,161 human cancer samples from 14 types of cancer,
DHS profiles and SNP distributions were mapped to link to promoter
activity, some of which were involved in differential nucleotide
excision repair (NER) and resulted in carcinogenesis (48). Consistent with this finding,
genome-wide maps of NER regions show that the repair ability of
nucleotide excision was decreased with mutation at the DHS of gene
promoter regions.
Jin et al (33)
reported that thousands of tumor-specific DHSs were identified on
cells dissected from follicular thyroid carcinoma samples fixed on
formalin-fixed paraffin-embedded slides. Numerous DHSs have been
reported to be correlated with the development of thyroid cancer
(33). A de novo mutation
(chr18:52417839 G>C) at a DHS located downstream of the TXNL1
gene is associated with the formation of thyroid carcinoma. It was
reported that rs62331150 located at promoter region and rs73838678
located at the enhancer region of the gene, increased the risk of
breast cancer. These two SNP sites were found to be in linkage
disequilibrium with rs9790517 of the adjacent TET2 gene (55). It was also found that, in samples with
mutation at the rs12309362 and rs9970827 sites, the risk of being
affected with HCC decreased significantly (57).
Ten DHSs were identified as being mutated with
abnormal expression of target genes in breast cancer (55). Mutation at the DHS
chr5:1325957-1328153, was present at a high frequency in the cancer
cells, resulting in the high level of transcription of certain
genes close to it, including TERT, CLPTM1L, TRIP13 and LPCAT1,
which has been confirmed to be associated with cancer (52,53,65).
Mutations at DHSs chr5:1325957-1328153 were found to result in the
high expression of TRIM27, and certain mutations in this region
caused abnormal accessibility of DHS chr6:28948439-28951450, which
are associated with cancer.
The pattern of DHSs can be stimulated by hormones
through regulation of the binding capacity of AR and ESR1 in
prostate cancer cells and breast cancer cells. Following binding
with AR or ESR1, the DNase I-hypersensitivity of certain sequences
was found to be altered, and the regional nucleosome occupancy
changed for AR binding but not for ESR1b binding, which indicated
different interaction modes in AR and ESR1 regulation (59).
In Gene Ontology analysis, genes associated with
tumor-specific DHSs are abundant in biological processes, including
the regulation of GTPase activity and response to hypoxia, and
cancer-related pathways. Understanding the accessibility dynamics
of chromatin in the process of disease occurrence and development
can provide insights into how cell fate is regulated, and how
transcriptional systems are organized and regulated in different
tissues and how they are destroyed in disease states. In addition,
the application of DHSs in biomedical research can expand the field
of cell-selective gene regulation analysis, enabling the
identification of long-range regulatory patterns of the system and
previously undescribed phenomena, such as DHS activation patterns
and mutation rates in abnormal and immortal cells.
6. Discussion
Although DHSs occupy a small portion of human
genome, ~2% of the genome, a relatively large proportion of CREs
may be involved in the establishment of well-organized expression
networks in each cell type and thus contribute to the etiology of a
certain disease. The comprehensive delineation of distribution,
constituents and biological activities of DHSs help to map and
classify functional CREs. The identification of CREs is critical
for elucidating the mechanism of gene expression regulation
underlying biological events, and the development and progression
of certain diseases.
To date, the DNase-seq technique remains one of the
most efficient techniques for disclosing the known and unknown
regulatory elements of diverse target cells. Cooper et al
(32) released a detailed protocol in
Nature that may facilitate the spread of DNase-seq analysis.
However, the number of researchers able to utilize the
high-throughput DHS capture technique well is limited. In addition,
careful manipulation is required due to high background noise.
Difficulties in performing experiments are not the greatest
challenge for its wider application; bioinformatics analysis is
difficult for the majority of laboratories. An insufficient number
of bioinformatics technicians, particularly those with the required
programming skills and familiarity in this field, is the main
concern. The exploitation of software or online tools is required
to simplify bioinformatics analysis and enable easier understanding
by researchers.
Although evidence has identified an association
between DHSs and diseases, using DHSs as a marker for prediction,
prevention and pre-clinical diagnosis remains a challenge due to
the complexity of regulation of the DHS profile. The uncertainty of
genome-wide prediction of CREs and specific DHS related to gene
function may increase the challenge of its application in clinical
practice. Future work should focus on the investigation of more
delicate methods to locate DHSs precisely, help disclose the
mechanisms underlying gene expression differences, determine how to
modify chromatin accessibility, reveal how changes in transcription
factor binding are driven by genetic variations, and guide how to
integrate DHSs in clinical practice.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81671473), the Key Talents
of Jiangsu Province (grant nos. WSW-108 and FRC201754) and the
Innovation Team Project of Wuxi (grant no. CXTDJS003).
Availability of data and materials
Not applicable.
Authors' contributions
YC conceived and wrote the manuscript with some
assistance from AC.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Elgin SC: DNAase I-hypersensitive sites of
chromatin. Cell. 27:413–415. 1981.
|
|
2
|
Meisterernst M, Roy AL, Lieu HM and Roeder
RG: Activation of class II gene transcription by regulatory factors
is potentiated by a novel activity. Cell. 66:981–993.
1991.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Weintraub H and Groudine M: Chromosomal
subunits in active genes have an altered conformation. Science.
193:848–856. 1976.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Felsenfeld G, Boyes J, Chung J, Clark D
and Studitsky V: Chromatin structure and gene expression. Proc Natl
Acad Sci USA. 93:9384–9388. 1996.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wu C: The 5' ends of Drosophila heat shock
genes in chromatin are hypersensitive to DNase I. Nature.
286:854–860. 1980.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Martinez-Balbas MA, Dey A, Rabindran SK,
Ozato K and Wu C: Displacement of sequence-specific transcription
factors from mitotic chromatin. Cell. 83:29–38. 1995.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hebbes TR, Clayton AL, Thorne AW and
Crane-Robinson C: Core histone hyperacetylation co-maps with
generalized DNase I sensitivity in the chicken beta-globin
chromosomal domain. EMBO J. 13:1823–1830. 1994.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kleff S, Andrulis ED, Anderson CW and
Sternglanz R: Identification of a gene encoding a yeast histone H4
acetyltransferase. J Biol Chem. 270:24674–24677. 1995.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Peterson CL and Tamkun JW: The SWI-SNF
complex: A chromatin remodeling machine? Trends Biochem Sci.
20:143–146. 1995.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Roeder RG: The role of general initiation
factors in transcription by RNA polymerase II. Trends Biochem Sci.
21:327–335. 1996.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim YJ, Bjorklund S, Li Y, Sayre MH and
Kornberg RD: A multiprotein mediator of transcriptional activation
and its interaction with the C-terminal repeat domain of RNA
polymerase II. Cell. 77:599–608. 1994.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Koleske AJ and Young RA: An RNA polymerase
II holoenzyme responsive to activators. Nature. 368:466–469.
1994.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Dynlacht BD, Hoey T and Tjian R: Isolation
of coactivators associated with the TATA-binding protein that
mediate transcriptional activation. Cell. 66:563–576.
1991.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Verrijzer CP and Tjian R: TAFs mediate
transcriptional activation and promoter selectivity. Trends Biochem
Sci. 21:338–342. 1996.PubMed/NCBI
|
|
15
|
Lefevre P, Witham J, Lacroix CE, Cockerill
PN and Bonifer C: The LPS-induced transcriptional upregulation of
the chicken lysozyme locus involves CTCF eviction and noncoding RNA
transcription. Mol Cell. 32:129–139. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ishii H, Du H, Zhang Z, Henderson A, Sen R
and Pazin MJ: Mi2beta shows chromatin enzyme specificity by erasing
a DNase I-hypersensitive site established by ACF. J Biol Chem.
284:7533–7541. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zeng WP and McFarland MM: Rapid and
unambiguous detection of DNase I hypersensitive site in rare
population of cells. PLoS One. 9(e85740)2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Trynka G, Sandor C, Han B, Xu H, Stranger
BE, Liu XS and Raychaudhuri S: Chromatin marks identify critical
cell types for fine mapping complex trait variants. Nat Genet.
45:124–130. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Frank CL, Manandhar D, Gordan R and
Crawford GE: HDAC inhibitors cause site-specific chromatin
remodeling at PU.1-bound enhancers in K562 cells. Epigenetics
Chromatin. 9(15)2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Choi YC and Chae CB: DNA hypomethylation
and germ cell-specific expression of testis-specific H2B histone
gene. J Biol Chem. 266:20504–20511. 1991.PubMed/NCBI
|
|
21
|
Ngo V, Gourdji D and Laverriere JN:
Site-specific methylation of the rat prolactin and growth hormone
promoters correlates with gene expression. Mol Cell Biol.
16:3245–3254. 1996.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang T, Marand AP and Jiang J: PlantDHS:
A database for DNase I hypersensitive sites in plants. Nucleic
Acids Res. 44:D1148–D1153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Deng T, Zhu ZI, Zhang S, Postnikov Y,
Huang D, Horsch M, Furusawa T, Beckers J, Rozman J, Klingenspor M,
et al: Functional compensation among HMGN variants modulates the
DNase I hypersensitive sites at enhancers. Genome Res.
25:1295–1308. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kodama Y, Nagaya S, Shinmyo A and Kato K:
Mapping and characterization of DNase I hypersensitive sites in
Arabidopsis chromatin. Plant Cell Physiol. 48:459–470.
2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Boyle AP, Davis S, Shulha HP, Meltzer P,
Margulies EH, Weng Z, Furey TS and Crawford GE: High-resolution
mapping and characterization of open chromatin across the genome.
Cell. 132:311–322. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Crawford GE, Davis S, Scacheri PC, Renaud
G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG and
Collins FS: DNase-chip: A high-resolution method to identify DNase
I hypersensitive sites using tiled microarrays. Nat Methods.
3:503–509. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Crawford GE, Holt IE, Mullikin JC, Tai D,
Blakesley R, Bouffard G, Young A, Masiello C, Green ED, Wolfsberg
TG, et al: Identifying gene regulatory elements by genome-wide
recovery of DNase hypersensitive sites. Proc Natl Acad Sci USA.
101:992–997. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Crawford GE, Holt IE, Whittle J, Webb BD,
Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, et al:
Genome-wide mapping of DNase hypersensitive sites using massively
parallel signature sequencing (MPSS). Genome Res. 16:123–131.
2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Thurman RE, Rynes E, Humbert R, Vierstra
J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H,
Vernot B, et al: The accessible chromatin landscape of the human
genome. Nature. 489:75–82. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Vierstra J, Rynes E, Sandstrom R, Zhang M,
Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R,
et al: Mouse regulatory DNA landscapes reveal global principles of
cis-regulatory evolution. Science. 346:1007–1012. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Morin A, Kwan T, Ge B, Letourneau L, Ban
M, Tandre K, Caron M, Sandling JK, Carlsson J, Bourque G, et al:
Immunoseq: The identification of functionally relevant variants
through targeted capture and sequencing of active regulatory
regions in human immune cells. BMC Med Genomics.
9(59)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cooper J, Ding Y, Song J and Zhao K:
Genome-wide mapping of DNase I hypersensitive sites in rare cell
populations using single-cell DNase sequencing. Nat Protoc.
12:2342–2354. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren
G, Ni B, Sklar J, Przytycka TM, Childs R, et al: Genome-wide
detection of DNase I hypersensitive sites in single cells and FFPE
tissue samples. Nature. 528:142–146. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lu F, Liu Y, Inoue A, Suzuki T, Zhao K and
Zhang Y: Establishing chromatin regulatory landscape during mouse
preimplantation development. Cell. 165:1375–1388. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gross DS and Garrard WT: Nuclease
hypersensitive sites in chromatin. Annu Rev Biochem. 57:159–197.
1988. View Article : Google Scholar
|
|
36
|
Gaszner M and Felsenfeld G: Insulators:
Exploiting transcriptional and epigenetic mechanisms. Nat Rev
Genet. 7:703–713. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li Q, Harju S and Peterson KR: Locus
control regions: Coming of age at a decade plus. Trends Genet.
15:403–408. 1999.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Huang WY and Liew CC: A conserved GATA
motif in a tissue-specific DNase I hypersensitive site of the
cardiac alpha-myosin heavy chain gene. Biochem J. 325:47–51.
1997.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bell O, Tiwari VK, Thoma NH and Schubeler
D: Determinants and dynamics of genome accessibility. Nat Rev
Genet. 12:554–564. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Pan Z, Lichtler AC and Upholt WB: DNase I
hypersensitive sites in the chromatin of the chicken Msx2 gene
differ in anterior and posterior limb mesenchyme, calvarial
osteoblasts and embryonic fibroblasts. Biochem Mol Biol Int.
46:549–557. 1998.PubMed/NCBI
|
|
41
|
Grünweller A, Purschke WG, Kügler S, Kruse
C and Müller PK: Chicken vigilin gene: A distinctive pattern of
hypersensitive sites is characteristic for its transcriptional
activity. Biochem J. 326:601–607. 1997.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Heintzman ND, Stuart RK, Hon G, Fu Y,
Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et
al: Distinct and predictive chromatin signatures of transcriptional
promoters and enhancers in the human genome. Nat Genet. 39:311–318.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
43
|
Sheffield NC, Thurman RE, Song L, Safi A,
Stamatoyannopoulos JA, Lenhard B, Crawford GE and Furey TS:
Patterns of regulatory activity across diverse human cell types
predict tissue identity, transcription factor binding, and
long-range interactions. Genome Res. 23:777–788. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu Y, Ding D, Liu H and Sun X: The
accessible chromatin landscape during conversion of human embryonic
stem cells to trophoblast by bone morphogenetic protein 4. Biol
Reprod. 96:1267–1278. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dong X, Wang X, Zhang F and Tian W:
Genome-wide identification of regulatory sequences undergoing
accelerated evolution in the human genome. Mol Biol Evol.
33:2565–2575. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mokry M, Harakalova M, Asselbergs FW, de
Bakker PI and Nieuwenhuis EE: Extensive association of common
disease variants with regulatory sequence. PLoS One.
11(e0165893)2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
D'Antonio M and Ciccarelli FD: Integrated
analysis of recurrent properties of cancer genes to identify novel
drivers. Genome Biol. 14(R52)2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Perera D, Poulos RC, Shah A, Beck D,
Pimanda JE and Wong JW: Differential DNA repair underlies mutation
hotspots at active promoters in cancer genomes. Nature.
532:259–263. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Peterson RE, Cai N, Bigdeli TB, Li Y,
Reimers M, Nikulova A, Webb BT, Bacanu SA, Riley BP, Flint J and
Kendler KS: The genetic architecture of major depressive disorder
in han chinese women. JAMA Psychiatry. 74:162–168. 2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Turner TN, Hormozdiari F, Duyzend MH,
McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K,
Stessman HA, et al: Genome sequencing of autism-affected families
reveals disruption of putative noncoding regulatory DNA. Am J Hum
Genet. 98:58–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yuen RK, Merico D, Cao H, Pellecchia G,
Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T,
et al: Genome-wide characteristics of de novo mutations in autism.
NPJ Genom Med. 1:160271–1602710. 2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wang K, Sturt-Gillespie B, Hittle JC,
Macdonald D, Chan GK, Yen TJ and Liu ST: Thyroid hormone receptor
interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic
checkpoint-silencing protein. J Biol Chem. 289:23928–23937.
2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Rafnar T, Sulem P, Stacey SN, Geller F,
Gudmundsson J, Sigurdsson A, Jakobsdottir M, Helgadottir H,
Thorlacius S, Aben KK, et al: Sequence variants at the TERT-CLPTM1L
locus associate with many cancer types. Nat Genet. 41:221–227.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
54
|
Abdelzaher E and MostafaM F:
Lysophosphatidylcholine acyltransferase 1 (LPCAT1) upregulation in
breast carcinoma contributes to tumor progression and predicts
early tumor recurrence. Tumour Biol. 36:5473–5483. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
D Antonio M, Weghorn D, D
Antonio-Chronowska A, Coulet F, Olson KM, DeBoever C, Drees F,
Arias A, Alakus H, Richardson AL, et al: Identifying DNase I
hypersensitive sites as driver distal regulatory elements in breast
cancer. Nat Commun. 8(436)2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Guo X, Long J, Zeng C, Michailidou K,
Ghoussaini M, Bolla MK, Wang Q, Milne RL, Shu XO, Cai Q, et al:
Fine-scale mapping of the 4q24 locus identifies two independent
loci associated with breast cancer risk. Cancer Epidemiol
Biomarkers Prev. 24:1680–1691. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Jiang T, Du F, Qin N, Lu Q, Dai J, Shen H
and Hu Z: Systematical analyses of variants in DNase I
hypersensitive sites to identify hepatocellular carcinoma
susceptibility loci in a Chinese population. J Gastroenterol
Hepatol. 32:1887–1894. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Nakaoka H, Gurumurthy A, Hayano T,
Ahmadloo S, Omer WH, Yoshihara K, Yamamoto A, Kurose K, Enomoto T,
Akira S, et al: Allelic imbalance in regulation of ANRIL through
chromatin interaction at 9p21 endometriosis risk locus. PLoS Genet.
12(e1005893)2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
He HH, Meyer CA, Chen MW, Jordan VC, Brown
M and Liu XS: Differential DNase I hypersensitivity reveals
factor-dependent chromatin dynamics. Genome Res. 22:1015–1025.
2012.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wei X, Yu L, Jin X, Song L, Lv Y and Han
Y: Identification of open chromosomal regions and key genes in
prostate cancer via integrated analysis of DNase-seq and RNA-seq
data. Mol Med Rep. 18:2245–2252. 2018.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kallioniemi A: Bone morphogenetic protein
4-a fascinating regulator of cancer cell behavior. Cancer Genet.
205:267–277. 2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Ampuja M, Rantapero T, Rodriguez-Martinez
A, Palmroth M, Alarmo EL, Nykter M and Kallioniemi A: Integrated
RNA-seq and DNase-seq analyses identify phenotype-specific BMP4
signaling in breast cancer. BMC Genomics. 18(68)2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
de Boer B, Prick J, Pruis MG, Keane P,
Imperato MR, Jaques J, Brouwers-Vos AZ, Hogeling SM, Woolthuis CM,
Nijk MT, et al: Prospective isolation and characterization of
genetically and functionally distinct AML subclones. Cancer Cell.
34:674–689. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Stergachis AB, Neph S, Reynolds A, Humbert
R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R,
et al: Developmental fate and cellular maturity encoded in human
regulatory DNA landscapes. Cell. 154:888–903. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Wei C and Dong X, Lu H, Tong F, Chen L,
Zhang R, Dong J, Hu Y, Wu G and Dong X: LPCAT1 promotes brain
metastasis of lung adenocarcinoma by up-regulating PI3K/AKT/MYC
pathway. J Exp Clin Cancer Res. 38(95)2019.PubMed/NCBI View Article : Google Scholar
|