Introduction
Perry syndrome (PS; Mendelian inheritance in man ID,
168605) is a rare hereditary progressive neurodegenerative disease
first described in 1975 by Perry et al (1) in Canada. PS is characterized by early
onset parkinsonism, central hypoventilation, severe weight loss and
neuropsychiatric features (mainly apathy and depression), which
begin at ~48 years of age. Psychiatric alterations and parkinsonism
are typically the first symptoms to appear, followed by weight loss
(2). Post-mortem brain examination
of PS cases revealed a severe degeneration in the substantia nigra
with the presence of TAR DNA binding protein (TDP)-43 inclusions in
the basal ganglia and brain stem (3). However, the observed inclusion pattern
was different from those reported in other neurological diseases
involving TDP-43 proteinopathy (4).
PS is caused by pathogenic variants in the dynactin
subunit 1 (DCTN1) gene, located at locus 2p13.1 (5,6). The
DCTN1 gene was characterized in 1994(7) and encodes the p150glued
protein, the largest subunit of the macromolecular complex,
dynactin. Dynactin has 10 subunits and is involved in a number of
diverse cellular functions, including endoplasmic
reticulum-to-Golgi transport, centripetal movement of lysosomes and
endosomes, spindle formation, chromosome movement, nuclear
positioning and axonogenesis (7,8). PS is
inherited as an autosomal dominant trait (5,9), with
an established age-dependent penetrance and a certain variability
in clinical features (10). Most
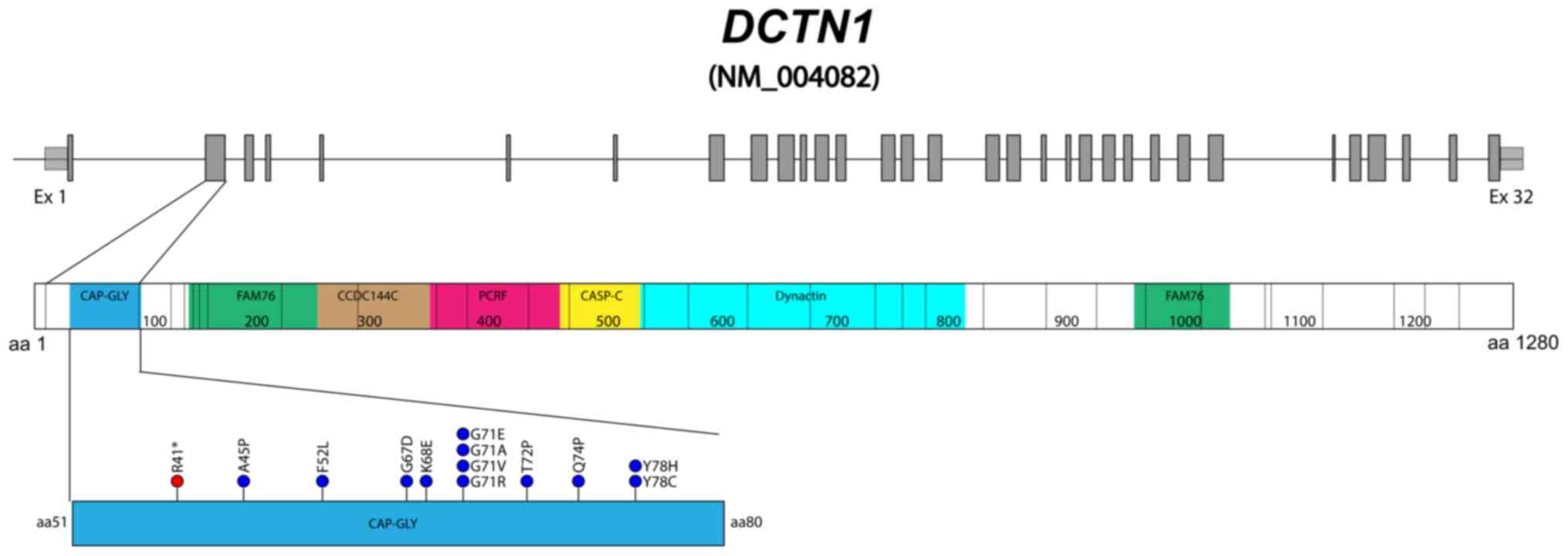
pathogenic variants associated with PS are located on exon 2 of the
gene encoding the cytoskeleton-associated protein Gly rich
(CAP-Gly) domain of p150glued. Pathogenic variants of
DCTN1 also cause distal hereditary motor neuronopathy type
7B (also known as distal spinal and bulbar muscular atrophy)
(11), progressive supranuclear
palsy (PSP)-like phenotype frontotemporal dementia (12) and motor neuron disease/amyotrophic
lateral sclerosis. Certain individuals also present with
overlapping phenotypes (10).
PS has no cure and requires multidisciplinary
clinical management, depending on the clinical features of each
case. For instance, parkinsonism is treated with dopaminergic
therapy, psychiatric manifestations are treated with
antidepressants and weight loss is treated with hypercaloric and
hyperproteic diets. In addition, ventilatory support (invasive or
non-invasive) improves the survival rate of patients with central
hypoventilation (9,10). However, affected individuals
typically die from respiratory complications and the average
survival rate is 5 years following diagnosis (10).
The present study described the first (to the best
of our knowledge) Mexican family diagnosed with PS. Molecular
confirmation of this diagnosis was obtained using a mini-exome
sequencing panel. The potential impact of the identified
DCTN1 mutation on the structure and function of the
p150glued protein was assessed using in silico
analysis.
Patient and methods
Case report
Clinical evaluation was performed in August 2016 at
the Department of Medical Genetics of the National Medical Center,
‘La Raza’ (Mexico City, Mexico), which is part of the Mexican
Social Security Institute. The proband was a Mexican mestizo male
aged 55 years at the time of consultation. The proband's family was
originally from the state of Hidalgo, Mexico. The proband's main
symptoms were chronic fatigue for 6 years and weight loss in the
previous year at a rate of 1 kg per month, the body mass index at
presentation was 14.66 kg/m2 (normal range, 18.5-24.9
kg/m2), which is considered severely underweight. The
proband also reported difficulty performing daily activities due to
bradykinesia, gait disturbances, dyskinesias and tremors. Physical
examination revealed that the proband looked older than their
chronological age with several facial wrinkles and gray hair. The
proband also had a steppage gait, marked reduction in subcutaneous
fat, expressionless facies and scanning speech. Facial
hypertrichosis, horizontal saccadic eye movements, slight blinking
and stiffness in the upper and lower extremities were also
observed. The proband showed no clinical signs of depression and
did not report any suicide attempts.
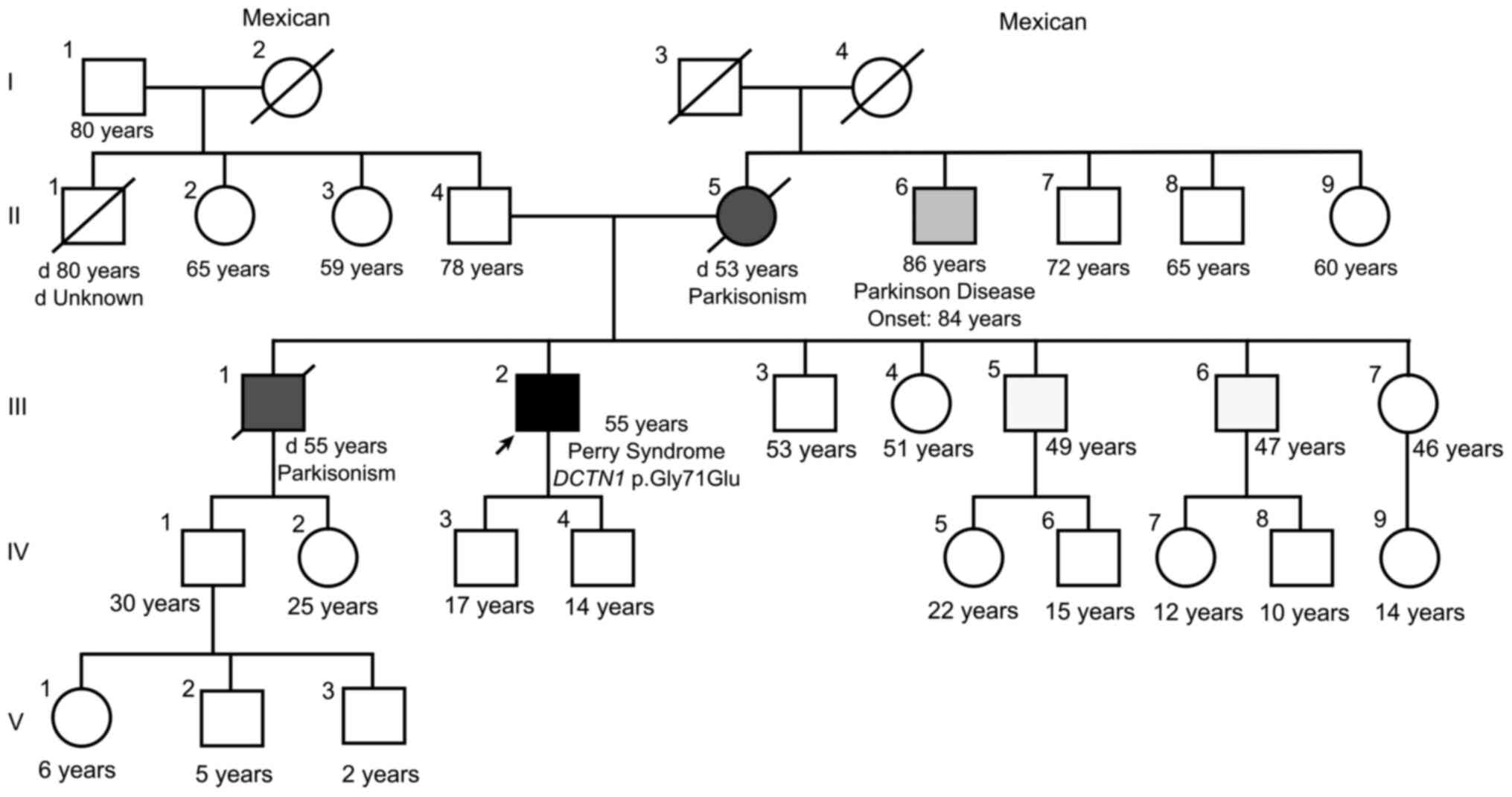
The family history (Fig.
1) indicated that the proband's mother (II.5) and one of the
proband's brothers (III.1) had died from complications of
Parkinson's disease (PD) at the ages of 53 and 55 years,
respectively. A maternal uncle had also developed late-onset PD
(II.6). The proband had two sons (IV.3 and IV.4), aged 17 and 14
years at the time of the present study. The clinical data and
family history of the proband led to the suspicion of PS. In
addition, a polysomnography study corroborated the presence of
central alveolar hypoventilation and alterations in sleep
patterns.
Mini-exome sequencing
Following genetic counseling, the proband signed an
informed consent form for molecular analysis. The analysis was
conducted at the Laboratory of Genomic Diagnosis at the National
Institute of Genomic Medicine (Mexico City, Mexico). For this,
genomic DNA was obtained from EDTA-peripheral blood. DNA was
extracted using an AS1010 cartridge and the Maxwell 16 System
(Promega Corp.) following the manufacturer's instructions. The
quality and integrity of the DNA were evaluated using a NanoDrop
1000 spectrophotometer (Thermo Fisher Scientific, Inc.) and Qubit
fluorometer (Thermo Fisher Scientific, Inc.). Exome sequencing was
performed using the TruSight One Sequencing Panel kit (Illumina,
Inc.) following the manufacturer's instructions. This kit allowed
for simultaneous sequencing of exons and splicing regions of 4,813
genes, including the DCTN1 gene and other genes associated
with autosomal dominant PD. Sequencing was performed using the
MiSeq Instrument and v3 reagents (Illumina, Inc.). A paired-end
read of 150 bp and a library pool concentration of 12.5 pM
quantified by a Qubit fluorometer (Thermo Fisher Scientific, Inc.)
were used. A 100x mean coverage of the targeted regions was
obtained. Data analyses were performed using the Trimmomatic tool
(13), Burrows-Wheeler
Aligner-Maximal Exact Matches (14)
and the Genome Analysis Toolkit algorithm (15).
Prediction of the functional impact of
the DCTN1 variant
The Sorting Intolerant From Tolerant (SIFT)
(16), Polymorphism Phenotyping v2
(PolyPhen-2) (17) and
MutationTaster (18) algorithms
were used to predict the functional effects of the identified
DCTN1 variant. Population databases such as the Genome
Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/), 1000 Genomes
Project (https://www.internationalgenome.org/) and ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) and the
literature in PubMed (https://pubmed.ncbi.nlm.nih.gov/) were also reviewed
(the search terms used in the databases are provided in the
supplementary material).
Computational modeling of the
variant
In total, four different models of
p150glued bearing the mutation were generated using
SWISS-MODEL (19). Models 1, 2, 3
and 4 were generated using the Protein Data Bank (PDB) structures:
Solution structure of the CAP-Gly domain in human dynactin 1
(2COY), crystal structure of the C-terminal domain of human EB1 in
complex with the CAP-Gly domain of human dynactin-1 (p150-Glued)
(2HKQ), three-dimensional structure of cap-gly domain assembled on
microtubules determined by magic angle spinning (MAS) NMR
spectroscopy (2MPX) and crystal structure of the zink-knuckle 2
domain of human CLIP-170 in complex with p150-Glued (3E2U) as
templates. The target sequence bearing the (p.Gly71Glu) variant was
defined in each case by aligning the reference sequence of the
human p150glued protein (accession no. NP_004073.2) with
that of the template molecule. The models were visualized and
analyzed using PyMOL software version 1.8 (Schrödinger, LLC)
(20). The Voronoia 1.0 program
suite was used to detect the internal cavities of the models
(21).
Protein electrostatic potential
Using the default settings, the electrostatic
potential maps of the mutant model structures and the structures
used as templates were constructed using Swiss-PdbViewer
4.1.0(22).
Results
Mini-exome sequencing analysis
Following exome sequencing, the
NM_004082.4:c.212G>A variant was identified in exon 2 of
DCTN1, causing a Gly substitution for Glu at position 71 of
the protein, NP_004073.2:(p.Gly71Glu). Gly71 belongs to the
Gly-Lys-Asn-Asp-Gly (GKNDG) motif, which is highly conserved in
numerous CAP-Gly domains (23). The
Gly71Glu substitution was predicted to be deleterious by the SIFT
algorithm (score 0), probably damaging by the PolyPhen-2 algorithm
(HumVar score, 0.9) and a cause of disease by the MutationTaster
(score, 0.999999997834239) algorithm. This variant has been
submitted to ClinVar three times (ClinVar ID: 34242) but was absent
from the exome and genome sequencing data submitted to gnomAD
(24) and from the Mexican database
available through the Franklin by Genoox platform (https://franklin.genoox.com/clinical-db/home). No
pathogenic or probable pathogenic variants were identified in genes
associated with autosomal dominant PD or other neurological
diseases with similar clinical manifestations.
None of the other family members of the proband
accepted medical examination or molecular analysis. Since the
proband's sons were minors at the time of the study, molecular
testing was not offered as per the recommendations of the National
Society of Genetic Counselors, the American Academy of Pediatrics,
the American College of Medical Genetics and Genomics (ACMG) and
the American Society of Human Genetics, which indicate that a
pre-symptomatic diagnosis should not be performed on minors
(25). At present, both sons remain
asymptomatic (last contact March, 2024).
Modeling and in silico evaluation of
the Gly71Glu variant
Based on the different reported structures of
p150glued, four models of p150glued Gly71Glu
were obtained (Figs. 2A and
S1). Fig. 2A shows the structural alignment of
the wild-type CAP-Gly domain (PDB: 2COY) and the Gly71Glu CAP-Gly
domain in Model 1. The spatial orientation and atomic contacts of
Glu71 in Model 1 point toward the solvent and occupy a cavity
delimited by the side chains of Asp70, Thr72, Lys77, Tyr78, Phe79
and Thr80, as well as the peptide backbone atoms of Tyr78 (Fig. 2A-C). This cavity was empty in the
wild-type protein (Fig. 2B). In the
structural context of Model 1, Glu71 establishes a pair of H-bonds
between its γ carboxylate group, the hydroxyl group of Thr80 and
the peptide backbone amide of residue Phe79. A comparison of Models
2 and 4 is provided in Fig. S1.
Computational analysis showed that Model 1 had five internal
cavities, which was similar to the template (Fig. 2D). A comparison of the cavities in
Models 2 and 4 is provided in Fig.
S2.
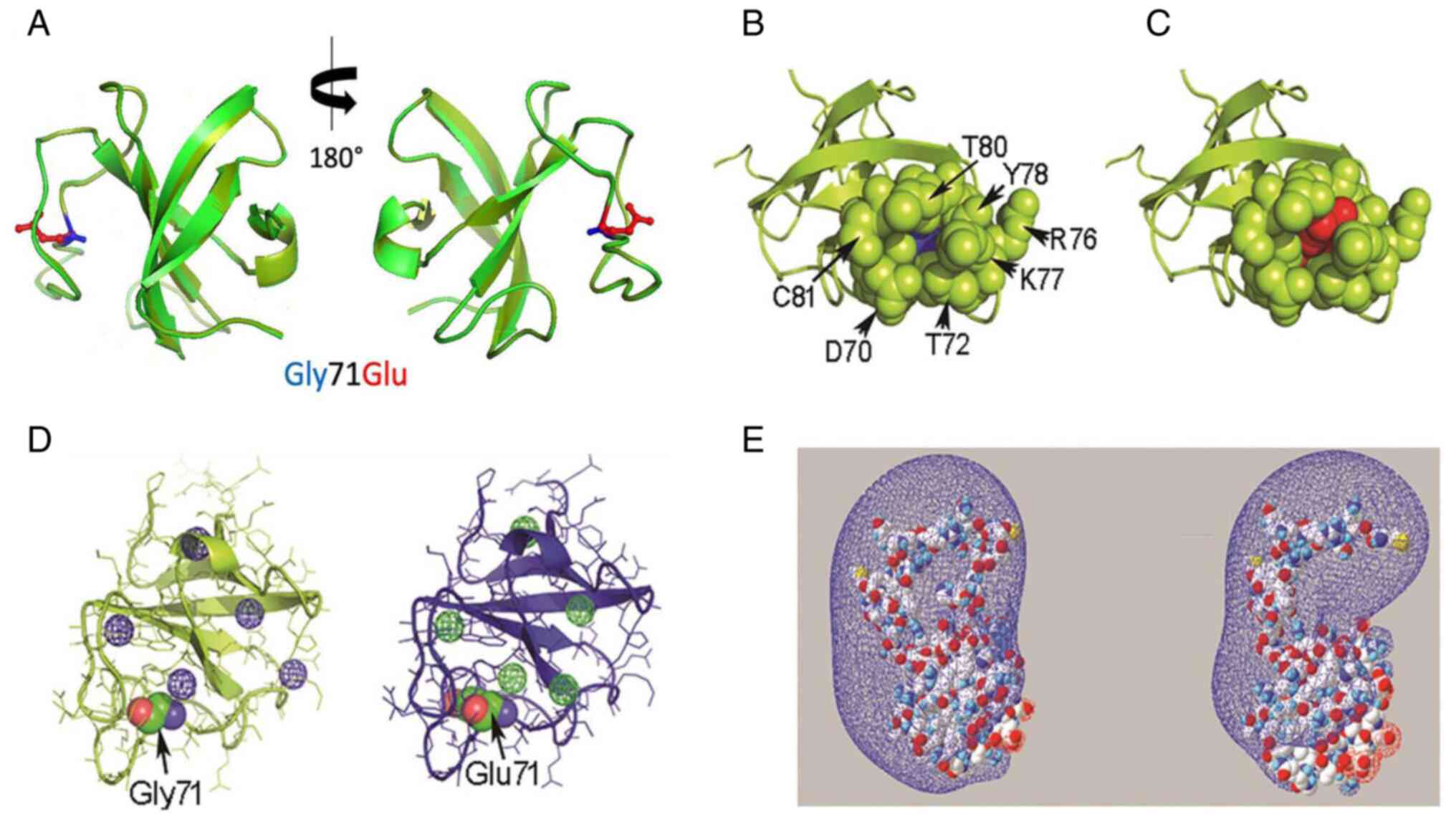 | Figure 2In silico evaluation of the
Gly71Glu variant-spatial relationship of the residue at position 71
of the CAP-Gly domain. (A) Structural alignment of the PDB 2COY
template (light green) and Model 1 (dark green). The side chains of
Gly71 and Glu71 are displayed in blue and red, respectively. (B)
Representative image of residues located spatially close (4 Å) to
Gly71 in the wild-type CAP-Gly domain in the free state (PDB 2COY).
(C) Equivalent representative image of Glu71 in Model 1. Gly71 and
Glu71 are represented as ‘spheres’ in blue and red, respectively.
The residues in the proximity of position 71 are shown as green
spheres. (D) Different views of the cavities found in PDB 2COY and
Model 1 using the Voronoia program suite. The cavities are
represented as spheres in a mesh format, in blue or green. (E)
Different perspectives of the electrostatic potential map of the
PDB 2COY and Model 1 protein structures constructed using
Swiss-PdbViewer 4-1-0. The conventional code represents the charge
distribution, where blue and red represent the electron-poor and
electron-rich regions, respectively. The molecules are represented
in a space-filled format, using the CPK color code: Carbon is gray,
oxygen is red, nitrogen is blue, sulfur is yellow and hydrogen is
white. PDB, protein data bank; CAP-Gly, cytoskeleton-associated
protein glycine-rich domain; CPK, Corey-Pauling-Koltun. |
Protein electrostatic potential
The electrostatic potential map of the CAP-Gly
domain structure in Model 1 is shown in Fig. 2E. The Glu71 variant expanded the
electron-rich region in the modeled structure compared with the
template. The electrostatic potential maps for Models 2 and 4 are
shown in Fig. S3.
Discussion
PS is a rare disease, to the best of our knowledge,
the proband reported in this study was the first to be identified
in Mexico and the third in Latin America. A summary of the reported
PS cases and the identified mutation is shown in Table SI. The first identified case in
Latin America harbored the (p.Gly71Arg) pathogenic variant and was
reported in Colombia in 2014 by Pretelt et al (26). In 2022, the (p.Gly67Asp) variant was
identified in an Argentine family by Silva et al (27). The other confirmed PS cases were
mainly of European or Asian origin (28).
The molecular testing using exome sequencing
identified the NM_004082:c.212G>A (p.Gly71Glu) pathogenic
variant in the DCTN1 gene, confirming the diagnosis of PS in
the proband. The (p.Gly71Glu) variant was first identified by
Farrer et al (5) in 2009 in
a French family with PS. There were no data suggesting French
ancestry in the present case; therefore, the variant likely
resulted from a de novo mutation in this family.
The proband reported in the present study had three
of the four cardinal criteria proposed by Wider and Wszolek
(9): Autosomal dominant
parkinsonism, weight loss and central hypoventilation. No
depression, apathy, social withdrawal or suicidal attempts were
reported at the first medical appointment or during the following 2
years. However, a formal psychiatric evaluation of the proband was
never performed; therefore, the absence of psychiatric symptoms
must be considered with caution. Table
SII lists the clinical features of other PS patients with the
Gly71Glu variant, as documented in the literature. The clinical
features are similar among patients with the same variant.
Considering the proband's chronic fatigue in the 6 years prior to
diagnosis, the onset of symptoms was likely in the fifth decade of
life (~49 years of age), similar to other reported patients
harboring the same mutation.
Before the identification of the DCTN1 gene
and the possibility of molecular testing, diagnostic confirmation
of PS was impossible. The syndrome was classified as
Parkinsonism-plus due to alterations in extraocular movements and
the presence of pyramidal dysfunction and respiratory disorders
(29). The availability of
molecular testing using multigene next-generation sequencing-based
panels contributes to an earlier diagnosis, confirming the clinical
suspicion, identifying the precise molecular defect and ruling out
other autosomal dominant parkinsonism diseases (such as variants in
MAPT). The identification of the molecular defect allows for
more accurate genetic counseling and identification of
pre-symptomatic carriers among at-risk asymptomatic relatives
(12). Early disease diagnosis may
also improve the quality of life of the patient by reducing
complications and allowing for timely interventions, such as using
non-invasive mechanical ventilation or inserting a diaphragmatic
pacemaker, avoiding unnecessary medical and diagnosis procedures
for other affected family members.
However, the early detection of PS is impaired by
several factors: PS is a rare disease and certain clinical and
pathological features overlap with other more common and well-known
sporadic neurodegenerative disorders, limiting the rise of clinical
suspicion. In these instances, patients would be diagnosed with
autosomal dominant parkinsonism or PSP until a marked weight loss
is observed, central hypoventilation develops or respiratory
failure and death occur (2). For
example, two relatives of the proband presented with early
parkinsonism (individual II.5, deceased at 53 years of age; and
individual III.1, deceased at 55 years of age) and both had been
diagnosed with PD. No additional studies were performed to
establish the cause of the familial PD and the possibility of PS
was not suggested to the family by the attending physician at the
time. Additionally, PS may be accompanied by phenotypic
heterogeneity among the affected individuals in the same family
(30). Caroppo et al
(12) reported a family with the
(p.Gly71Glu) mutation, in which the affected individuals presented
with PS or PSP-like or frontotemporal dementia.
Pathogenic variants of DCTN1 are also
associated with different clinical phenotypes. Most variants
associated with PS affect the GKNDG motif of the CAP-Gly domain
(31), particularly residues 67,
71, 72, 74 and 78. Recently, two new reports of genetic variants
outside the CAP-Gly domain in exon 2 were reported. The first case
was identified in a female patient without a family history of PS.
The variant was demonstrated to be pathogenic based on the clinical
features and autopsy findings (32). The second was a 65-year-old female
patient who presented with difficulties in falling asleep and
personality changes that progressed over four years. Exome
sequencing revealed a novel splicing site mutation, c.279+1G>T,
in the DCTN1 gene. The variant was classified as pathogenic
according to the ACMG Standards and Guidelines (33).
The missense variant (p.Phe52Leu) is associated with
late-onset PD, hypoventilation and frontotemporal atrophy (34). By contrast, a variant affecting
Gly59 has been associated with distal hereditary motor neuronopathy
type VII (11), whereas variants
Ile935Met, Ile196Val and Ser111Cys have been associated with
susceptibility to amyotrophic lateral sclerosis (35). Certain variants associated with
PSP-like syndrome are the same as those identified in PS (5); however, other alleles have also been
associated with this phenotype (36).
Gly71 appears to be a recurrent site of mutation in
DCTN1, since four out of the 14 PS causal variants
identified thus far affect residue 71 (p.Gly71Arg/Glu/Val/Ala)
(Fig. 3), according to the Human
Gene Mutation Database (as of March 2024). The (p.Gly71Glu) variant
has been identified in at least three additional patients with PS
from different ethnic backgrounds, confirming its pathogenicity
(37). In the present study, to
clarify the impact of (p.Gly71Glu) on the structure and function of
the CAP-Gly domain, the Glu71 mutated CAP-Gly domain was modeled
and compared with the wild-type. The model indicated that the
presence of Glu induced local changes in the conformation and
environment of the CAP-Gly domain. The larger and more charged side
chain of Glu resulted in positive volume variation and a negative
charge gain at physiological pH at the mutated site. This change
imposed a conformational restriction on the peptide backbone,
affecting its ability to rearrange appropriately when the CAP-Gly
domain interacts with its functional partners (38). It has been shown that the
conformational flexibility of the CAP-Gly domain is crucial for the
efficient function of p150glued (39). Several regions of the CAP-Gly domain
participate in the binding of the p150glued protein
partners (26). Among these
regions, the conserved GKNDG motif interacts with the
acidic-aromatic sequence motifs located at the C-terminus of the
protein partners (5).
In the present study, in Models 2 and 4, Gly71 was
oriented towards the core of the domain, contacting Phe79, which
connected Gly71 with the conserved cluster of aromatic residues
(Tyr46, Phe52, Trp57, Tyr78, Phe79 and Phe88), stabilizing the
native folding of the CAP-Gly domain. These contacts with Glu71
resulted in the rearrangement of Phe79 in Model 2, which would
destabilize the domain by perturbing the interaction network of the
conserved aromatic residues present in the folding core. In Model
3, substitution with Glu71 led to less compact folding,
characterized by several empty cavities. It has been shown that
cavities resulting from packing defects typically destabilize
proteins (40) and promote their
aggregation (41). As a protein
becomes thermodynamically less stable, the probability of adopting
non-native conformations prone to aggregation increases. TDP-43
proteinopathy and dynactin aggregates have been detected in all of
the examined PS post-mortem brains reported (3-5).
In addition, in cells coexpressing the (p.Gly71Ala) mutant and
TDP-43, cytoplasmic mislocalization of TDP-43 and aggregates of
variable sizes were detected in the cytoplasm and neurites,
indicating that mutated DCTN1 induced mislocalization and
aggregation of wild-type TDP-43 protein in human neurons (42). In the present study, the constructed
electrostatic potential map indicated that Glu71 modified the
electrostatic potential of the CAP-Gly domain. However, the effect
on charge distribution differed from one model to another. Since
the electrostatic potential is a factor that regulates
protein-protein interactions (43),
the overall change in charge distribution induced by Glu71 would
also contribute to disrupting the binding of the CAP-Gly domain to
the acidic-aromatic sequence motifs of binding partners (23). In summary, the contribution of the
(p.Gly71Glu) variant to the pathogenesis of PS is most likely
related to the processes that support the function of proteins,
such as folding efficiency, folding stability, protein dynamics,
conformational fitting ability and the affinity of interactions
with the binding partners (44).
The present study reported the first case of PS
identified in Mexico; however, certain limitations hindered a more
comprehensive disease description in the proband and their family:
Brain images were not available for any of the affected family
members and post-mortem brain evaluation was unfeasible. Variant
segregation in healthy and symptomatic family members was not
possible at the time of the study; however, individual III.5
(living outside of Mexico) was also recently diagnosed and
molecularly confirmed with PS (personal communication with a family
member).
In conclusion, the present study revealed for the
first time that PS is present in the Mexican population, expanding
the geographical and ethnic prevalence of this disease and
enriching the global dataset of Perry syndrome cases. The clinical
features of the proband and the family history of early
parkinsonism suggested a diagnosis of PS, which was confirmed by
the identification of the pathogenic DCTN1 variant. Unlike
the PS cases previously reported, the proband of the present study
lacks psychiatric manifestations such as depression and apathy at
diagnosis and during the clinical follow-up period. In addition,
the in silico modeling of the (p.Gly71Glu) variant allowed
us to provide new insight into how this change influences the
structural and functional integrity of the cytoskeleton-associated
protein complex. We hope this work also raises awareness of rare
neurological disorders that are often overlooked in low- and
medium-income populations due to the limited availability of
genetic testing.
Supplementary Material
Terms used for searching in databases
and literature
Spatial relationship of the residue at
position 71 of the CAP-Gly domain. Images show the residues located
spatially close (4 Å) to Gly71 in the wild-type CAP-Gly domain
(left) and the equivalent representation of residues close to Glu71
in the mutant models (right). Gly71 and Glu71 are represented as
blue and red spheres (2MPX/Model 3), respectively, or by element
(2HKQ/Model 2 and 3E2U/Model 4). In 2MPX/Model 3, the residues in
the proximity of position 71 are shown as green spheres. The
remaining images are shown in the stick format. The remaining
regions of the molecules are represented in cartoon format. The
residues of interest are identified using the one letter code. In
Model 2, Glu71 is oriented in the opposite direction compared with
Model 1 shown in Fig. 1C. Glu71
points toward the β3 strand, which is part of the structural core
of the domain. Consequently, both Phe79 and Cys81 rearrange to
create room for Glu71. In Model 3, Glu71 is oriented towards the
domain surface, as in Model 1, but it is more solvent-exposed in
Model 3. Such variation in the environment of Glu71 reflects
differences in the loop conformation connecting the β3 and β4
strands between these models, resulting in a less compact
environment of Glu71 in Model 3. In Model 4, Glu71 adopts a spatial
positioning similar to that adopted in Model 2, but in Model 4, the
Glu71 side chain is rotated nearly 90˚, respective to Model 2. Å,
Angstrom; CAP-Gly, cytoskeleton-associated protein glycine-rich
domain; 2MPX, three-dimensional structure of cap-gly domain
assembled on microtubules determined by Magic Angle Spinning NMR
spectroscopy; 2HKQ, crystal structure of the C-terminal domain of
human EB1 in complex with the CAP-Gly domain of human dynactin-1
(p150-Glued); 3E2U, crystal structure of the zink-knuckle 2 domain
of human CLIP-170 in complex with p150-Glued.
Cavities in the structures of the
2HKQ/Model 2, 2MPX/Model 3 and 3E2U/Model 4. The analysis was
performed using the Voronoia program suite (21). The cavities are represented as
spheres with mesh format, in blue or green. In the protein
structures, the peptide backbone is shown in the cartoon format and
the lines indicate residues, except position 71 (Gly or Glu), which
is shown as a sphere. The image was generated by PyMOL (20) using data in the cav PDB file
generated by Voronoia and the PDB file of respective structures.
PDB, protein data bank; 2HKQ, crystal structure of the C-terminal
domain of human EB1 in complex with the CAP-Gly domain of human
dynactin-1 (p150-Glued); 2MPX, three-dimensional structure of
cap-gly domain assembled on microtubules determined by Magic Angle
Spinning NMR spectroscopy; 3E2U, crystal structure of the
zink-knuckle 2 domain of human CLIP-170 in complex with p150-Glued
PDB entries.
Electrostatic potential maps of the
2HKQ/Model 2, 2MPX/Model 3 and 3E2U/Model 4 protein structures,
constructed using Swiss-PdbViewer 4-1-0 software (22). The conventional code represents the
charge distribution, where blue and red represent electron-poor and
electron-rich regions, respectively. The molecules are represented
in a space-filled format, using the CPK color code: Carbon is gray,
oxygen is red, nitrogen is blue, sulfur is yellow and hydrogen is
white. 2HKQ, crystal structure of the C-terminal domain of human
EB1 in complex with the CAP-Gly domain of human dynactin-1
(p150-Glued); 2MPX, three-dimensional structure of cap-gly domain
assembled on microtubules determined by Magic Angle Spinning NMR
spectroscopy; 3E2U, crystal structure of the zink-knuckle 2 domain
of human CLIP-170 in complex with p150-Glued; CPK,
Corey-Pauling-Koltun.
(A) Prediction of
aggregation/amyloidogenic-prone sequences in the CAP-Gly domain of
p150glued protein. The analysis was performed using the web-based
tool AmylPred2, a consensus method for predicting amyloid
propensity (21). Consensus-5
indicates that the sequence was predicted to be prone to aggregate
as amyloid by at least 5 of the 10 algorithms applied by AmylPred2.
The three segments identified as consensus-5 are highlighted in
dark blue, red and light gray in the CAP-Gly domain sequence. The
pattern of secondary structure elements in the protein is shown at
the top. The regions folded as β strands are represented as arrows
and the short helix near the C-terminus is represented as a short
cylinder. The downward red and blue arrows indicate the Gly59 and
Gly71 residues, respectively. The conserved GKNDG motif is
highlighted with a black bar. The result of each algorithm is shown
below consensus-5. (B) Location of the consensus-5 sequences
identified by AmylPred2 analysis in the CAP-Gly domain of p150glued
protein. The regions are shown in the same color pattern as
aforementioned in A. The β-strands 1-4 and Gly59 and Gly71 residues
are indicated by arrows. The image on the right side is rotated
180˚, respective to the left image. Graphical images were generated
by PyMOL (20) using the
coordinates contained in protein data bank no. 3E2U. CAP-Gly,
cytoskeleton-associated protein glycine-rich domain; GKNDG motif,
Glycine-Lysine-Asparagine-Aspartic acid-Glycine motif; Amyl Patt,
amyloidogenic pattern; Av P Densit, average packing density; β-str
Cont, β-strand contiguity; Hexa C Ener, hexapeptide configuration
energy; N-term, N-terminus; C-term, C-terminus.
Genetic variants affecting the
‘dynactin subunit 1’ gene reported in Perry syndrome.
Clinical characteristics of patients
with Perry syndrome with Gly71Glu (G71E) reported in varous
countries.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are not
publicly available due to restrictions in the informed consent
form, which does not allow for the unrestricted disclosure of
sequencing data, but may be requested from the corresponding
author. The information related to the identified DCTN1
variant has been submitted to the ClinVar database (variation ID,
21390; submission ID, 14421829).
Authors' contributions
LFL and ERM performed the clinical evaluations. KCS,
CMG and MJO performed the sequencing. CAV, LFL and JGS interpreted
the sequencing data and wrote the manuscript. LDPY, URC and GAH
analyzed the protein structure and wrote the manuscript. All
authors read and approved the final version of the manuscript. CAV,
LFL and JGS confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was reviewed and approved by the
Ethical Committee of the National Institute of Genomic Medicine in
Mexico City, Mexico (approval no. CEI 2018/37) and conducted in
accordance with the Code of Ethics of the World Medical Association
(Declaration of Helsinki). The proband provided informed consent
for molecular testing.
Patient consent for publication
The proband signed an informed consent form for
publication of this case.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Perry TL, Bratty PJ, Hansen S, Kennedy J,
Urquhart N and Dolman CL: Hereditary mental depression and
Parkinsonism with taurine deficiency. Arch Neurol. 32:108–113.
1975.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tsuboi Y, Mishima T and Fujioka S: Perry
disease: Concept of a new disease and clinical diagnostic criteria.
J Mov Disord. 14:1–9. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wider C, Dickson DW, Stoessl AJ, Tsuboi Y,
Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan
M, et al: Pallidonigral TDP-43 pathology in Perry syndrome.
Parkinsonism Relat Disord. 15:281–286. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mishima T, Koga S, Lin WL, Kasanuki K,
Castanedes-Casey M, Wszolek ZK, Oh SJ, Tsuboi Y and Dickson D:
Perry Syndrome: A Distinctive Type of TDP-43 Proteinopathy. J
Neuropathol Exp Neurol. 76:676–682. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Farrer MJ, Hulihan MM, Kachergus JM,
Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier
B, Chapon F, et al: DCTN1 mutations in Perry syndrome. Nat Genet.
41:163–165. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Holzbaur EL and Tokito MK: Localization of
the DCTN1 gene encoding p150Glued to human chromosome 2p13 by
fluorescence in situ hybridization. Genomics. 31:398–399.
1996.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Holzbaur EL and Vallee RB: DYNEINS:
Molecular structure and cellular function. Annu Rev Cell Biol.
10:339–372. 1994.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gauthier LR, Charrin BC, Borrell-Pagès M,
Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME,
Lessmann V, Humbert S and Saudou F: Huntingtin controls
neurotrophic support and survival of neurons by enhancing BDNF
vesicular transport along microtubules. Cell. 118:127–138.
2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wider C and Wszolek ZK: Rapidly
progressive familial parkinsonism with central hypoventilation,
depression and weight loss (Perry syndrome)-a literature review.
Parkinsonism Relat Disord. 14:1–7. 2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Konno T, Ross OA, Teive HAG, Sławek J,
Dickson DW and Wszolek ZK: DCTN1-related neurodegeneration: Perry
syndrome and beyond. Parkinsonism Relat Disord. 41:14–24.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Puls I, Oh SJ, Sumner CJ, Wallace KE,
Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer
A, Powers R, et al: Distal spinal and bulbar muscular atrophy
caused by dynactin mutation. Ann Neurol. 57:687–694.
2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Caroppo P, Le Ber I, Clot F,
Rivaud-Péchoux S, Camuzat A, De Septenville A,
Boutoleau-Bretonnière C, Mourlon V, Sauvée M, Lebouvier T, et al:
DCTN1 mutation analysis in families with progressive supranuclear
palsy-like phenotypes. JAMA Neurol. 71:208–215. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1781. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–289. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Waterhouse A, Bertoni M, Bienert S, Studer
G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C,
Bordoli L, et al: SWISS-MODEL: Homology modelling of protein
structures and complexes. Nucleic Acids Res. 46(W1):W296–W303.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rigsby RE and Parker AB: Using the PyMOL
application to reinforce visual understanding of protein structure.
Biochem Mol Biol Educ. 44:433–437. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Rother K, Hildebrand PW, Goede A, Gruening
B and Preissner R: Voronoia: Analyzing packing in protein
structures. Nucleic Acids Res. 37(Database issue):D393–D395.
2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Guex N and Peitsch MC: SWISS-MODEL and the
Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis. 18:2714–2723. 1997.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Weisbrich A, Honnappa S, Jaussi R,
Okhrimenko O, Frey D, Jelesarov I, Akhmanova A and Steinmetz MO:
Structure-function relationship of CAP-Gly domains. Nat Struct Mol
Biol. 14:959–967. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pretelt F, Castañeda Cardona C, Tacik P,
Ross OA and Wszolek ZK: Latin America's first case of Perry
syndrome and a new treatment option for respiratory insufficiency.
J Neurol. 261:620–621. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Silva E, Itzcovich T, Niikado M, Caride A,
Fernández E, Vázquez JC, Romorini L, Marazita M, Sevlever G,
Martinetto H and Surace EI: Perry disease in an Argentine family
due to the DCTN1 p.G67D variant. Parkinsonism Relat Disord.
97:63–64. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tacik P, Fiesel FC, Fujioka S, Ross OA,
Pretelt F, Castañeda Cardona C, Kidd A, Hlavac M, Raizis A, Okun
MS, et al: Three families with Perry syndrome from distinct parts
of the world. Parkinsonism Relat Disord. 20:884–888.
2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Denson MA and Wszolek ZK: Familial
parkinsonism: Our experience and review. Parkinsonism Relat Disord.
1:35–46. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dulski J, Koga S, Prudencio M, Tipton PW,
Ali S, Strongosky AJ, Rose JH, Parrales ZA, Dunmore JA, Jansen-West
K, et al: Perry syndrome: Novel DCTN1 mutation in a large kindred
and first observation of prodromal disease. Parkinsonism Relat
Disord. 112(105481)2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mishima T, Ishikawa T, Imamura K, Kondo T,
Koshiba Y, Takahashi R, Takahashi J, Watanabe A, Fujii N, Tsuboi Y
and Inoue H: Cytoplasmic aggregates of dynactin in iPSC-derived
tyrosine hydroxylase-positive neurons from a patient with Perry
syndrome. Parkinsonism Relat Disord. 30:67–72. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dulski J, Koga S, Liberski PP, Sitek EJ,
Butala AA, Sławek J, Dickson DW and Wszolek ZK: Perry disease:
Expanding the genetic basis. Mov Disord Clin Pract. 10:1136–1142.
2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tian W, Yao L, Shi G, Dai R and Cao L: A
novel DCTN1 mutation causing perry syndrome leads to abnormal
splicing of mRNA: Genetic and functional analyses. Acta Neurol
Belg. 124:661–663. 2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Araki E, Tsuboi Y, Daechsel J, Milnerwood
A, Vilarino-Guell C, Fujii N, Mishima T, Oka T, Hara H, Fukae J and
Farrer MJ: A novel DCTN1 mutation with late-onset parkinsonism and
frontotemporal atrophy. Mov Disord. 29:1201–1204. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Cady J, Allred P, Bali T, Pestronk A,
Goate A, Miller TM, Mitra RD, Ravits J, Harms MB and Baloh RH:
Amyotrophic lateral sclerosis onset is influenced by the burden of
rare variants in known amyotrophic lateral sclerosis genes. Ann
Neurol. 77:100–113. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gustavsson EK, Trinh J, Guella I, Szu-Tu
C, Khinda J, Lin CH, Wu RM, Stoessl J, Appel-Cresswell S, McKeown
M, et al: DCTN1 p.K56R in progressive supranuclear palsy.
Parkinsonism Relat Disord. 28:56–61. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dulski J, Cerquera-Cleves C, Milanowski L,
Kidd A, Sitek EJ, Strongosky A, Vanegas Monroy AM, Dickson DW, Ross
OA, Pentela-Nowicka J, et al: Clinical, pathological and genetic
characteristics of Perry disease-new cases and literature review.
Eur J Neurol. 28:4010–4021. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hayashi I, Wilde A, Mal TK and Ikura M:
Structural basis for the activation of microtubule assembly by the
EB1 and p150Glued complex. Mol Cell. 19:449–460. 2005.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yan S, Zhang H, Hou G, Ahmed S, Williams
JC and Polenova T: Internal dynamics of dynactin CAP-Gly is
regulated by microtubules and plus end tracking protein EB1. J Biol
Chem. 290:1607–1622. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zwarycz AS, Fossat M, Akanyeti O, Lin Z,
Rosenman DJ, Garcia AE, Royer CA, Mills KV and Wang C: V67L
mutation fills an internal cavity to stabilize RecA Mtu Intein.
Biochemistry. 56:2715–2722. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fernández A and Berry RS: Proteins with
H-bond packing defects are highly interactive with lipid bilayers:
Implications for amyloidogenesis. Proc Natl Acad Sci USA.
100:2391–2396. 2003.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Deshimaru M, Kinoshita-Kawada M, Kubota K,
Watanabe T, Tanaka Y, Hirano S, Ishidate F, Hiramoto M, Ishikawa M,
Uehara Y, et al: DCTN1 Binds to TDP-43 and Regulates TDP-43
Aggregation. Int J Mol Sci. 22(3985)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhang Z, Witham S and Alexov E: On the
role of electrostatics in protein-protein interactions. Phys Biol.
8(035001)2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vendruscolo M, Zurdo J, MacPhee CE and
Dobson CM: Protein folding and misfolding: A paradigm of
self-assembly and regulation in complex biological systems. Philos
Trans A Math Phys Eng Sci. 361:1205–1222. 2003.PubMed/NCBI View Article : Google Scholar
|