Introduction
Congenital hypothyroidism (CH) is a common neonatal
endocrine disorder that is characterized by irreversible
neurodevelopmental and growth retardation due to insufficient
biosynthesis of thyroid hormones. The incidence of CH is estimated
to be between 1:2,000 and 1:4,000(1), based on newborn screening programs for
CH that measure thyroid-stimulating hormone (TSH) and/or thyroxine
(T4) levels. In China, the incidence of CH is estimated to be 5.77
per 10,000 live births (2).
Previous studies have indicated an increase in the diagnosis of CH,
particularly in cases of glands in situ (GIS) (3,4), which
can be attributed to lower cut-off values for TSH during newborn
screening (5). However, the
etiology of CH remains unclear.
The majority of CH cases (80-85%) are attributed to
thyroid dysgenesis (TD), which may manifest as athyreosis,
hypoplasia, ectopic thyroid tissue, or a small thyroid gland. The
remaining CH cases (15-20%) are attributed to thyroid
dyshormonogenesis (DH), which presents with a normally located
intact thyroid gland and, in some cases, compensatory goiter
(6). Genetic defects in
DUOX2, TG, TPO, SLC26A4, SLC5A5,
DUOXA2 and IYD have been associated with inadequate
thyroid hormone biosynthesis (7,8).
Although TD is typically regarded as a sporadic disease, variants
in 5 genes (GLIS3, TSHR, NKX2-1, PAX8
and FOXE1) have been reported as monogenic causes of TD
(9). Additionally, genetic factors
play a significant role in the etiology of some familial forms of
TD (10-12).
Furthermore, isolated central CH is associated with genes involved
in hypothalamic-pituitary-thyroid axis regulation, including
TRHR, TSHB and IGSF1 (13).
Next-generation sequencing (NGS) has enabled the
genetic screening of patients with CH and comprehensive analysis of
CH-related genes, which may reveal the complex genetic etiology and
inheritance patterns of CH. Variants in CH-related genes have been
identified in populations of different ethnicities from different
regions (14-16).
In China, multigenic screening of patients with CH and systematic
analysis of genotype-phenotype correlations have been conducted in
several provinces (17-19).
However, little is known about the variant characteristics of
CH-related genes in Foshan, China.
In the present study, an NGS panel containing 30
candidate genes was established for multigenic screening of 105
patients with CH, diagnosed through newborn screening programs in
Foshan, China. Variant frequencies and the variant spectrum of CH
in the neonatal population of Foshan were evaluated.
Materials and methods
Patients
The present retrospective study included 105
unrelated patients with CH and 138 controls at the Foshan Women and
Children Hospital between December 2018 and September 2022 (Foshan,
China). The inclusion and exclusion criteria for patients were as
follows: Subjects with elevated TSH levels and decreased free
thyroxine (FT4) levels who were born to non-consanguineous parents.
The control subjects were healthy newborns with no detectable
inherited metabolic disorders at newborn screening. Of the 105
patients, 56 were males and 49 were females. The median age of the
patient cohort at diagnosis was 16 days, with a range of 6 to 355
days. CH diagnosis was based on TSH levels and FT4 levels in the
neonatal screening program. Heel prick samples were collected on
filter paper and TSH levels were analyzed by a time-resolved
fluorescence-based assay using an Auto TRFIA-4 automatic
fluorescence immunoassay analyzer (Guangzhou Fenghua Biotechnology
Co., Ltd.; http://www.bio-fenghua.com/index.asp). Patients with
high TSH (≥10 µIU/ml) levels were called back for further testing.
Subsequently, serum TSH, FT4, free triiodothyronine, T4 and
triiodothyronine levels were measured on the Roche Cobas e602
analyzer (Roche Diagnostics GmbH) using electrochemiluminescence
immunoassays. Thyroid ultrasonography was performed to assess
thyroid morphology whenever possible. The present study was
approved (approval no. FSFY-MEC-2021-041) by the Medical Ethics
Committee of the Foshan Women and Children Hospital, and written
informed consent was obtained from the parents of all patients in
accordance with the Declaration of Helsinki.
DNA extraction and sequencing
Genomic DNA was extracted from blood spot cards
using Nucleic Acid Isolation or Purification Reagent (cat. nos
DR-HS-004; Guangzhou Darui Biotechnology Co., Ltd.) according to
the manufacturer's protocols. The NGS panel consisted of 30
candidate genes (BCHE, DUOX2, EZH2,
GLI3, GLIS3, IYD, IGSF1, KAT6B,
NEFL, NEFM, NKX2-1, NKX2-5,
NSD1, PAX8, PHTF1, POU1F1,
SERPINA7, TG, UBR1, SH2B3,
SLC26A4, SLC5A5, SECISBP2, TPO,
DUOXA2, FOXE1, LHX4, TRHR, TSHB
and TSHR). Custom primers were designed to generate 687
amplicons. The target gene exon regions comprised 396 regions, and
all exons along with 20 bp of the flanking introns of these regions
were amplified by multiplex PCR. The total coverage of the target
genes was >98% (Table SI). The
Ion AmpliSeq Library Kit Plus and Ion Xpress Barcode Adapters Kits
(cat. nos. A35907 and 4474517, respectively; both from Thermo
Fisher Scientific, Inc.) were used to prepare DNA libraries for
sequencing. The library was then quantified using the Equalbit 1X
dsDNA HS Assay Kit (cat. no. EQ121-01; Vazyme, Biotech Co., Ltd.)
and the Qubit Fluorometer 3.0 (Thermo Fisher Scientific, Inc.).
Targeted sequencing was performed using a single-end 250 bp
sequencing method on the DA8600 sequencer (Guangzhou Darui
Biotechnology Co., Ltd.) with the Universal Sequencing Kit
(semiconductor sequencing; cat. no. DR-CX-A001; Guangzhou Darui
Biotechnology Co., Ltd.).
Bioinformatic analysis and
classification of variants
The raw data generated by sequencing were analyzed
using Torrent Suite Software (v.4.4.3) (Thermo Fisher Scientific,
Inc.). Reads were aligned to the human reference genome (hg19)
using the Torrent Mapping Alignment Program (v.4.4.11) (Thermo
Fisher Scientific, Inc.). The coverage analysis plugin (v.4.4.2.2)
(Thermo Fisher Scientific, Inc.) was used to assess the level of
sequence coverage and overall quality of the targeted regions. The
Variant Caller plugin (v.4.4.3.3) (Thermo Fisher Scientific, Inc.)
was used to evaluate variants, and the called variants were
annotated using Ensemble's Variant Effect Predictor (v.102)
(grch37.ensembl.org/info/docs/tools/vep) based on the
1000 Genomes Project (http://www.1000genomes.org), dbSNP (http://www.ncbi.nlm.nih.gov/), Exome Aggregation
Consortium (http://exac.broadinstitute.org/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/), Genome Aggregation
Database (http://gnomad.broadinstitute.org/), ESP6500
(http://evs.gs.washington.edu/EVS/),
Ensembl (http://plants.ensembl.org/index.html), Refseq
(http://www.ncbi.nlm.nih.gov/refseq/rsg), OMIM
(https://omim.org/), and UCSC (https://genome.ucsc.edu). Functional predictions of
the variants were evaluated using dbNSFP (v.4.1) (https://sites.google.com/site/jpopgen/dbNSFP) to
obtain prediction scores based on Sorting Intolerant from Tolerant
(http://sift-dna.org), Mutation Taster (http://www.mutationtaster.org/), Mutation
Assessor (http://mutationassessor.org/r3/), ClinPred (https://sites.google.com/site/clinpred/), CADD
(https://cadd.gs.washington.edu/),
Polymorphism Phenotyping-2 (http://genetics.bwh.harvard.edu/pph2/), FATHMM
(http://fathmm.biocompute.org.uk/), REVEL
(https://sites.google.com/site/revelgenomics/), MetaSVM
(http://genomics.usc.edu/members/15-member-detail/36-coco-dong),
PROVEAN (http://provean.jcvi.org/index.php), LRT (http://www.genetics.wustl.edu/jflab/lrt_query.html),
GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/),
and SpliceAI (https://spliceailookup.broadinstitute.org/). The
detected variants were classified into 5 categories according to
the guidelines of the American College of Medical Genetics and
Genomics, namely pathogenic, likely pathogenic, variants of
uncertain significance, likely benign, or benign. Each pathogenic
criterion was weighted as very strong (PVS1), strong (PS1-4),
moderate (PM1-6), or supporting (PP1-5), while each benign
criterion was weighted as stand-alone (BA1), strong (BS1-4), or
supporting (BP1-6). The criteria were selected based on the
evidence observed for the variants and then combined to choose a
classification from the five categories (20). Variants classified as likely benign
or benign were excluded from the subsequent analysis.
Results
Variant frequency in the study
population
In total, 91 variants were identified in 78 of the
105 cases (74.29%). These variants were distributed across 16 genes
(DUOX2, TG, TPO, DUOXA2,
SLC26A4, SLC5A5, IYD, TSHR,
GLIS3, KAT6B, NKX2-5, LHX4,
POU1F1, SECISBP2, IGSF1 and TRHR). The
most frequently mutated gene was DOUX2 (50.55%, 46/91),
followed by TG (10.99%; 10/91), TSHR (8.79%; 8/91)
and TPO (6.59%; 6/91). Two variants [p.Lys530Ter
(DUOX2) and p.Gly132Arg (TSHR)] were homozygous,
while the other variants were heterozygous (Table I). The clinical, biochemical and
variant information of the 78 patients is shown in Table SII.
 | Table ISpectrum of 91 variants in 16
genes. |
Table I
Spectrum of 91 variants in 16
genes.
| Gene | Nucleotide
change | Amino acid
change | Exon/Intron
position | dbSNP number | Variant type | Status | Zygosity | Allele frequency
(gnomAD) | Evidence of
classification | Variant
classification | Numbers of
patients | (Refs.) |
|---|
| DUOX2 | c.1588A>T | p.Lys530 Ter | Exon 14 | rs180671269 | Stop gained | Known | Het/Hom | 0.0092 | PVS1_VeryStrong +
PM2_Supporting | LP | 20 | (21) |
| DUOX2 | c.3329G>A | p.Arg1110 Gln | Exon 25 | rs368488511 | Missense | Known | Het | 0.00244645 | PM3_VeryStrong +
PM2_Supporing + PP1 + PS3_Supporting + PP3_Moderate | P | 8 | (22) |
| DUOX2 | c.3693+
1G>T | IVS28+ 1G>T | Intron 28 | rs200717240 | Splice donor | Known | Het | 0.001413 | PM3_VeryStrong +
PM2_Supporting + PM3_Strong | P | 5 | (23) |
| DUOX2 | c.2654G>A | p.Arg885 Gln | Exon 20 | rs181461079 | Missense | Known | Het | 0.0011 | PM3_Strong +
PM2_Supporting + PM5_Moderate + PP3 + PP1 + PS3 Supporting | P | 3 | (21) |
| DUOX2 | c.596del | p.Ser199
TrpfsTer122 | Exon 6 | rs766103168 | Frame-shift | Known | Het | 0.0002 | PVS1_VeryStrong +
PM2_Supporting | LP | 2 | (24) |
| DUOX2 | c.3616G>A | p.Ala1206 Thr | Exon 28 | rs762588205 | Missense | Known | Het | 0.0002 | PM3_Strong +
PM2_Supporting + PS3_Supporting | P | 1 | (25) |
| DUOX2 | c.1300C>T | p.Arg434 Ter | Exon 12 | rs119472026 | Stop gained | Known | Het | 0.00010873 | PVS1_VeryStrong +
PM3_VeryStrong + PM2_Supporting | P | 1 | (26) |
| DUOX2 | c.2101C>T | p.Arg701 Ter | Exon 17 | rs201109959 | Stop gained | Known | Het | 0.00010874 | PVS1_VeryStrong +
PM3_Strong + PM2_Supporting | P | 1 | (26) |
| DUOX2 | c.1708C>T | p.Gln570 Ter | Exon 15 | rs1332668133 | Stop gained | Known | Het | 0.0003 | PVS1_VeryStrong +
PM2_Supporting | LP | 1 | (27) |
| DUOX2 | c.477del | p.Glu160
ArgfsTer16 | Exon 5 | rs1480917996 | Frame-shift | Known | Het | NA | PVS1_VeryStrong +
PM2_Supporting | LP | 1 | (28) |
| DUOX2 | c.602dup | p.Gln202
ThrfsTer99 | Exon 6 | rs567500345 | Frame shift | Known | Het | 0/0.001285 | PVS1_VeryStrong +
PM3_VeryStrong + PM2_Supporting + PP1 | P | 1 | (29) |
| DUOX2 | c.2635G>A | p.Glu879Lys | Exon 20 | rs774556391 | Missense | Known | Het | 0.00092421 |
PM3_Strong+PM2_Supporting + PP3_Moderate +
PP1 + Supporting + PS3_Supporting | LP | 3 | (21) |
| DUOX2 | c.2104_2106del | p.Gly702del | Exon 17 | rs779340990 | Inframe
deletion | Knowna | Het | 0.001033 | PM2_Supporting +
PM4 | VUS | 2 | NA |
| DUOX2 | c.3285_3286del | p.Ile1097
LeufsTer24 | Exon 25 | NA | Frame-shift | Novel | Het | NA | PVS1_VeryStrong +
PM2_Supporting | LP | 1 | NA |
| DUOX2 | c.2048G>T | p.Arg683 Leu | Exon 17 | rs8028305 | Missense | Known | Het | 0.00462107 | BS1_Strong | VUS | 10 | (17) |
| DUOX2 | c.1268C>T | p.Thr423Ile | Exon 12 | rs201197899 | Missense | Known | Het | 0.0016 | PM2_Supporting | VUS | 3 | (30) |
| DUOX2 | c.3632G>A | p.Arg1211 His | Exon 28 | rs141763307 | Missense | Known | Het | 0.0003262 | PP3_Strong +
PM2_Supporting | VUS | 3 | (23) |
| DUOX2 | c.1310G>C | p.Gly437Ala | Exon 12 | rs769796932 | Missense | Known | Het | 0.0017 | PP3 + PM2
Supporting | VUS | 3 | (19) |
| DUOX2 | c.3689C>T | p.Ala1230 Val | Exon 28 | rs557220354 | Missense | Knowna | Het | 0.0002175 | PM2_Supporting +
BP4g | VUS | 2 | NA |
| DUOX2 | c.505C>T | p.Arg169Trp | Exon 5 | rs201590426 | Missense | Known | Het | 0.001935 | PM2_Supporting +
PM5 | VUS | 2 | (31) |
| DUOX2 | c.364C>A | p.Pro122Thr | Exon 5 | rs200265605 | Missense | Known | Het | 0.0004 | PM2_Supporting +
BP4g | VUS | 1 | (19) |
| DUOX2 | c.1428C>A | p.Asn476 Lys | Exon 13 | rs199918362 | Missense | Known | Het | 0.0053 | BS1_Strong +
BP4g | VUS | 1 | (16) |
| DUOX2 | c.4537G>C | p.Gly1513 Arg | Exon 34 | rs748262140 | Missense | Known | Het | NA/0.000002052 | PP3_Strong +
PM2_Supporting | VUS | 1 | (23) |
| DUOX2 | c.2291G>A | p.Arg764 Gln | Exon 18 | rs201884203 | Missense | Known | Het | 0.00010873 | PM2_Supporting | VUS | 1 | (27) |
| DUOX2 | c.4561G>T | p.Gly1521 Ter | Exon 34 | rs765781255 | Stop gained | Known | Het | 0.001 | PVS1_Moderate +
PM2_Supporting | VUS | 1 | (23) |
| DUOX2 | c.1946C>A | p.Ala649Glu | Exon 17 | rs748793969 | Missense | Known | Het | 5.44E-05 | PM3_Strong +
PM2_Supporting | VUS | 1 | (21) |
| DUOX2 | c.1295G>A | p.Arg432His | Exon 12 | rs530736554 | Missense | Known | Het | 0.0006 | PM2_Supporting | VUS | 1 | (23) |
| DUOX2 | c.3251G>A | p.Arg1084 Gln | Exon 25 | rs558919433 | Missense | Known | Het | 0.0004 | PP3_Moderate +
PM2_Supporting | VUS | 1 | (32) |
| DUOX2 | c.4348T>C | p.Tyr1450 His | Exon 32 | rs753591292 | Missense | Known | Het | 0.0004 | PP3 +
PM2_Supporting | VUS | 1 | (33) |
| DUOX2 | c.3595C>G | p.Leu1199 Val | Exon 28 | NA | Missense | Novel | Het | NA | PM2_Supporting | VUS | 1 | NA |
| DUOX2 |
c.2335-25T>C | IVS18-25T>C | Intron 18 | NA | Intron | Novel | Het | NA | BP7_Supporting +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.1304A>G | p.Asp435 Gly | Exon 12 | rs772040742 | Missense | Known | Het | 0.00043492 | PP3 +
PM2_Supporting | VUS | 1 | (23) |
| DUOX2 | c.4408C>T | p.Arg1470 Trp | Exon 33 | rs200785525 | Missense | Known | Het | 0.0022 | PM2_Supporting +
PP3_Moderate | VUS | 1 | (18) |
| DUOX2 | c.1855G>T | p.Val619Leu | Exon 16 | rs768447406 | Missense | Knowna | Het | 0.0007068 | BP4g +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.903G>T | p.Trp301Cys | Exon 8 | rs568196384 | Missense | Known | Het | 0.0003263 | PP3 + PM2
Supporting | VUS | 1 | (17) |
| DUOX2 | c.2412C>G | p.Cys804 Trp | Exon 19 | NA | Missense | Novel | Het | NA | PP3 +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.655_656
delinsTC | p.Leu219 Ser | Exon 6 | NA | Missense | Novel | Het | NA | PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.646_647 insTTTCC
CCCG | p.Gln216 delinsLeu
SerProGlu | Exon 6 | rs1894400616 | Protein
altering | Knowna | Het | NA/0.000016 | PM4 +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.647_658 del |
p.Gln216_Leu219del | Exon 6 | NA | Inframe
deletion | Novel | Het | NA | PM4 +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.2921G>A | p.Arg974His | Exon 22 | rs778216481 | Missense | Known | Het | 0.0013 | PP3 +
PM2_Supporting | VUS | 1 | (19) |
| DUOX2 | c.1127G>A | p.Arg376 Gln | Exon 10 | rs778729877 | Missense | Knowna | Het | 0.00005437 | PM5 +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.514-2A>G | IVS5-2A>G | intron 5 | NA | Splice
acceptor | Known | Het | NA | PVS1_VeryStrong +
PM2_Supporing | LP | 1 | (28) |
| DUOX2 | c.3374A>G | p.Asp1125 Gly | Exon 25 | NA | Missense | Novel | Het | NA | PP3_Moderate +
PM2_Supporting | VUS | 1 | NA |
| DUOX2 | c.4375G>A | p.Asp1459 Asn | Exon 32 | rs199546504 | Missense | Known | Het | 0.0005 | PM2_Supporting | VUS | 1 | (27) |
| DUOX2 | c.2894C>T | p.Ser965Leu | Exon 22 | rs144153950 | Missense | Known | Het | 0.0005 | PM2_Supporting | VUS | 1 | (18) |
| DUOX2 | c.1393C>A | p.Pro465Thr | Exon 12 | rs774177514 | Missense | Knowna | Het | NA | BP4 +
PM2_Supporting | VUS | 1 | NA |
| DUO-
XA2 | c.738C>G | p.Tyr246Ter | Exon 5 | rs4774518 | Stop gained | Known | Het | 0.0019 | PVS1_Strong +
PM3_Strong + PM2_Supporting + PS3_Supporting | P | 1 | (34) |
| DUO
XA2 | c.413dup | p.Tyr138Ter | Exon 4 | rs778410503 | Frame-shift | Known | Het | 0.0033 | PVS1_VeryStrong +
PM2_Supporing | LP | 1 | (35) |
| GLIS3 | c.1982del | p.Lys661
SerfsTer145 | Exon 6 | NA | Frameshift | Novel | Het | NA | PVS1_VeryStrong +
PM2_Supporing | LP | 2 | NA |
| GLIS3 | c.1843G>A | p.Ala615Thr | Exon 5 | rs752946704 | Missense | Knowna | Het | 0.000054371 | PM2_Supporting | VUS | 1 | NA |
| GLIS3 | c.2723C>T | p.Ala908Val | Exon 11 | rs140101069 | Missense | Known | Het | 0.004245 | BP4 +
PM2_Supporting | VUS | 1 | (36) |
| IGSF1 | c.584G>C | p.Gly195Ala | Exon 5 | rs745841814 | Missense | Knowna | Het | 0.0003 | PM2_Supporting | VUS | 1 | NA |
| IYD | c.688-7G>A | IVS4-7G>A | Intron 4 | rs1778273239 | Splice region | Knowna | Het | NA/0.000001446 |
BP4+PM2_Supporting | VUS | 1 | NA |
| IYD | c.380C>T | p.Pro127Leu | Exon 3 | rs372196319 | Missense | Knowna | Het | 0.0001089 | PP3_Moderate +
PM2_Supporting | VUS | 1 | NA |
| KAT6B | c.1025T>C | p.Ile342Thr | Exon 7 | rs182392778 | Missense | Knowna | Het | 0.0028 | BS1_Strong | VUS | 2 | NA |
| LHX4 | c.970G>A | p.Ala324Thr | Exon 6 | rs544059210 | Missense | Known | Het | 0.00005437 | PM2_Supporting | VUS | 1 | (37) |
| LHX4 | c.1127C>T | p.Thr376Ile | Exon 6 | rs1334926032 | Missense | Knowna | Het | NA | PM2_Supporting | VUS | 1 | NA |
| NKX2-5 | c.773G>C | p.Gly258Ala | Exon 2 | NA | Missense | Novel | Het | NA | PP2 +
PM2_Supporting | VUS | 1 | NA |
| POU
1F1 | c.744-6C>A | IVS5-6C>A | Intron 5 | NA | Intron | Novel | Het | NA | PP3_Moderate +
PM2_Supporting | VUS | 1 | NA |
| SECIS
BP2 | c.1212+
4C>T | IVS8+4C>T | Intron8 | NA | Intron | Novel | Het | NA | BP4 +
PM2_Supporting | VUS | 1 | NA |
| SLC26
A4 | c.919-2A>G | IVS7-2A>G | Intron 7 | rs111033313 | Splice
acceptor | Known | Het | 0.0048 | PVS1_VeryStrong +
PM3_VeryStrong + PM2_Supporting + PP1 + PS3_Supporting | P | 1 | (38) |
| SLC26
A4 | c.2086C>T | p.Gln696Ter | Exon 18 | rs752807925 | Stop gained | Known | Het | 0.0002 | PVS1_VeryStrong +
PM3_VeryStrong + PM2_Supporting | P | 1 | (39) |
| SLC26
A4 | c.269C>T | p.Ser90Leu | Exon 3 | rs370588279 | Missense | Known | Het | 0.00005437 | PM3_VeryStrong +
PM1 + PP2 + PM2_Supporting + PP3 + PS3_Supporting | P | 1 | (40) |
| SLC26
A4 | c.-3-46C>T | IVS1-46C> T | Intron 1 | NA | Intron | Novel | Het | NA | BP7 +
PM2_Supporting | VUS | 1 | NA |
| SLC26
A4 | c.697G>C | p.Val233Leu | Exon 6 | rs397516431 | Missense | Known | Het | 0.0014135 | PM3_Strong + PM1 +
PP2 + PM2_Supporting + PP3 | VUS | 1 | (41) |
| SLC5A5 | c.1499C>T | p.Pro500Leu | Exon 12 | rs531134045 | Missense | Knowna | Het | 0.0003 | PM2_Supporting | VUS | 1 | NA |
| TG | c.5512del | p.Asp1838
ThrfsTer14 | Exon 29 | NA | Frame-shift | Novel | Het | NA | PVS1_VeryStrong +
PM2_Supporing | LP | 1 | NA |
| TG | c.5854C>T | p.Arg1952 Trp | Exon 31 | rs369705913 | Missense | Knowna | Het | 0.00010903 | PM2_Supporting | VUS | 1 | NA |
| TG | c.3641G>A | p.Arg1214 Gln | Exon 17 | rs200877580 | Missense | Knowna | Het | 0.0002 | BP4 +
PM2_Supporting | VUS | 1 | NA |
| TG | c.635A>G | p.Asn212 Ser | Exon 5 | rs187737243 | Missense | Knowna | Het | 0.0021 | BP4 | VUS | 1 | NA |
| TG | c.1597G>A | p.Gly533 Arg | Exon 9 | NA | Missense | Novel | Het | NA | PM2_Supporting | VUS | 1 | NA |
| TG | c.958C>T | p.Arg320 Cys | Exon 8 | rs138561283 | Missense | Knowna | Het | 0.0008 | PM2_Supporting | VUS | 1 | NA |
| TG | c.7753C>T | p.Arg2585 Trp | Exon 44 | rs114211101 | Missense | Known | Het | 0.00513651 | BS1 | VUS | 1 | (18) |
| TG | c.8205del | p.Gln2736
SerfsTer10 | Exon 48 | rs758002273 | Frame-shift | Knowna | Het | 0.0014 | PVS1_Moderate +
PM2_Supporing | VUS | 1 | NA |
| TG | c.925A>G | p.Thr309Ala | Exon 8 | rs199712883 | Missense | Known | Het | 0.001 | BP4 +
PM2_Supporting | VUS | 1 | (16) |
| TG | c.3040G>A | p.Asp1014 Asn | Exon 12 | rs114772213 | Missense | Known | Het | 0.0005 | BP4 +
PM2_Supporting | VUS | 1 | (42) |
| TPO | c.2268dup | p.Glu757Ter | Exon 13 | rs770781635 | Frame-shift | Known | Het | 0.0016 | PVS1_VeryStrong +
PM3_VeryStrong + PM2_Supporting | P | 1 | (43) |
| TPO | c.2146G>T | p.Glu716Ter | Exon 12 | NA | Stop gained | Novel | Het | NA | PVS1_VeryStrong +
PM2_Supporing | LP | 1 | NA |
| TPO | c.2017G>A | p.Glu673 Lys | Exon 12 | rs201193196 | Missense | Known | Het | 0.0007 | PP3 +
PM2_Supporting | VUS | 1 | (44) |
| TPO | c.2603C>T | p.Thr868 Met | Exon 15 | rs201576336 | Missense | Knowna | Het | 0.0009 | PM2_Supporting | VUS | 1 | NA |
| TPO | c.2536C>T | p.Arg846 Trp | Exon 15 | rs28913014 | Missense | Known | Het | 0.0017 | BS1 +
BS2_Supporting | VUS | 1 | (45) |
| TPO | c.1367G>A | p.Arg456 Lys | Exon 9 | rs1329337261 | Missense | Knowna | Het | 0 | PM2_Supporting | VUS | 1 | NA |
| TRHR | c.504T>G | p.Ile168Met | Exon 2 | rs13306060 | Missense | Known | Het | 0.0024 |
BP4+PM2_Supporting | VUS | 1 | (46) |
| TSHR | c.2272G>A | p.Glu758 Lys | Exon 10 | rs746522401 | Missense | Known | Het | 0.0003262 | PM2_Supporting | VUS | 1 | (32) |
| TSHR | c.740T>C | p.Val247Ala | Exon 2 | NA | Missense | Novel | Het | NA | BP4 +
PM2_Supporting | VUS | 1 | NA |
| TSHR | c.394G>C | p.Gly132 Arg | Exon 5 | rs760874290 | Missense | Known | Hom | 0.00054413 | PS4+PM2_Supporting
+ PP3_Supporting | VUS | 1 | (47) |
| TSHR | c.823G>A | p.Ala275Thr | Exon 9 | rs180762551 | Missense | Known | Het | 0.0003262 | PP3 +
PM2_Supporting | VUS | 1 | (48) |
| TSHR | c.915T>A | p.Ser305Arg | Exon 10 | rs142122217 | Missense | Known | Het | 0.0033 | NA | VUS | 1 | (48) |
| TSHR | c.1492G>A | p.Gly498Ser | Exon 10 | rs1376842882 | Missense | Known | Het | NA | PM2_Supporting +
PP3_Moderate + PM1 | VUS | 1 | (49) |
| TSHR | c.694G>C | p.Asp232His | Exon 9 | rs752791414 | Missense | Knowna | Het | 0.00005437 | PP3_Moderate +
PM2_Supporting | VUS | 1 | NA |
| TSHR | c.1960A>T | p.Ile654Phe | Exon 10 | rs767239688 | Missense | Knowna | Het | 0.0002 | PP3_Strong + PS4 +
PM2_Supporting | P | 1 | NA |
Biallelic variants were identified in 24.76%
(26/105) of patients with CH: In DUOX2 (24 cases),
TSHR (1 case) and TPO (1 case). In addition,
monoallelic variants were identified in 28.57% (30/105) of the
patients: In DUOX2 (18 cases), TSHR (3 cases),
TG (2 cases), GLIS3 (2 cases), SLC5A5 (1
case), DUOXA2 (1 case), IYD (1 case), TPO (1
case) and KAT6B (1 case). Oligogenic variants were
identified in 19.05% (20/105) of the patients. The most common
variant combinations of oligogenic variants were DUOX2 and
TG (2 cases), and DUOX2 and SLC26A4 (2 cases).
In total, 2 patients harbored tri-allelic and tetra-allelic
variants in DUOX2, respectively. Notably, 71.43% (75/105) of
the patients harbored variants in at least one gene involved in
thyroid hormone biosynthesis (9)
(DUOX2, TG, TPO, DUOXA2,
SLC26A4, SLC5A5, IYD and TSHR)
(Table SII).
Analysis of pathogenicity
Of the 91 variants across 16 genes, 16 were novel
variants, while 75 variants had been previously reported in
literature and databases. A total of 13 of the known variants were
classified as pathogenic, including 7 variants in DUOX2
(p.Arg1110Gln, IVS28+1G>T, p.Arg885Gln, p.Ala1206Thr,
p.Arg434Ter, p.Arg701Ter and p.Gln202ThrfsTer99), 3 in
SLC26A4 (IVS7-2A>G, p.Gln696Ter and p.Ser90Leu), 1 in
TPO (p.Glu757Ter), 1 in TSHR (p.Ile654Phe) and 1 in
DUOXA2 (p.Tyr246Ter). A total of 7 known variants were
likely pathogenic, including 6 DUOX2 variants (p.Lys530Ter,
p.Ser199TrpfsTer122, p.Gln570Ter, p.Glu160ArgfsTer16, p.Glu879Lys
and IVS5-2A>G) and 1 of DUOXA2 (p.Tyr138Ter). A total of
4 novel variants were likely pathogenic [p.Ile1097LeufsTer24
(DUOX2), p.Lys661SerfsTer145 (GLIS3),
p.Asp1838ThrfsTer14 (TG) and p.Glu716Ter (TPO)].
Furthermore, 67 variants were classified as variants of uncertain
significance (Table I).
The types of variants identified in our cohort are
shown in Fig. 1. Most variants were
missense variants (69.23%; 63/91), followed by frameshift variants
(9.89%; 9/91), stop gained variants (8.79%; 8/91) and intron
variants (4.40%; 4/91). Inframe deletion, splice acceptor, protein
altering, splice donor and splice region variants were also
identified in the present study. In addition, 7 known missense
variants in DUOX2 [p.Arg683Leu (n=10), p.Arg1110Gln (n=8),
p.Arg885Gln (n=3), p.Glu879Lys (n=3), p.Thr423Ile (n=3),
p.Arg1211His (n=3) and p.Gly437Ala (n=3)] were highly prevalent in
the cohort (Table I). A total of 20
patients harbored a stop gained variant in DUOX2
(p.Lys530Ter). A known splice donor variant (IVS28 + 1G>T) in
DUOX2 was detected in 5 patients (Table SII).
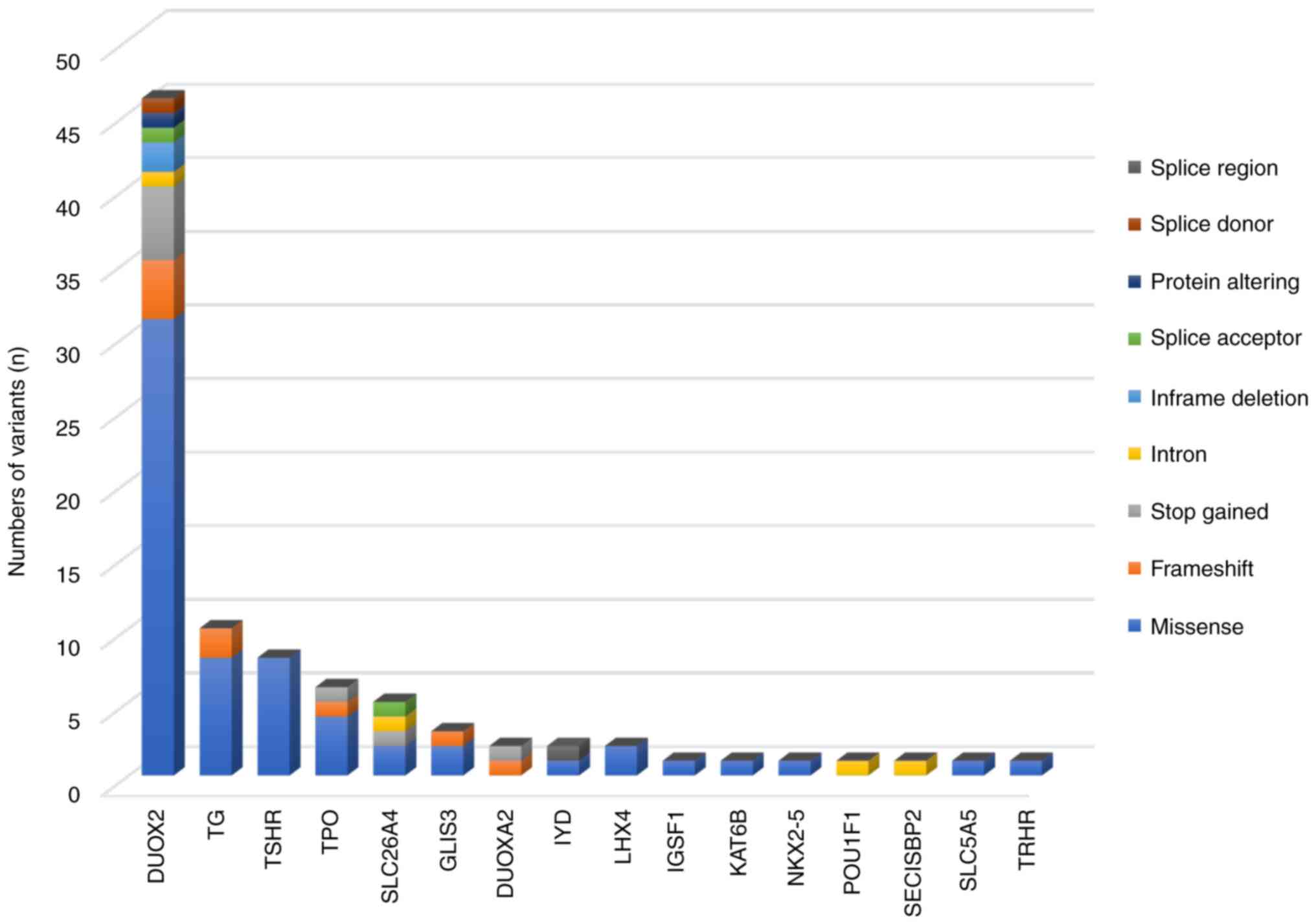 | Figure 1Distribution of 91 variants and their
variant type across 16 genes. Of the 91 variants, 46 were detected
in DUOX2, 10 in TG, 8 in TSHR, 6 in
TPO, 5 in SLC26A4, 3 in GLIS3, 2 each in
DUOXA2, IYD and LHX4 and 1 each in
IGSF1, KAT6B, NKX2-5, POU1F1,
SECISBP2, SLC5A5 and TRHR. Missense,
frameshift, stop gained, intron, in-frame deletion, splice
acceptor, splice donor, splice region and protein-altering variants
were identified in the present study. |
Genotype-phenotype correlation in
patients with CH
Of the 78 patients (34 females and 44 males) with
variants, 73 patients had normal or goitrous thyroid glands, which
may be associated with GIS. Only 1 patient had athyreosis with a
monoallelic GLIS3 mutation (p.Ala908Val). Thyroid morphology
was not detected in 4 patients. A total of 7 patients (patients
5,12, 30, 37, 40, 62 and 63) were preterm infants with normal
thyroid glands, 5 of whom harbored rare variants in SLC5A5,
GLIS3, POU1F1, DUOXA2, or KAT6B.
Furthermore, 10 patients had a family history of thyroid disease,
of which 7 (patients 8, 29, 48, 50, 64, 73 and 74) harbored
biallelic or monoallelic DUOX2 variants with eutopic thyroid
glands of normal size, and goiter was noted in only 1 case (patient
10) with monoallelic TG mutations (p.Asn212Ser). Notably,
patient 54 harbored tetra-allelic variants in DUOX2
(p.Arg683Leu, p.Leu219Ser, p.Gln216delinsLeuSerProGlu and
p.Gln216_Leu219del).
Discussion
In the present study, a cohort of 105 patients with
CH was comprehensively screened using NGS to analyze the variant
frequencies and variant spectrum of CH in the neonatal population
of Foshan. Variants in CH-related genes were identified in 74.29%
(78/105) of patients. DUOX2, TG, TSHR and
TPO were the most frequently mutated genes, similar to a
previous study in Chinese patients with CH (48).
Variants in DUOX2 have been reported to be a
common cause of CH in patients of Chinese and East Asian
ethnicities, often resulting in DH with a normal-sized eutopic or
goitrous thyroid gland owing to decreased
H2O2 production in the thyroid (33,50).
Among the 105 patients, 46 different DUOX2 variants were
identified in 61 patients (58.10%; 61/105), reflecting the high
prevalence of DUOX2 variants in patients in the present
study, which is consistent with previous studies in Chinese
populations (17,19). Moreover, 4 known DUOX2
variants, p.Lys530Ter, p.Arg683Leu, p.Arg1110Gln, and IVS28 +
1G>T, were highly recurrent in the cohort of the present study.
The most common variant detected in the present study was
p.Lys530Ter. Consistent with the findings of the present study, Tan
et al (28) and Fu et
al (17) reported p.Lys530Ter
to be the most common variant in Chinese populations. In the
present study, 25.71% (27/105) of the patients had monoallelic
variants in DUOX2, 29.52% (31/105) had biallelic variants in
DUOX2 and 1.90% (2/105) had tri-allelic variants in
DUOX2 (patients 13 and 69). In addition, 1 patient (patient
54) with tetra-allelic variants in DUOX2, whose brother was
also diagnosed with CH was also identified. However, additional
information regarding patients 13, 54 and 69 has not been
collected, which limited the evaluation of their types or
inheritance patterns of CH.
TG variants have been reported to affect the
synthesis and storage of thyroid hormones, resulting in
hypothyroidism with compensatory goiter (51). The variant frequency for TG
in the cohort of the present study was 9.52% (10/105), which was
higher than that reported for Japanese patients (2.82%) (52). In total, 8/10 patients with
TG variants in the cohort harbored monoallelic heterozygous
TG variants in combination with other CH-related genes,
suggesting that oligogenic involvement may contribute to the
genetic etiology of some patients with CH. The TG variant
p.Arg2585Trp has been reported in both Chinese and Japanese
populations (24,53).
Inactivating variants in TSHR cause TSH
resistance, which negatively affects thyroid growth, and stimulates
thyroid hormone synthesis and release (7). Monoallelic TSHR variants were
identified in 6 patients (5.71%; 6/105), and DUOX2 or
TG variants were found along with TSHR variants in
3/6 patients with TSHR variants. Biallelic variants in
TSHR were identified in 1 patient whose sister also had CH.
In contrast to Chinese populations, TSHR variants are the
most common genetic defects in patients with CH (10.9%) in Saudi
Arabia (54). In the present study,
TSHR variants-p.Glu758Lys, p.Ala275Thr and p.Ser305Arg-were
reported in the Chinese population (32,48,55).
p.Gly132Arg has been frequently reported in patients with CH of
Chinese (50), Japanese (47) and Korean (14) ethnicities. In total, 1 patient in
the cohort has a homozygous TSHR variant p.Gly132Arg.
p.Gly498Ser has been previously reported in the Japanese
population, and its low expression is likely to affect the
functions of the TSH receptor (49).
TPO plays an important role in thyroid
hormone biosynthesis and variants in TPO have been reported
to be highly prevalent in patients with DH-associated CH of
Caucasian (56) and
Malaysian-Chinese (57)
ethnicities. In the cohort of the present study, the variant rate
for TPO was ~4.76% (5/105), which was lower than that
reported in other populations. The stop gained variant p.Glu757Ter
identified in the cohort of the present study has been reported as
a common cause of CH in Taiwanese (43).
In addition, no definite pathogenic or likely
pathogenic variants were identified in IYD, SLC5A5,
LHX4, IGSF1, KAT6B, NKX2-5,
POU1F1, SECISBP2, or TRHR in the cohort.
Moreover, variants in these genes appear to be rare: Only 1 or 2
variants were identified for each of these genes, and are usually
associated with variants in other genes, especially DUOX2 or
TG.
A previous study reported that oligogenic variants
are common in sporadic CH (16),
suggesting that a combination of rare variations in CH-related
genes may underlie the complex genetic etiology of CH. In the
present study, oligogenic variants were detected in 19.05% (20/105)
of the patients, and the combination of DUOX2, TG,
TPO and TSHR variants was noted frequently. There is
some evidence that suggests that patients with tri-allelic variants
have permanent CH (17). However,
one of the limitations of the present study is that the clinical
phenotypes of all patients with CH in the cohort of the present
study were not clearly elucidated; therefore, the association
between the function of oligogenic variants and the hypothyroid
phenotype remains unclear.
In conclusion, DUOX2, TG, TSHR
and TPO variants were the most common genetic defects in
patients with CH in the neonatal population of Foshan.
Specifically, biallelic DUOX2 variants were highly prevalent
in the study population. Based on the findings of the present
study, the authors suggest that oligogenic variants in CH-related
genes may contribute to the complex genetic etiology of CH.
Further, the present investigation provides a detailed variant
spectrum of CH-related genes and identifies novel variants, which
may allow for an improved understanding of the underlying genetic
etiology of CH and provide evidence for further molecular
epidemiological investigations that can guide preventive and
therapeutic programs in Foshan, China.
Supplementary Material
List and the coverage of the 30
candidate genes included in the NGS panel.
Clinical features and genotypical data
in 78 patients with congenital hypothyroidism.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Scientific
Research Fund of Women and Children's Medical Research Center of
Foshan Women and Children Hospital (grant no. FEYJZX-2020-003) and
the Foshan Genetic Metabolic Disease Molecular Diagnosis and
Treatment Engineering Technology Research Center (grant no.
2120001009239).
Availability of data and materials
The data generated in the present study may be
found in the Genome Sequence Archive for Human under accession
number HRA008474 or at the following URL: https://ngdc.cncb.ac.cn/gsa-human/s/U36l4c36.
Authors' contributions
WC collected the patients' blood samples. WC, SW
and WY performed the experiments. XH, QS and JT analyzed and
interpreted the data. SW, XY and XH drafted and wrote the
manuscript. XH, XY and SW participated in discussing and revising
the manuscript. XS and XY conceived and designed the study. XS, XH
and JT contributed to overall senior mentorship and guidance and
support to the project. XH and SW confirm the authenticity of all
the raw data. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical
Ethics Committee of the Foshan Women and Children Hospital (Foshan,
China) on 12 March 2021 (approval no. FSFY-MEC-2021-041), and the
renewal date of the ethics approval was 22 May 2025. Written
informed consent was obtained from the parents/guardians of all
patients and controls in accordance with the Declaration of
Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rastogi MV and LaFranchi SH: Congenital
hypothyroidism. Orphanet J Rare Dis. 5(17)2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yao Y, Deng K, Zhu J, Xiang L, Yuan X, Li
Q, Liu L and Xu W: Increased incidence of congenital hypothyroidism
in China: An analysis of 119 million screened newborns. Eur J
Pediatr. 182:4477–4486. 2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Corbetta C, Weber G, Cortinovis F,
Calebiro D, Passoni A, Vigone MC, Beck-Peccoz P, Chiumello G and
Persani L: A 7-year experience with low blood TSH cutoff levels for
neonatal screening reveals an unsuspected frequency of congenital
hypothyroidism (CH). Clin Endocrinol (Oxf). 71:739–745.
2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mitrovic K, Vukovic R, Milenkovic T,
Todorovic S, Radivojcevic J and Zdravkovic D: Changes in the
incidence and etiology of congenital hypothyroidism detected during
30 years of a screening program in central Serbia. Eur J Pediatr.
175:253–259. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu L, He W, Zhu J, Deng K, Tan H, Xiang
L, Yuan X, Li Q, Huang M, Guo Y, et al: Global prevalence of
congenital hypothyroidism among neonates from 1969 to 2020: A
systematic review and meta-analysis. Eur J Pediatr. 182:2957–2965.
2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stoupa A, Kariyawasam D, Carré A and Polak
M: Update of thyroid developmental genes. Endocrinol Metab Clin
North Am. 45:243–254. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Szinnai G: Clinical genetics of congenital
hypothyroidism. In: Szinnai G (ed). Endocrine Development. Vol. 26.
S. Karger AG, pp60-78, 2014.
|
|
8
|
Grasberger H and Refetoff S: Genetic
causes of congenital hypothyroidism due to dyshormonogenesis. Curr
Opin Pediatr. 23:421–428. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Peters C, Van Trotsenburg ASP and
Schoenmakers N: Diagnosis of endocrine disease: Congenital
hypothyroidism: Update and perspectives. Eur J Endocrinol.
179:R297–R317. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Castanet M, Lyonnet S, Bonaïti-Pellié C,
Polak M, Czernichow P and Léger J: Familial forms of thyroid
dysgenesis among infants with congenital hypothyroidism. N Engl J
Med. 343:441–442. 2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Léger J, Marinovic D, Garel C,
Bonaïti-Pellié C, Polak M and Czernichow P: Thyroid developmental
anomalies in first degree relatives of children with congenital
hypothyroidism. J Clin Endocrinol Metab. 87:575–580.
2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Castanet M, Polak M, Bonaïti-Pellié C,
Lyonnet S, Czernichow P and Léger J: AFDPHE (Association Française
pour le Dépistage et la Prévention des Handicaps de l'Enfant).
Nineteen years of national screening for congenital hypothyroidism:
Familial cases with thyroid dysgenesis suggest the involvement of
genetic factors. J Clin Endocrinol Metab. 86:2009–2014.
2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Schoenmakers N, Alatzoglou KS, Chatterjee
VK and Dattani MT: Recent advances in central congenital
hypothyroidism. J Endocrinol. 227:R51–R71. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lee ST, Lee DH, Kim JY, Kwon MJ, Kim JW,
Hong YH, Lee YW and Ki CS: Molecular screening of the TSH receptor
(TSHR) and thyroid peroxidase (TPO) genes in Korean patients with
nonsyndromic congenital hypothyroidism. Clin Endocrinol (Oxf).
75:715–721. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Löf C, Patyra K, Kuulasmaa T, Vangipurapu
J, Undeutsch H, Jaeschke H, Pajunen T, Kero A, Krude H, Biebermann
H, et al: Detection of novel gene variants associated with
congenital hypothyroidism in a finnish patient cohort. Thyroid.
26:1215–1224. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
De Filippis T, Gelmini G, Paraboschi E,
Vigone MC, Di Frenna M, Marelli F, Bonomi M, Cassio A, Larizza D,
Moro M, et al: A frequent oligogenic involvement in congenital
hypothyroidism. Hum Mol Genet. 26:2507–2514. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fu C, Zhang S, Su J, Luo S, Zheng H, Wang
J, Qin H, Chen Y, Shen Y, Hu X, et al: Mutation screening of DUOX2
in Chinese patients with congenital hypothyroidism. J Endocrinol
Invest. 38:1219–1224. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jiang H, Wu J, Ke S, Hu Y, Fei A, Zhen Y,
Yu J and Zhu K: High prevalence of DUOX2 gene mutations among
children with congenital hypothyroidism in central China. Eur J Med
Genet. 59:526–531. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB,
Han B, Zhang LL, Wan YY, Ye XP, Ma YR, et al: The genetic
characteristics of congenital hypothyroidism in China by
comprehensive screening of 21 candidate genes. Eur J Endocrinol.
178:623–633. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Maruo Y, Takahashi H, Soeda I, Nishikura
N, Matsui K, Ota Y, Mimura Y, Mori A, Sato H and Takeuchi Y:
Transient congenital hypothyroidism caused by biallelic mutations
of the dual oxidase 2 gene in Japanese patients detected by a
neonatal screening program. J Clin Endocrinol Metab. 93:4261–4267.
2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ohye H, Fukata S, Hishinuma A, Kudo T,
Nishihara E, Ito M, Kubota S, Amino N, Ieiri T, Kuma K and Miyauchi
A: A novel homozygous missense mutation of the dual oxidase 2
(DUOX2) gene in an adult patient with large goiter. Thyroid.
18:561–566. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen X, Kong X, Zhu J, Zhang T, Li Y, Ding
G and Wang H: Mutational spectrum analysis of seven genes
associated with thyroid dyshormonogenesis. Int J Endocrinol.
2018(8986475)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Maruo Y, Nagasaki K, Matsui K, Mimura Y,
Mori A, Fukami M and Takeuchi Y: Natural course of congenital
hypothyroidism by dual oxidase 2 mutations from the neonatal period
through puberty. Eur J Endocrinol. 174:453–463. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang
L, Liu S and Yan S: Genotypes and phenotypes of congenital goitre
and hypothyroidism caused by mutations in dual oxidase 2 genes.
Clin Endocrinol (Oxf). 81:452–457. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Moreno JC, Bikker H, Kempers MJ, van
Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T and Ris-Stalpers C:
Inactivating mutations in the gene for thyroid oxidase 2 (THOX2)
and congenital hypothyroidism. N Engl J Med. 347:95–102.
2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang F, Zang Y, Li M, Liu W, Wang Y, Yu X,
Li H, Wang F and Liu S: DUOX2 and DUOXA2 variants confer
susceptibility to thyroid dysgenesis and gland-in-situ with
congenital hypothyroidism. Front Endocrinol (Lausanne).
11(237)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tan M, Huang Y, Jiang X, Li P, Tang C, Jia
X, Chen Q, Chen W, Sheng H, Feng Y, et al: The prevalence,
clinical, and molecular characteristics of congenital
hypothyroidism caused by DUOX2 mutations: A population-based cohort
study in Guangzhou. Horm Metab Res. 48:581–588. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Muzza M, Rabbiosi S, Vigone MC, Zamproni
I, Cirello V, Maffini MA, Maruca K, Schoenmakers N, Beccaria L,
Gallo F, et al: The clinical and molecular characterization of
patients with dyshormonogenic congenital hypothyroidism reveals
specific diagnostic clues for DUOX2 defects. J Clin Endocrinol
Metab. 99:E544–E553. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang F, Xiaole L, Ma R, Zhao D and Liu S:
Dual oxidase system genes defects in children with congenital
hypothyroidism. Endocrinology. 162(bqab043)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ye Z, Huang Y, Zheng C, Wang Y, Lu J, Wang
H, Wu B, Wang X, Zhang R and Wang J: Clinical and genetic spectrum
of children with congenital diarrhea and enteropathy in China.
Genet Med. 21:2224–2230. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Fu C, Wang J, Luo S, Yang Q, Li Q, Zheng
H, Hu X, Su J, Zhang S, Chen R, et al: Next-generation sequencing
analysis of TSHR in 384 Chinese subclinical congenital
hypothyroidism (CH) and CH patients. Clin Chim Acta. 462:127–132.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Park KJ, Park HK, Kim YJ, Lee KR, Park JH,
Park JH, Park HD, Lee SY and Kim JW: DUOX2 mutations are frequently
associated with congenital hypothyroidism in the Korean population.
Ann Lab Med. 36:145–153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zamproni I, Grasberger H, Cortinovis F,
Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S,
Persani L and Weber G: Biallelic inactivation of the dual oxidase
maturation factor 2 (DUOXA2) gene as a novel cause of congenital
hypothyroidism. J Clin Endocrinol Metab. 93:605–610.
2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yi RH, Zhu WB, Yang LY, Lan L, Chen Y,
Zhou JF, Wang J and Su YQ: A novel dual oxidase maturation factor 2
gene mutation for congenital hypothyroidism. Int J Mol Med.
31:467–470. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Awata T, Yamashita H, Kurihara S,
Morita-Ohkubo T, Miyashita Y, Katayama S, Kawasaki E, Tanaka S,
Ikegami H, Maruyama T, et al: A low-frequency GLIS3 variant
associated with resistance to Japanese type 1 diabetes. Biochem
Biophys Res Commun. 437:521–525. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lu YC, Huang LY, Yan JM, Zhang Y and Li
DZ: Whole-exome sequencing identifies compound heterozygous LHX4
mutations in a fetus with early-onset growth restriction. Eur J
Obstet Gynecol Reprod Biol. 211:225–227. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li Q, Zhu QW, Yuan YY, Huang SS, Han DY,
Huang DL and Dai P: Identification of SLC26A4 c.919-2A>G
compound heterozygosity in hearing-impaired patients to improve
genetic counseling. J Transl Med. 10(225)2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Huang S, Han D, Yuan Y, Wang G, Kang D,
Zhang X, Yan X, Meng X, Dong M and Dai P: Extremely discrepant
mutation spectrum of SLC26A4 between Chinese patients with isolated
Mondini deformity and enlarged vestibular aqueduct. J Transl Med.
9(167)2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz
S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, et al: Origins and
frequencies of SLC26A4 (PDS) mutations in east and south Asians:
global implications for the epidemiology of deafness. J Med Genet.
40:242–248. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hu H, Wu L, Feng Y, Pan Q, Long Z, Li J,
Dai H, Xia K, Liang D, Niikawa N and Xia J: Molecular analysis of
hearing loss associated with enlarged vestibular aqueduct in the
mainland Chinese: A unique SLC26A4 mutation spectrum. J Hum Genet.
52:492–497. 2007.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yang R, Lu Y, Yang C, Wu X, Feng J, Zhu L,
Shu Q and Jiang P: Case report: Expanding the digenic variants
involved in thyroid hormone synthesis-10 new cases of congenital
hypothyroidism and a literature review. Front Genet.
12(694683)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Niu DM, Hwang B, Chu YK, Liao CJ, Wang PL
and Lin CY: High prevalence of a novel mutation (2268 insT) of the
thyroid peroxidase gene in Taiwanese patients with total iodide
organification defect, and evidence for a founder effect. J Clin
Endocrinol Metab. 87:4208–4212. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Makretskaya N, Bezlepkina O, Kolodkina A,
Kiyaev A, Vasilyev EV, Petrov V, Kalinenkova S, Malievsky O, Dedov
II and Tiulpakov A: High frequency of mutations in
‘dyshormonogenesis genes’ in severe congenital hypothyroidism. PLoS
One. 13(e0204323)2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhang RJ, Sun F, Chen F, Fang Y, Yan CY,
Zhang CR, Ying YX, Wang Z, Zhang CX, Wu FY, et al: The TPO mutation
screening and genotype-phenotype analysis in 230 Chinese patients
with congenital hypothyroidism. Mol Cell Endocrinol.
506(110761)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wang H, Kong X, Pei Y, Cui X, Zhu Y, He Z,
Wang Y, Zhang L, Zhuo L, Chen C and Yan X: Mutation spectrum
analysis of 29 causative genes in 43 Chinese patients with
congenital hypothyroidism. Mol Med Rep. 22:297–309. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Narumi S, Muroya K, Abe Y, Yasui M,
Asakura Y, Adachi M and Hasegawa T: TSHR mutations as a cause of
congenital hypothyroidism in Japan: A population-based genetic
epidemiology study. J Clin Endocrinol Metab. 94:1317–1323.
2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Long W, Lu G, Zhou W, Yang Y, Zhang B,
Zhou H, Jiang L and Yu B: Targeted next-generation sequencing of
thirteen causative genes in Chinese patients with congenital
hypothyroidism. Endocr J. 65:1019–1028. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Nagashima T, Murakami M, Onigata K,
Morimura T, Nagashima K, Mori M and Morikawa A: Novel inactivating
missense mutations in the thyrotropin receptor gene in Japanese
children with resistance to thyrotropin. Thyroid. 11:551–559.
2001.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jin HY, Heo SH, Kim YM, Kim GH, Choi JH,
Lee BH and Yoo HW: High frequency of DUOX2 mutations in transient
or permanent congenital hypothyroidism with eutopic thyroid glands.
Horm Res Paediatr. 82:252–260. 2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Targovnik HM, Citterio CE and Rivolta CM:
Iodide handling disorders (NIS, TPO, TG, IYD). Best Pract Res Clin
Endocrinol Metab. 31:195–212. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Tanase-Nakao K, Iwahashi-Odano M, Sugisawa
C, Abe K, Muroya K, Yamamoto Y, Kawada Y, Mushimoto Y, Ohkubo K,
Kinjo S, et al: Genotype-phenotype correlations in 30 Japanese
patients with congenital hypothyroidism attributable to TG defects.
J Clin Endocrinol Metab. 109:2358–2365. 2024.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Hu X, Chen R, Fu C, Fan X, Wang J, Qian J,
Yi S, Li C, Luo J, Su J, et al: Thyroglobulin gene mutations in
Chinese patients with congenital hypothyroidism. Mol Cell
Endocrinol. 423:60–66. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zou M, Alzahrani AS, Al-Odaib A, Alqahtani
MA, Babiker O, Al-Rijjal RA, BinEssa HA, Kattan WE, Al-Enezi AF, Al
Qarni A, et al: Molecular analysis of congenital hypothyroidism in
Saudi Arabia: SLC26A7 mutation is a novel defect in thyroid
dyshormonogenesis. J Clin Endocrinol Metab. 103:1889–1898.
2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Zhang RJ, Yang GL, Cheng F, Sun F, Fang Y,
Zhang CX, Wang Z, Wu FY, Zhang JX, Zhao SX, et al: The mutation
screening in candidate genes related to thyroid dysgenesis by
targeted next-generation sequencing panel in the Chinese congenital
hypothyroidism. Clin Endocrinol (Oxf). 96:617–626. 2022.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Avbelj M, Tahirovic H, Debeljak M,
Kusekova M, Toromanovic A, Krzisnik C and Battelino T: High
prevalence of thyroid peroxidase gene mutations in patients with
thyroid dyshormonogenesis. Eur J Endocrinol. 156:511–519.
2007.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Lee CC, Harun F, Jalaludin MY, Heh CH,
Othman R and Junit SM: Prevalence of c.2268dup and detection of two
novel alterations, c.670_672del and c.1186C>T, in the TPO gene
in a cohort of Malaysian-Chinese with thyroid dyshormonogenesis.
BMJ Open. 5(e006121)2015.PubMed/NCBI View Article : Google Scholar
|














