Introduction
Neurofibromatosis (NF) is a genetic disorder
manifested by tumor formation in the central or peripheral nervous
system. It comprises three types and NF type 1 (NF1; Online
Mendelian Inheritance in Man no. 613113) is an autosomal dominant
genetic disorder exhibiting a range of manifestations such as
café-au-lait spots, skinfold freckling, cutaneous neurofibromas and
Lisch nodules in the iris, also known as von Recklinghausen's
disease (1). Von Recklinghausen, in
1882, was the first to recognize that the tumor arises from nervous
tissue. The prevalence of NF1 is estimated to be ~1 in 3,000
individuals worldwide, making it one of the most common genetic
disorders (2). The NF1 gene, which
codes for neurofibromin, is positioned on chromosome 17 at the
locus 17q11.2(3). Neurofibromin, a
tumor suppressor protein, is synthesized in neurons,
oligodendrocytes and Schwann cells, where it acts as a Ras-GTPase
activating protein. NF1 has almost 100% penetrance but variable
expression and 50% of cases are sporadic (4). NF1 is typically diagnosed based on
clinical criteria established by the National Institutes of Health
(NIH). The diagnosis frequently involves the presence of specific
clinical features such as café-au-lait spots, neurofibromas,
freckling, Lisch nodules and a family history of NF1. Genetic
testing can also be used to confirm the diagnosis by identifying
pathogenic mutations in the NF1 gene. More than half of patients
with NF1 also have plexiform neurofibroma (PN). Individuals with
NF1 have a significantly increased risk of malignancy and a reduced
life expectancy compared to the general population (5). Early and accurate diagnosis of NF1 is
crucial for appropriate management and monitoring of the disorder
and its potential complications.
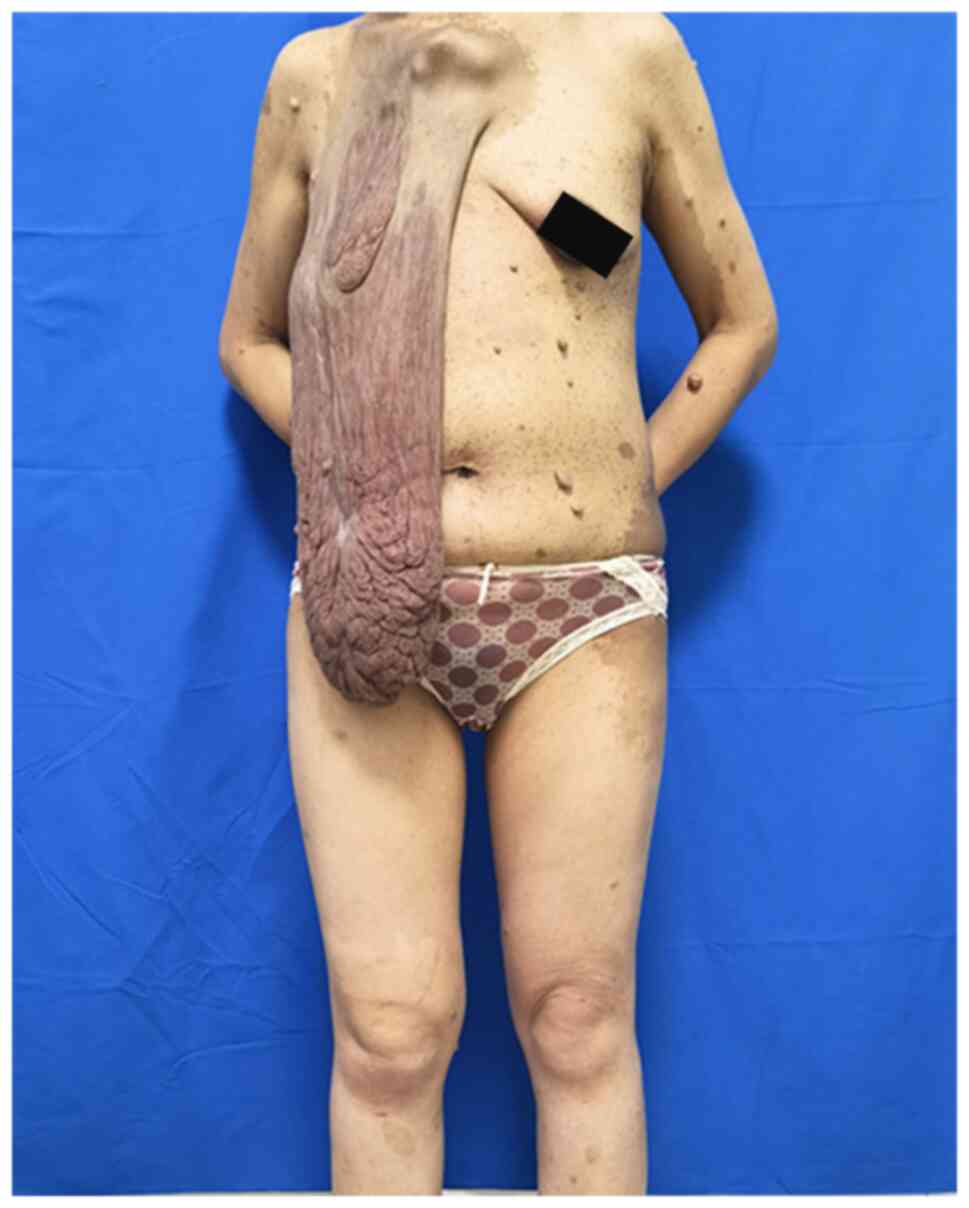
The present study reported on the case of a
36-year-old female patient with a sizable mass on the right chest
wall. The mass's base was located above the right breast, extending
to the patient's thigh when standing. The patient, facing mental
stress and financial challenges, had not sought any medical care.
Eventually, the patient presented at our department due to the
substantial impact of the mass on her daily life.
Case report
In July 2022, a 36-year-old female with giant
cutaneous neoplasm on the right breast skin presented at the
surgical outpatient department of the Affiliated Tumor Hospital of
Xinjiang Medical University (Urumqi, China), requesting the
excision of a large neurofibroma. Upon examination, a large tumor
was observed extending downward from the skin on the right side
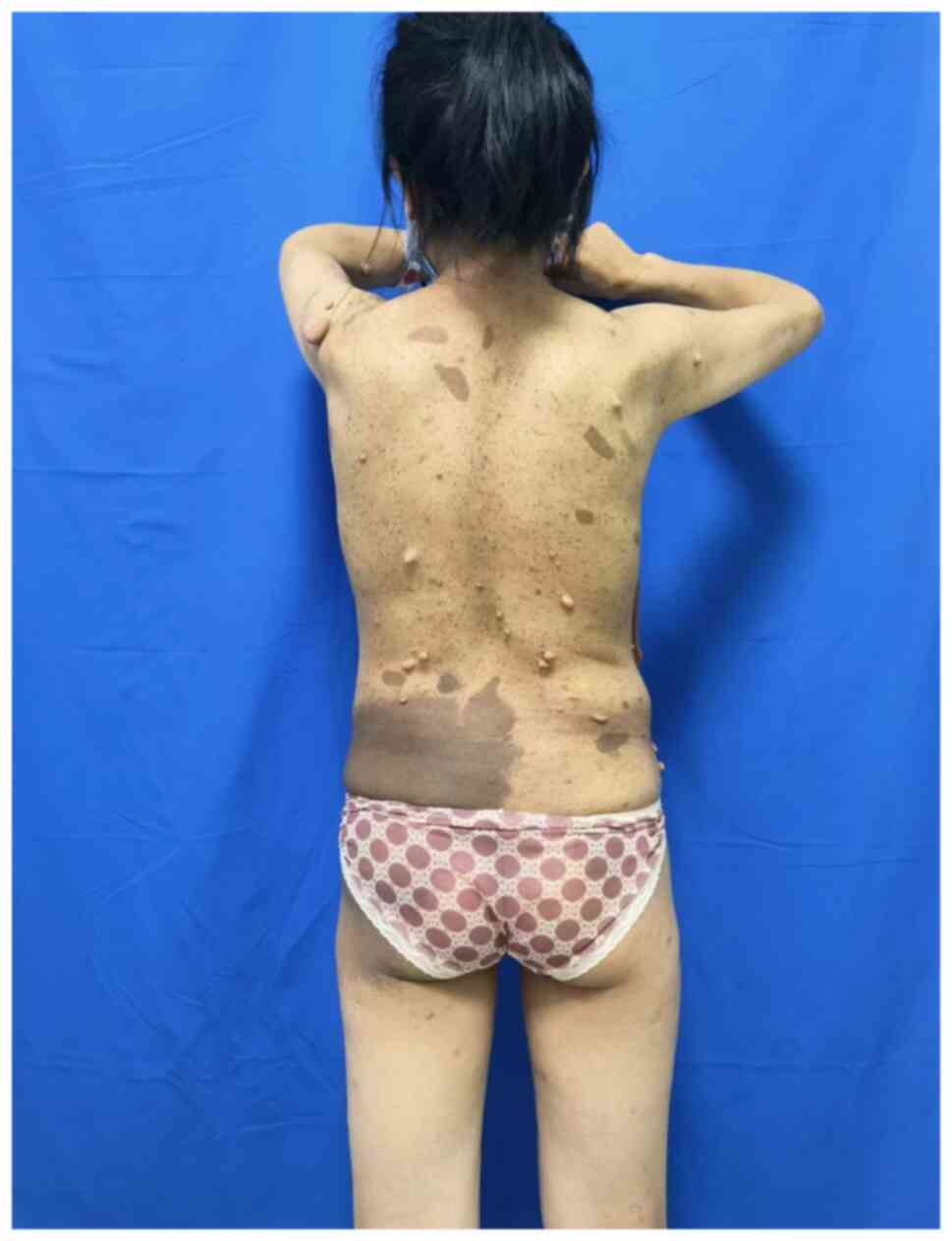
above the breast and there were numerous soft, fleshy and
non-tender nodules of varying sizes, along with brown-pigmented
macules mainly distributed on the chest, waist and upper arms. The
patient could only report that she had had them for as long as she
could remember and the neoplasm on the right breast skin had
enlarged markedly after her giving birth to her four children. The
clinical features corresponded to a diagnosis of NF1. The patient
has a family history of related diseases, as the patient's father
has extensive cutaneous nodules and the patient's children are also
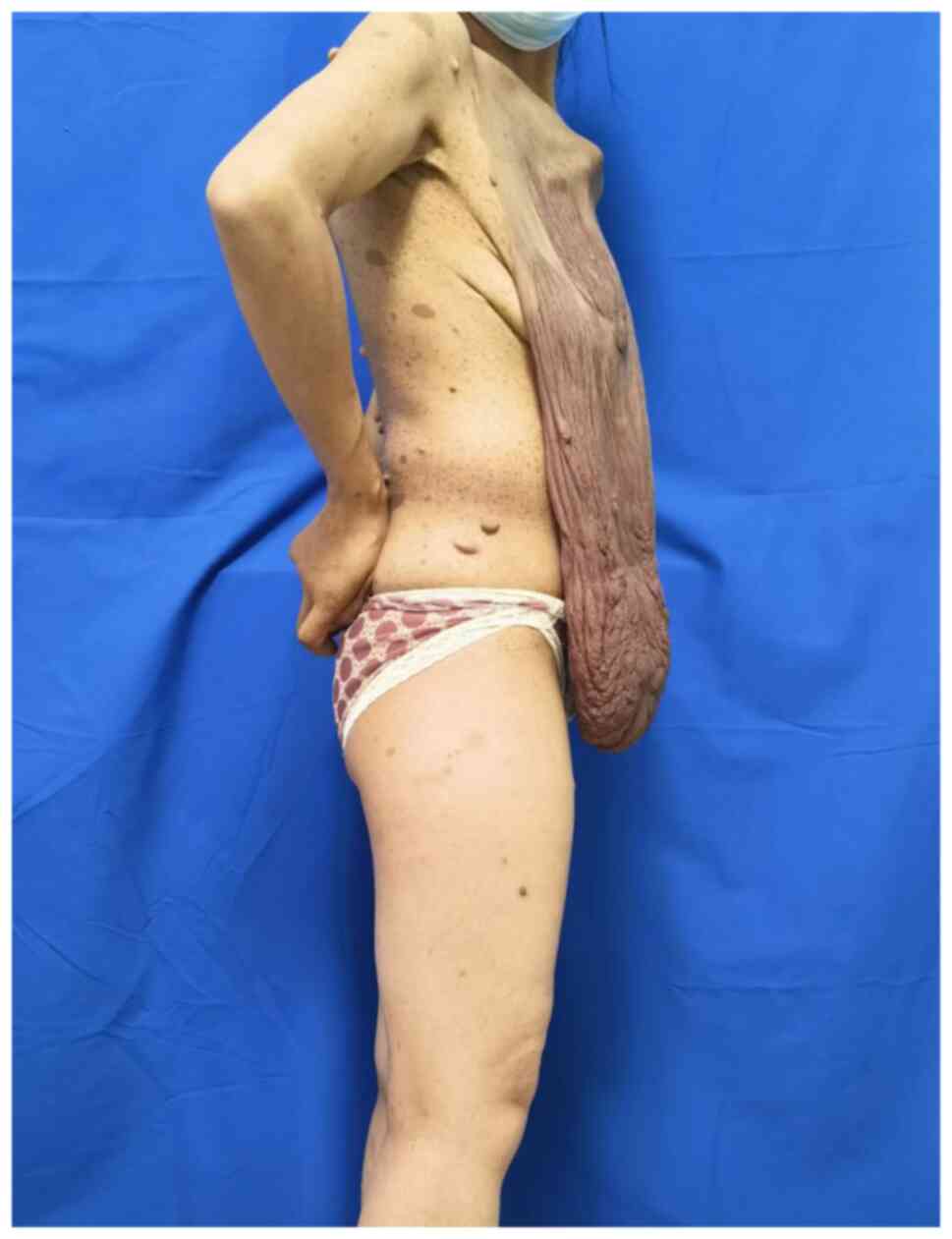
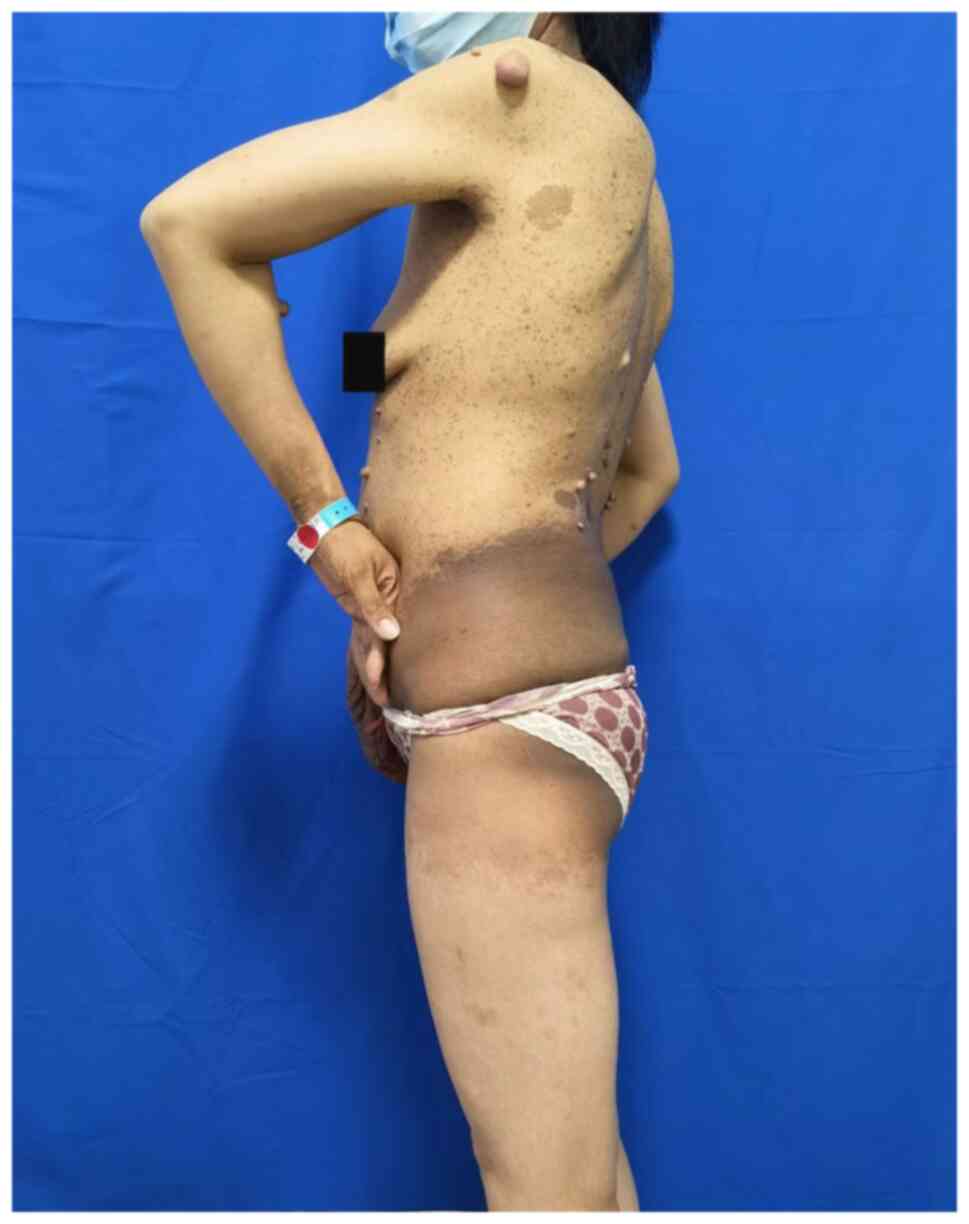
affected. Preoperative photographs are provided in Fig. 1, Fig.
2, Fig. 3 and Fig. 4.
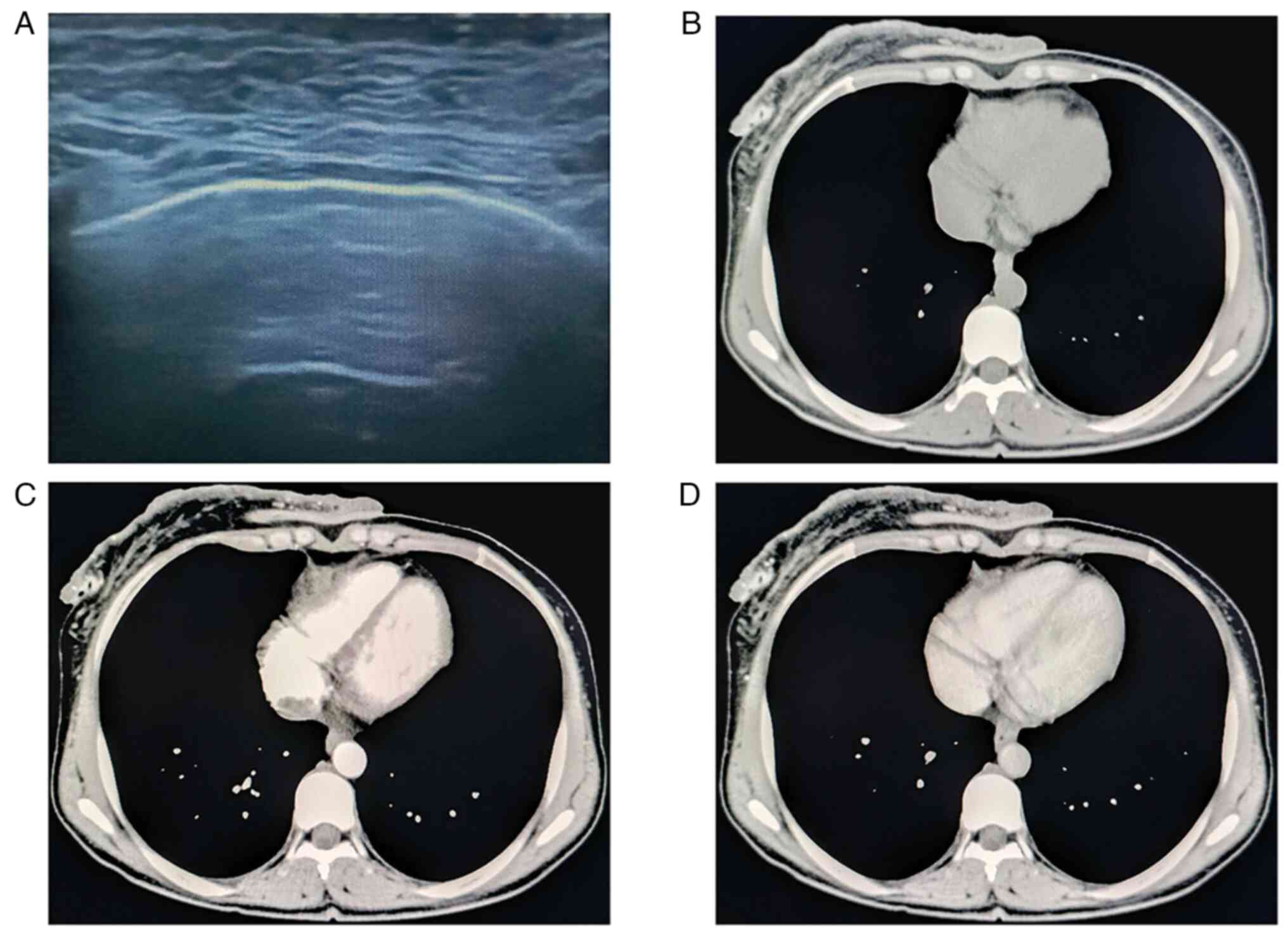
Auxiliary examinations were as follows: Ultrasonic
imaging revealed a large neoplasm on the skin above the right
breast with an indeterminate size and unclear demarcation between
the base and the skin. Thoracic CT scan indicated skin thickening
of the right anterior chest wall and thus, skin malignancy was
considered; multiple nodules were present in the right thoracic
muscular space, and thus, metastases were suspected; sternum
destruction and an abnormal shape and position of the sternum, with
forward protrusion, suggested a congenital developmental
abnormality; multiple nodules were observed on the skin surface and
subcutaneous tissue of the chest and back, and metastases were
suspected. Typical ultrasound images and CT scans are shown in
Fig. 5.
Surgical excision is the mainstay of treatment for
cutaneous neurofibromas, but local recurrence is possible (6). After completion of the auxiliary
examinations, the patient underwent a neurofibro-resection and the
skin of the chest was sutured using the oncoplastic technique in
July 2022, one week after her first presentation at surgical
outpatient department. An incision was made at the base of the skin
appendage on the right chest wall, extending through the skin and
subcutaneous tissue to the pectoralis muscle. Hemostasis was
meticulously maintained during the procedure, followed by excision
of the excess appendage for rapid pathological evaluation. The
rapid pathological results, produced according to standard
protocols, indicated a mesenchymal tumor with diffuse spindle cell
proliferation, displaying a mild morphology, partial stromal edema
and localized deposition of pigment granules, suggesting a probable
benign or borderline nature. The surgery lasted 120 min, with two
disposable drainage tubes placed intraoperatively - one in the
chest wall and the other in the right axilla. On the first
postoperative day, 150 ml of drainage from the axilla tube and 40
ml from the chest wall tube were recorded. At three days after the
surgery, the drainage from both of the patient's tubes decreased to
<10 ml, with the drainage fluid exhibiting a pale red color.
Subsequently, all drainage tubes were removed before the patient's
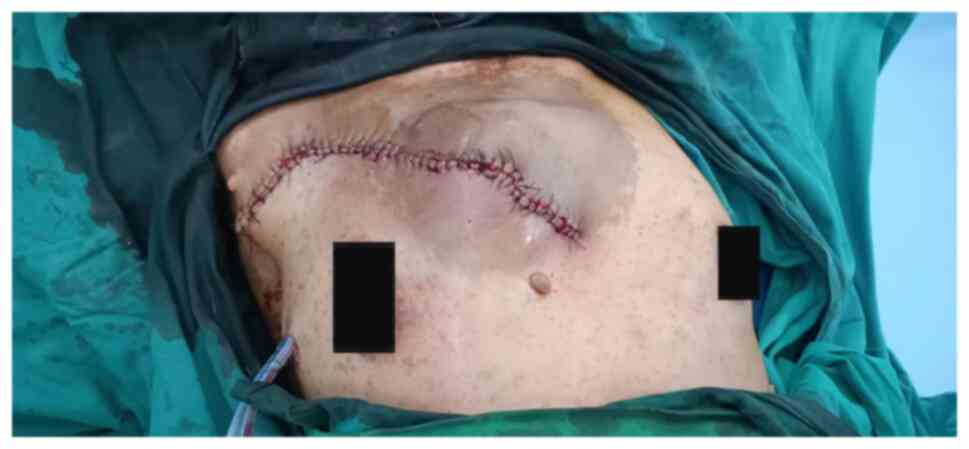
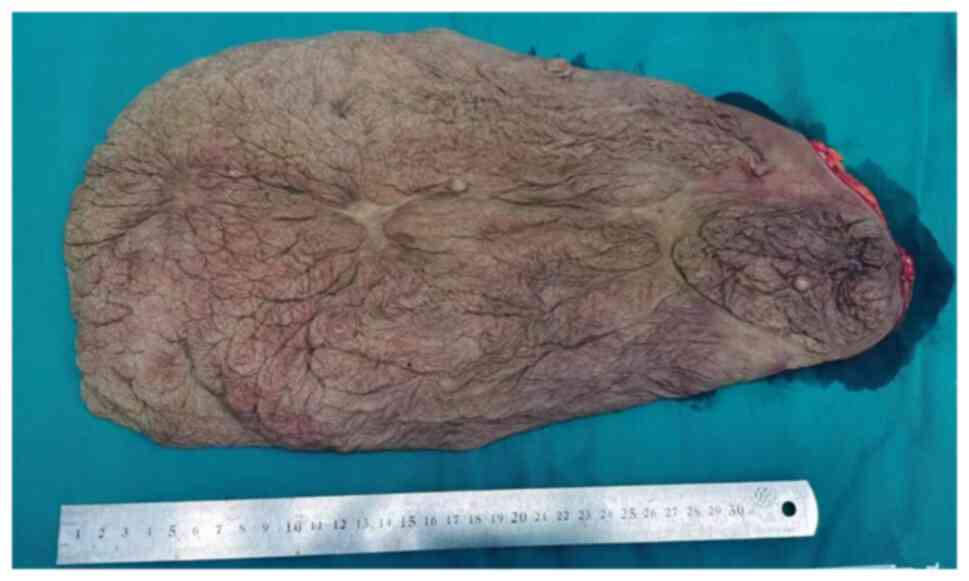
discharge. A tumor measuring 33x25x3.5 cm had been excised.
Postoperative photographs are provided in Figs. 6 and 7. Subsequent histopathological
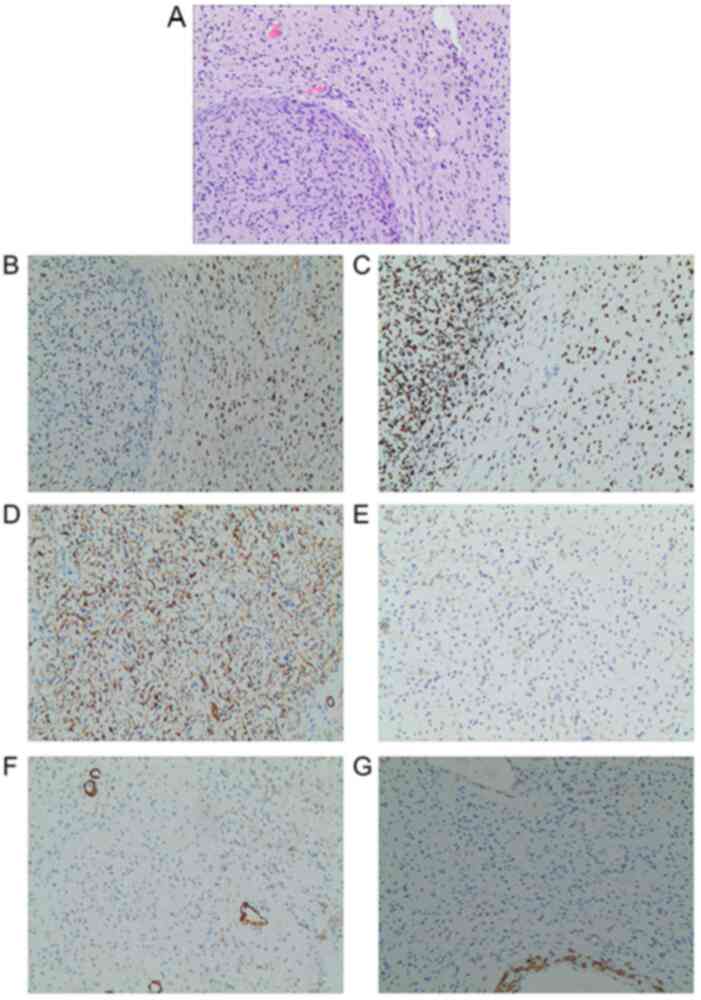
examination, performed according to standard protocols, of the
massive tumor on the right chest wall indicated the following:
Spindle cell tumor with focal myxoid change, consistent with
neurofibroma based on immunohistochemical staining and tissue
morphology; tumor tissue visible at the surgical margin and base.
Another smaller neurofibroma was excised during the surgery with
the dimensions of 3.0x2.5x1.5 cm. The immunohistochemistry results
were as follows: S-100 (+), SOX10 (+), CD34 (+), Ki-67 (+, <1%),
smooth muscle actin (SMA) (-), and Desmin (-) (7). The antibodies used according to
standard protocols were as follows: S-100 (cat. no. ab183979;
1:1,000 dilution; Abcam), SOX10 (cat. no. ab227680; 1:100 dilution;
Abcam), CD34 (cat. no. ab81289; 1:2,500 dilution; Abcam), Ki-67
(cat. no. ab16667; 1:200 dilution; Abcam), SMA (cat. no. ab5694;
1:100 dilution; Abcam) and Desmin (cat. no. ab32362; 1:2,000
dilution; Abcam) The histopathological results of the cutaneous
nodules were consistent with neurofibroma. Images from the
pathological examination are provided in Fig. 8.
The patient remained hospitalized for 6 days
postoperatively and was then discharged in August 2022, one week
after the operation. Due to the extensive scope of the lesion,
complete excision was not feasible. The patient and family were
informed of the situation before the surgery and consent was
obtained after full explanation to them. At one month after
discharge, the patient was followed up at the surgical outpatient
department of our hospital in September 2022. The patient made a
good recovery and the patient's life quality significantly
improved.
Discussion
There is currently no treatment available for the
underlying genetic defect that causes NF1. The treatment of
patients with NF1 is based on identifying manifestations of NF1 and
treating the complications induced by them (8). Approximately 30% of individuals with
NF1 exhibit symptoms associated with PN. PN are a common
manifestation of NF1, representing a histologically benign
neurofibroma (9). In the current
study, the case of a 36-year-old female patient with a PN on the
right anterior chest since birth, gradually increasing in size, was
presented. Due to financial hardship and psychological stress, the
patient delayed regular check-ups until the tumor significantly
impacted her daily life. Preoperatively, the patient underwent head
and chest CT scans, revealing sternal protrusion, suggesting a
congenital anomaly. Factors affecting PN symptoms and complications
include tumor location, size, nerve involvement and age. Typically,
younger patients experience faster tumor growth, while older
adolescents and adults often have slower progression (10). Symptoms vary based on PN location,
including impairments in vision or hearing, airway obstruction,
speech and swallowing difficulties, motor dysfunction,
gastrointestinal or bladder issues, deformities, as well as common
symptoms such as pain and reduced mobility (11).
Psychological aspects should not be overlooked in
addition to physical symptoms. Reports indicate that approximately
one-third of children and adolescents with PN may display anxiety,
depression and social withdrawal, impacting mental health and
quality of life (12). The patient
stated that the sizable mass caused significant mental distress,
impacting her social life and having deterred her from seeking
professional assistance for an extended period. PN have the
potential to evolve into atypical neurofibromas (AN). Rapid PN
growth in adults may suggest malignancy, with pain being a
prominent feature in patients with AN, necessitating comprehensive
evaluation for progressive severe pain in patients with PN
(10). All patients with NF1 should
undergo imaging studies, such as MRI and CT, to identify and
monitor PN growth (13). In the
case reported in the present study, the patient did not undergo MRI
due to financial constraints. A biopsy for histological PN
diagnosis is typically unnecessary unless malignancy is suspected
based on clinical or imaging findings (14). In this instance, the patient
exhibited no pain symptoms, with slow tumor growth, and was
postoperatively diagnosed with a spindle cell tumor. The extensive
size of the patient's tumor posed challenges for curative surgery,
given its invasion of the surrounding normal tissues. The family
was briefed on the pertinent surgical details prior to the
operation and they provided written consent for the surgical
intervention.
Genetic testing is particularly essential for
individuals with a familial history of NF1, as it can identify
genetic mutations and evaluate the likelihood of transmitting the
condition to subsequent generations. The analysis of mRNA and
genomic DNA enables the identification of 95% of pathogenic NF1
mutations in individuals meeting the diagnostic criteria
established by the NIH (4).
Specific correlations exist between mutant NF1 alleles and clinical
phenotypes, including whole NF1 gene deletions linked to severe
cognitive abnormalities and somatic overgrowth (15,16).
Regrettably, the patient did not undergo genetic testing due to
financial constraints, although she reported that her daughter
exhibits similar manifestations, including cafe-au-lait spots. When
contemplating treatment, factors such as patient age, the impact of
PN on morbidity or associated risks, as well as growth progression,
should be thoroughly assessed. Rapidly progressing PN with
potential morbidities may call for intervention, whereas
slow-growing tumors with no or minimal impact may warrant
observation (17). Surgical
intervention and medication are current treatment options for
patients with PN. Excision is frequently challenging due to the
tumor's impingement on adjacent nerves and structures, as well as
its characteristic extensive vascularity that may lead to
life-threatening hemorrhage. Personalized treatment plans should be
developed through multidisciplinary discussions to optimize
benefits and minimize risks (18).
In terms of patients who reject surgical treatment or whose tumors
are unresectable, annual imaging examination is important in case
of transformation to peripheral nerve-sheath tumors, as reported in
another rare NF1 case with giant cutaneous neoplasm (6). The patient failed to attend subsequent
follow-up appointments at the hospital post-surgery, and thus, it
was not possible to monitor the patient's ongoing condition.
In conclusion, the present study reported a case of
giant NF1 presented on the right breast skin. We reviewed the
choice of diagnosis and therapeutic options for this disease.
Family history, multiple café-au-lait spots and cutaneous fleshy
nodules throughout the body are the typical characteristics of
NF1(19). Numerous recent advances
have been made in the management of PN. In addition to
surveillance, symptomatic management and surgery, effective
targeted medical therapies have become available. A major challenge
going forward will be the identification of individualized
treatment schedules and therapeutic combinations that can provide
the best outcomes for all patients who require treatment for PN.
The clinical implementation of therapies for NF1 requires careful
consideration of multiple factors and should be performed with the
input of a multidisciplinary team experienced in NF1.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
CZ and YS drafted the manuscript. YL prepared all of
the figures and participated in drafting the manuscript. JO
conceived the idea of the study and supervised the workflow. CZ and
JO checked and confirmed the authenticity of the raw data. All
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The ethics committee at the Affiliated Tumor
Hospital of Xinjiang Medical University (Urumqi, China) does not
require authors to obtain ethics approval for case reports.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of this case report and corresponding
images.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Tamura R: Current understanding of
neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci.
22(5850)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Evans DG, Howard E, Giblin C, Clancy T,
Spencer H, Huson SM and Lalloo F: Birth incidence and prevalence of
tumor-prone syndromes: Estimates from a UK family genetic register
service. Am J Med Genet A. 152A:327–332. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ferner RE and Gutmann DH:
Neurofibromatosis type 1 (NF1): Diagnosis and management. Handb
Clin Neurol. 115:939–955. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huson SM, Compston DA, Clark P and Harper
PS: A genetic study of von recklinghausen neurofibromatosis in
south east Wales. I. Prevalence, fitness, mutation rate, and effect
of parental transmission on severity. J Med Genet. 26:704–711.
1989.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Korf BR: Plexiform neurofibromas. Am J Med
Genet. 89:31–37. 1999.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bayram T, Bayram D and Tireli H:
Neurofibromatosis type 1-related multiple plexiform neurofibromas:
A case report. Turk J Neurol. 26:42–46. 2020.
|
|
7
|
Loughran PA, Ross MA and St Croix CM:
Immunohistochemistry. Curr Protoc. 2(e549)2022.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Jett K and Friedman JM: Clinical and
genetic aspects of neurofibromatosis 1. Genet Med. 12:1–11.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ehara Y, Koga M, Imafuku S, Yamamoto O and
Yoshida Y: Distribution of diffuse plexiform neurofibroma on the
body surface in patients with neurofibromatosis 1. J Dermatol.
47:190–192. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Uusitalo E, Rantanen M, Kallionpää RA,
Pöyhönen M, Leppävirta J, Ylä-Outinen H, Riccardi VM, Pukkala E,
Pitkäniemi J, Peltonen S and Peltonen J: Distinctive cancer
associations in patients with neurofibromatosis type 1. J Clin
Oncol. 34:1978–1986. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cieza Rivera AM, Lobato Fuertes C,
Fernández-Villa T, Martín Sánchez V and Atallah I: Impact of
neurofibromatosis type 1 on quality of life using the skindex-29
questionnaire quality of life in NF1. Orphanet J Rare Dis.
19(85)2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Prause M, Schulz HJ and Wagler D:
Rechnergestützte führung von fermentationsprozessen, teil 2. Acta
Biotechnologica. 4:143–151. 1984.
|
|
13
|
Lin J and Martel W: Cross-sectional
imaging of peripheral nerve sheath tumors: Characteristic signs on
CT, MR imaging, and sonography. AJR Am J Roentgenol. 176:75–82.
2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Legius E, Messiaen L, Wolkenstein P,
Pancza P, Avery RA, Berman Y, Blakeley J, Babovic-Vuksanovic D,
Cunha KS, Ferner R, et al: Revised diagnostic criteria for
neurofibromatosis type 1 and legius syndrome: An international
consensus recommendation. Genet Med. 23:1506–1513. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jett K and Friedman JM: Clinical and
genetic aspects of neurofibromatosis 1. Genet Med. 12:1–11.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Napolitano F, Dell'Aquila M, Terracciano
C, Franzese G, Gentile MT, Piluso G, Santoro C, Colavito D, Patanè
A, De Blasiis P, et al: Genotype-phenotype correlations in
neurofibromatosis type 1: Identification of novel and recurrent
NF1 gene variants and correlations with neurocognitive
phenotype. Genes (Basel). 13(1130)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fisher MJ, Blakeley JO, Weiss BD, Dombi E,
Ahlawat S, Akshintala S, Belzberg AJ, Bornhorst M, Bredella MA, Cai
W, et al: Management of neurofibromatosis type 1-associated
plexiform neurofibromas. Neuro Oncol. 24:1827–1844. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Vaassen P, Dürr N, Röhrig A, Willing R and
Rosenbaum T: Trametinib induces neurofibroma shrinkage and enables
surgery. Neuropediatrics. 50:300–303. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lalor L, Davies OMT, Basel D and Siegel
DH: Café au lait spots: When and how to pursue their genetic
origins. Clin Dermatol. 38:421–431. 2020.PubMed/NCBI View Article : Google Scholar
|