Introduction
Non-small cell lung cancer (NSCLC) is a highly
aggressive disease with lung adenocarcinoma (LUAD), the most
diagnosed histological subtype of NSCLC, having a 5-year patient
survival rate of only 15%. Recent molecular advances in tumor
biology have identified epidermal growth factor receptor (EGFR) to
be highly expressed and/or mutated in NSCLC, and EGFR inhibition
[for example, tyrosine kinase inhibitors (TKIs)] has shown promise
in the treatment of patients with LUAD (1). Gefitinib, a first-generation EGFR-TKI,
shows effective antitumor activity in patients with EGFR-mutant
LUAD as compared with chemotherapy. Despite their initial response,
numerous EGFR-TKI-treated patients eventually acquire resistance.
EGFR-TKI resistance mechanisms include secondary EGFR mutations
(for example, T790M), bypass signaling activations (for example,
MET amplification) and phenotypic transformation [for example,
epithelial-mesenchymal transition (EMT)] (2). In addition, some patients with EGFR-TK
mutations do not respond to EGFR-TKIs (intrinsic resistance) while
others with wild type EGFR respond to this drug (3). Hence, it is likely that other
molecular factors beyond EGFR mutations determine the sensitivity
of lung cancer cells to EGFR-TKIs.
A major antitumor effect of EGFR targeted therapy is
through induction of apoptosis (4).
To date, the molecular mechanisms underlying EGFR-TKI-induced
apoptosis have not been fully elucidated. In addition to induction
and/or activation of the intrinsic mitochondrial pro-apoptotic
Bcl-2 family members such as BIM, downregulation of anti-apoptosis
effectors (for example, survivin) has been shown to promote
gefitinib sensitivity (5,6). While key survival signaling pathways
impacted by EGFR-TKIs are yet to be identified, very little
information is currently available on the effects of EGFR-TKIs on
gene transcription and epigenetic machineries. Undoubtedly,
EGFR-TKIs may alter transcriptional programs and induce
reprogramming to exert antitumor activity. Molecular
characterization of the transcriptional and epigenetic machineries
regulated by EGFR-TKIs will yield novel targets to potentiate the
antitumor effect of EGFR inhibition therapy.
In addition to genetic mutations, aberrant function
of epigenetic regulators contributes to EGFR-TKI resistance.
Dysfunction of epigenetic regulators in the form of transcriptional
coactivators and corepressors may result in altered DNA and histone
proteins through recruitment of chromatin remodelling enzymes to
target gene promoter and in transcriptional profiles that render
cancer cells resistant to targeted therapy. Importantly, with the
ability to impact multiple gene regulatory and cell signaling
pathways due to their interaction with diverse transcription
factors (TFs), transcription coregulators represent viable
molecular targets to circumvent drug resistance.
Transducin-like enhancer of split 1 (TLE1) is a
transcriptional corepressor that exerts a lung specific oncogenic
function. In a transgenic mouse model, overexpression of Grg1 gene
(the mouse homologue of TLE1) resulted in lung tumors that resemble
human lung adenocarcinoma (7).
Aligning with this in vivo data, our laboratory has
previously shown that the transcriptional corepressor TLE1
regulates a survival- and an EMT-promoting gene expression programs
in LUAD cells to promote anoikis resistance and
anchorage-independent growth in vitro and tumorigenicity
in vivo (8,9). The anti-apoptosis and pro-EMT function
of TLE1 in part involves inhibiting the tumor suppressive
Bcl-2-inhibitor of transcription 1 (Bit1) cell death pathway.
Despite its known function in orchestrating anti-apoptotic and EMT
transcriptional programs which are key determinants of drug
resistance, TLE1 effects on anticancer drug resistance particularly
in LUAD remain unknown. As a ‘master’ regulator of lung cancer cell
apoptosis resistance and EMT phenotype, it is hypothesized that
inhibiting the TLE1 nuclear function can block its oncogenic
function and its potential antagonistic effects on molecular
targeted therapy including EGFR-TKI. In the present study, it was
identified that TLE1 reduces gefitinib-induced growth inhibition
and apoptosis in LUAD A549 cells in part via downregulation of
E-cadherin, and TLE1 expression is upregulated in the
experimentally generated gefitinib-resistant A549 (A549GR) cell
line to promote EMT and gefitinib insensitivity.
Materials and methods
Cell culture and transduction
assays
The human LUAD cell line A549 (cat. no. CCL-185) and
HCC827 (cat. no. CRL 2868) were obtained from the American Type
Culture Collection. A549 was cultured in Dulbecco's modified
Eagle's medium (DMEM) with glutamine containing 10% fetal bovine
serum (FBS) and 1% penicillin-streptomycin. HCC827 was cultured in
RPMI-1640 with 10% FBS and penicillin-streptomycin (Gibco; Thermo
Fisher Scientific, Inc.). To express exogenous TLE1 in A549 cell
line, parental cells were transduced with lentiviral GFP-TLE1 or
the empty control GFP construct (Horizon Discovery) (8,9).
Briefly, the lentiviral products were produced using the
second-generation system by transfecting the 293T cell line with 1
µg of lentiviral plasmid, 1.2 µg of packaging plasmid, and 6.6 µl
of Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) in 250 µl OPTI-MEM (Invitrogen; Thermo Fisher
Scientific, Inc.) and incubated at 37˚C humidified incubator with
5% CO2. Following a 16-h incubation, culture medium was
harvested and clarified via centrifugation at 500 x g at 5 min
followed by 0.45-µm filtration. Lentivirus-containing culture
medium was used immediately, or stored at 4˚C. Lentiviral
transduction of the parental A549 cell line was performed in a
24-well plate using a 0.5 multiplicity of infection followed by
incubation at 37˚C in a humidified incubator with 5% CO2
for 48 h prior to selection and subsequent experiments. Two control
GFP clones and three distinct exogenous TLE1-expressing GFP-TLE1
clones were pooled together to generate the control GFP and
GFP-TLE1 pools, respectively. Meanwhile, to generate the stable
A549 TLE1 shRNA and control shRNA cells, the parental A549 cell
line was transfected with 0.5 µg of the control short hairpin RNA
(shRNA) or TLE1-specific shRNA construct (OriGene Technologies,
Inc.) cells in OPTI-MEM (Invitrogen; Thermo Fisher Scientific,
Inc.) using Lipofectamine 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and cultured at 37˚C humidified
incubator with 5% CO2. A total of 24 h
post-transfection, transfected cells were treated with 1 µg/ml
puromycin (Invitrogen; Thermo Fisher Scientific, Inc.) to select
for stable clones. The TLE1 shRNA is sense,
5'-GGAATGTGAGAAACTGGCAAGTGAA-3' and antisense,
5'-UUCACUUGCCAGUUUCUCACAUUCC-3'; and the control shRNA with sense,
5'-UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3' does not target any annotated human
genes. To restore TLE1 expression, TLE1 shRNA cells were
transfected with a TLE1 plasmid containing silent mutations in the
region targeted by the TLE1 shRNA. Lastly, the gefitinib-resistant
A549 (A549GR) cell line was established using a previous protocol
(10), wherein the parental A549
cells were continuously exposed to a concentration of 20 µM
gefitinib.
Chemical reagents, antibodies and
plasmids
The Cell Fractionation Kit (cat. no. ab109719) was
purchased from Abcam. The anti-COX IV (1:1,000; cat. no. 4850T) was
purchased from Cell Signaling Technology, Inc. The mouse monoclonal
anti-myc (1:1,000; cat. no. MAI-980) was from Invitrogen; Thermo
Fisher Scientific, Inc. The anti-E-cadherin (1:1,000; cat. no.
610181) and anti-vimentin (1:1,000; cat. no. 550513) were acquired
from BD Biosciences while anti-GFP (1:500; cat. no. sc-53882),
anti-β-actin (1:1,000; cat. no. sc-81178), anti-GAPDH (1:1,000;
cat. no. sc-47724) and anti-TLE1(1:200; cat. no. sc-137098)
antibodies were obtained from Santa Cruz Biotechnology, Inc. The
anti-AES antibody (1:1,000; cat. no. PA5-121149) was purchased from
Thermo Fisher Scientific, Inc. The EGFR-TKI gefitinib and z-VAD-fmk
were purchased from Selleck Chemicals. The Bit1-myc tagged
construct which encodes for the mitochondrial localized Bit1
protein was generated as previously described (8,9). The
GFP-TLE1 and the full-length E-cadherin encoding plasmids were
obtained from Origene Technologies, Inc.
Small interfering RNA (siRNA) and
plasmid transfection
For acute knockdown studies, control non-targeting
siRNA or pool of siRNAs specifically targeting TLE1 (Santa Cruz
Biotechnology, Inc.) or AES (Invitrogen; Thermo Fisher Scientific,
Inc.) were transfected into A549 cells (2x105) using the
Lipofectamine RNAiMAX transfection reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and incubated at 37˚C in a humidified
incubator with 5% CO2 for 24 h followed by subsequent
experimentation (8,9). The TLE1 siRNA pool consisted of 3
different siRNA duplexes: TLE1 siRNA1 sense,
5'-GGACCGGAUUAAAGAGGAATT-3' and antisense,
5'-UUCCUCUUUAAUCCGGUCCTT-3'; TLE1 siRNA2 sense,
5'-GGCACUAUGUGAUGUAUUATT-3' and antisense,
5'-UAAUACAUCACAUAGUGCCTT-3'; and TLE1 siRNA3 sense,
5'-GAAGGCUACAGUCUAUGAATT-3' and antisense,
5'-UUCAUAGACUGUAGCCUUCTT-3'. The AES siRNA pool consisted of two
different siRNA duplexes: AES siRNA1 sense,
5'-CAAAGACGAAUUUCAGCUATT-3' and antisense,
5'-GAACAUCGAGAUGCACAAATT-3'; and AES siRNA2 sense,
5'-GAACAUCGAGAUGCACAAATT-3' and antisense,
5'-UUUGUGCAUCUCGAUGUUCAA-3'. For siRNA experiments, the negative
control siRNA with no homology to any known human genes is sense,
5'-UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3'. For plasmid DNA constructs, transient
transfection assays were conducted using lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for A549 cells in
OPTI-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) as prescribed
by the manufacturer with the total amount of plasmid used
normalized with the corresponding empty vector construct.
Cell viability, apoptosis and
migration assays
Cells were treated with various concentrations of
gefitinib (0-5 µM) or osimertinib (0-1 µm) for 48 h and subjected
to the metabolic activity-based Alamar Blue assay to assess cell
viability as previously described (8,9).
Briefly, the number of metabolically active cells was measured
using the PrestoBlue Cell Viability Reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) with fluorescence reading at 485 nm
excitation wavelength and 520 nm emission wavelength in a
microplate reader. In parallel, cells treated with various
concentrations of gefitinib (0-5 µM) or osimertinib (0-1 µm) for 48
h were subjected to Cell Death Apoptosis ELISA (cat. no.
11774425001; Roche Molecular Diagnostics) to quantify the amount of
DNA histone fragments (8,9). The migratory ability of cells was
quantified with the use of a Boyden chamber cell migration assay as
previously described (8). Briefly,
cells (3x104) were added to the upper chamber of 24-well
plates (BD Falcon) that contained cell culture inserts (8.0-µm
pores); and 10% FBS was added to the lower chamber to serve as a
chemoattractant. After 18 h, cells that migrated through the
membrane and attached on the underside of the membrane were stained
with 0.1% crystal violet at room temperature for 1 h and counted
using a light microscope.
Protein preparation, western blotting
and subcellular fractionation assays
Protein preparation and western blotting were
performed as previously described (8,9).
Protein lysate was prepared using the Mem-PER Plus eukaryotic
membrane protein extraction reagent kit (Thermo Fisher Scientific,
Inc.). For western blot analysis, equal amounts of proteins (35 µg)
were resolved on 4-20% gradient Tris-glycine gels (Invitrogen;
Thermo Fisher Scientific, Inc.) and electrophoretically transferred
to nitrocellulose membrane. The membranes were then incubated with
primary antibodies overnight at 4˚C, followed by incubation with
appropriate secondary antibodies [Amersham ECL Rabbit IgG,
HRP-linked whole Ab (1:20,000; cat. no. NA934V; Cytiva); Amersham
ECL Mouse IgG, HRP-linked whole Ab (1:25,000; cat. no. NA931V;
Cytiva)]. Visualization of protein bands on the membranes was
performed using the ECL detection system (cat. no. RPN2232;
Cytiva), and band intensities were quantified by densitometric
analysis using ImageJ software (National Institutes of Health).
Preparation of the mitochondrial, cytoplasmic and nuclear
containing fractions was conducted using the Cell Fractionation Kit
(Abcam). The protein concentration in different fractions was
measured using the Bio-Rad protein assay kit (Bio-Rad Laboratories,
Inc.) with BSA (cat. no. 23208; Thermo Fisher Scientific, Inc.) as
the standard.
Total RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from 5x106
cultured cells using the Qiagen RNeasy miniprep kit (Qiagen
Sciences, Inc.) as prescribed by the manufacturer and the
quantified by spectrophotometry (NanoDrop 8000; Thermo Fisher
Scientific, Inc.). Total RNA was subjected to a one-step real-time
RT-qPCR using the iTaq Universal SYBR (Bio-Rad Laboratories, Inc.)
by RT-qPCR on the BIO-RAD CFX96 Touch Real-Time PCR Detection
System utilizing the following primers: human E-cadherin forward,
5'-AGGCTAGAGGGTCACCGCGTC-3' and reverse,
5'-GCTTTGCAGTTCCGACGCCAC-3'; and TLE1 forward,
5'-CCTCCTACACAGCAGCAGTT-3' and reverse, 5'-TCTGCATCGTGGTGCTTCTT-3'.
In parallel, human GAPDH forward, 5'-CCCACTCCTCCACCTTTGAC-3' and
reverse, 5'-TTGCTGTAGCCAAATTCGTTGT-3' were used as control. The
thermocycling conditions were as follows: reverse transcription
reaction was 10 min at 50˚C, polymerase activation and DNA
denaturation was 1 min at 95˚C, and then amplification;
denaturation for 10 sec at 95˚C, annealing/extension for 30 sec at
60˚C, and run for 40 cycles. The melt-curve analysis was following
65-95˚C (0.5˚C increment 5 sec/step). The relative levels of mRNAs
were analyzed using the ΔΔCq method (11).
Bioinformatics analysis
The Cancer Genome Atlas TCGA database (https://portal.gdc.cancer.gov/) was analysed to
compare TLE1 mRNA level in LUAD and lung squamous cell carcinoma
(LUSC) vs. normal lung tissues. Kaplan-Meier survival plots were
generated using the R Package survival (https://cran.r-project.org/web/packages/survival/) to
assess the prognostic significance of TLE1 expression. To examine
TLE1 expression in patients with EGFR-TKI resistant and sensitive
LUAD, the Gene Expression Omnibus (GEO) Datasets (http://www.ncbi.nlm.nih.gov/geo/gds), hosted by
the National Center for Biotechnology Information (NCBI), was
employed as the database for dataset retrieval. The search query
used was ‘[(LUAD) AND (EGFR-TKI resistance) OR (gefitinib
resistance) OR (osimertinib resistance)]’, with additional filters
set for the organism as ‘Homo sapiens’ and entry type as either
‘DataSets’ or ‘Series’. The resulting datasets were then screened
to include only samples derived directly from patients, excluding
those from cell lines. Ultimately, dataset GSE231938 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE231938)
was selected, comprising samples from one EGFR-TKI-sensitive
patient and two EGFR-TKI-resistant patients (12). The levels of TLE1 mRNA in these
samples were subsequently analyzed using Geo2R, with expression
levels reported in Transcripts Per Million. To assess possible
enrichment of TF binding motif in the TLE1 promoter region, the EPD
Eukaryotic promoter database was utilized as a source of the human
TLE1 promoter sequence (13). The
TLE1 promoter region (-1,000 to 100 base pair (bp) relative to
transcription start site (TSS) was scanned with a cut-off P-value
of 0.001.
Statistical analysis
Data are presented as the mean ± standard deviation
(SD) of at least three independent experiments. All calculations
were performed using the NCSS statistical software (NCSS, LLC).
Statistical analyses were performed using two-tailed Student's t
test for experiments with two groups and one-way ANOVA with Tukey's
post hoc test for comparisons among multiple groups.
*P<0.05 was considered to indicate a statistically
significant difference.
Results
TLE1 expression is upregulated and
functions as a poor prognosis factor in patients with LUAD
To determine clinical significance of TLE1 in NSCLC,
TLE1 mRNA level in LUAD and LUSC (two major NSCLC subtypes) was
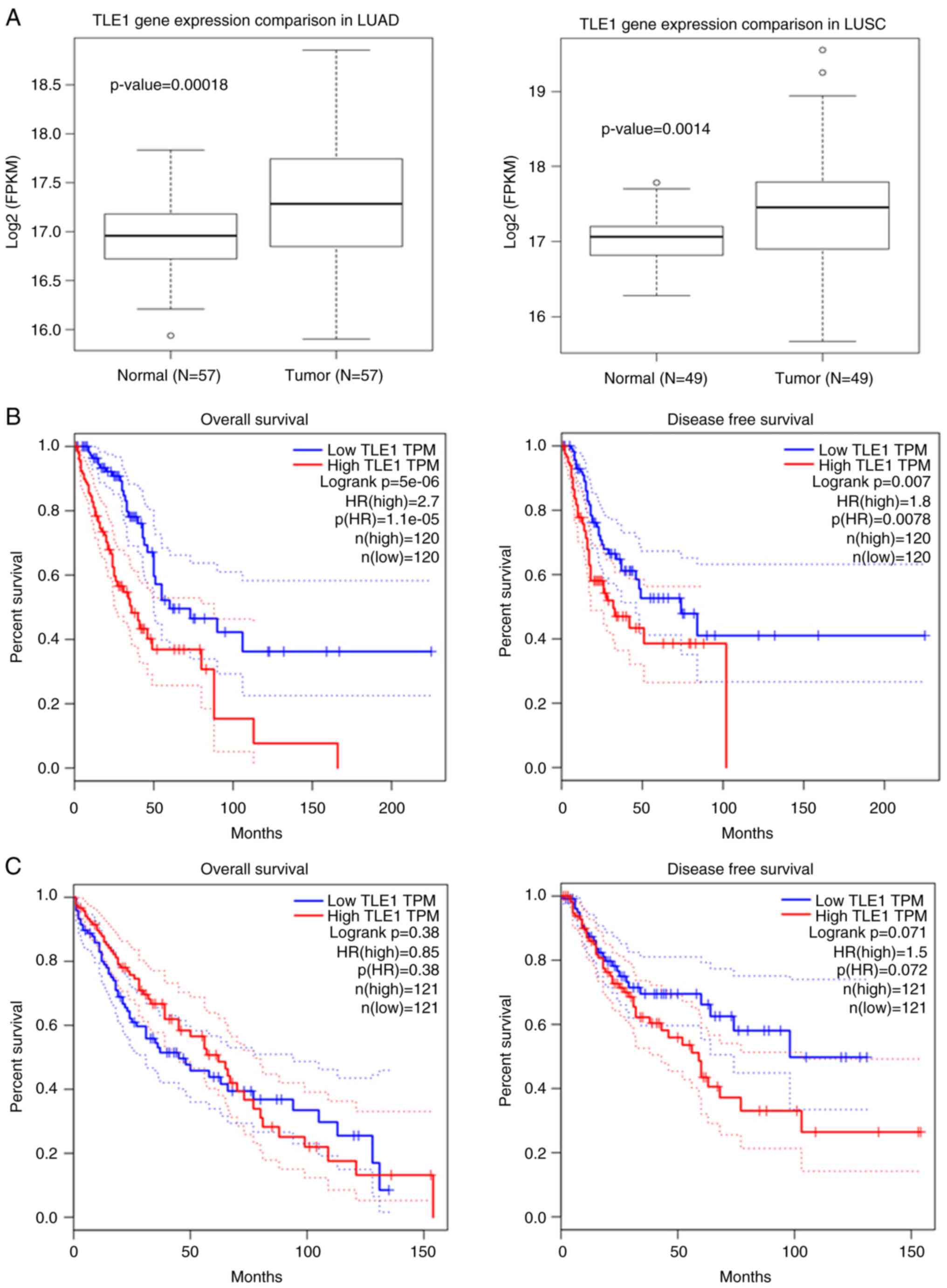
examined using the TCGA database. As shown in Fig. 1A, TLE1 expression was elevated in
both LUAD and LUSC as compared with normal counterparts.
Importantly, high TLE1 expression strongly associated with shorter
overall survival (OS) and disease-free survival in patients with
LUAD (Fig. 1B). As shown in
Fig. 1C, high TLE1 expression
failed to correlate with LUSC patient survival rates. Due to
crossing over of the OS curves in Fig.
1C, the period of analysis was restricted to exclude this
late-stage crossover event. Reanalysis failed to show statistically
significant difference in the survival rate between the high TLE1
expressing and low TLE1 expressing LUSC groups (Fig. S1B). These data are consistent with
TLE1 corepressor functioning as a molecular determinant of LUAD
aggressiveness and further indicate that TLE1 expression may serve
as a poor prognosis factor in patients with LUAD.
To assess the potential role of TLE1 in EGFR-TKI
resistance, one available GEO dataset GSE231938 was also examined
and the level of TLE1 mRNA was compared between EGFR-TKI sensitive
and resistant LUAD tumor samples. The two patients exhibiting
resistance to EGFR-TKI therapy demonstrated elevated levels of TLE1
compared with the patient who is sensitive to EGFR-TKI treatment
(Fig. S1A). However, due to the
limited sample size, statistical significance could not be
determined.
TLE1 modulates the sensitivity of A549
cells to EGFR-TKI gefitinib
In addition to tumor invasiveness, chemoresistance
is another hallmark of cancer aggressiveness. To address the
possibility that TLE1 may regulate drug resistance, the impact of
TLE1 expression on molecular targeting therapy against EGFR was
examined in LUAD. Since EMT is a known mechanism for EGFR-TKI
resistance in the human LUAD A459 cells (14) which were previously shown to develop
EMT phenotype upon exogenous TLE1 expression (8,9),
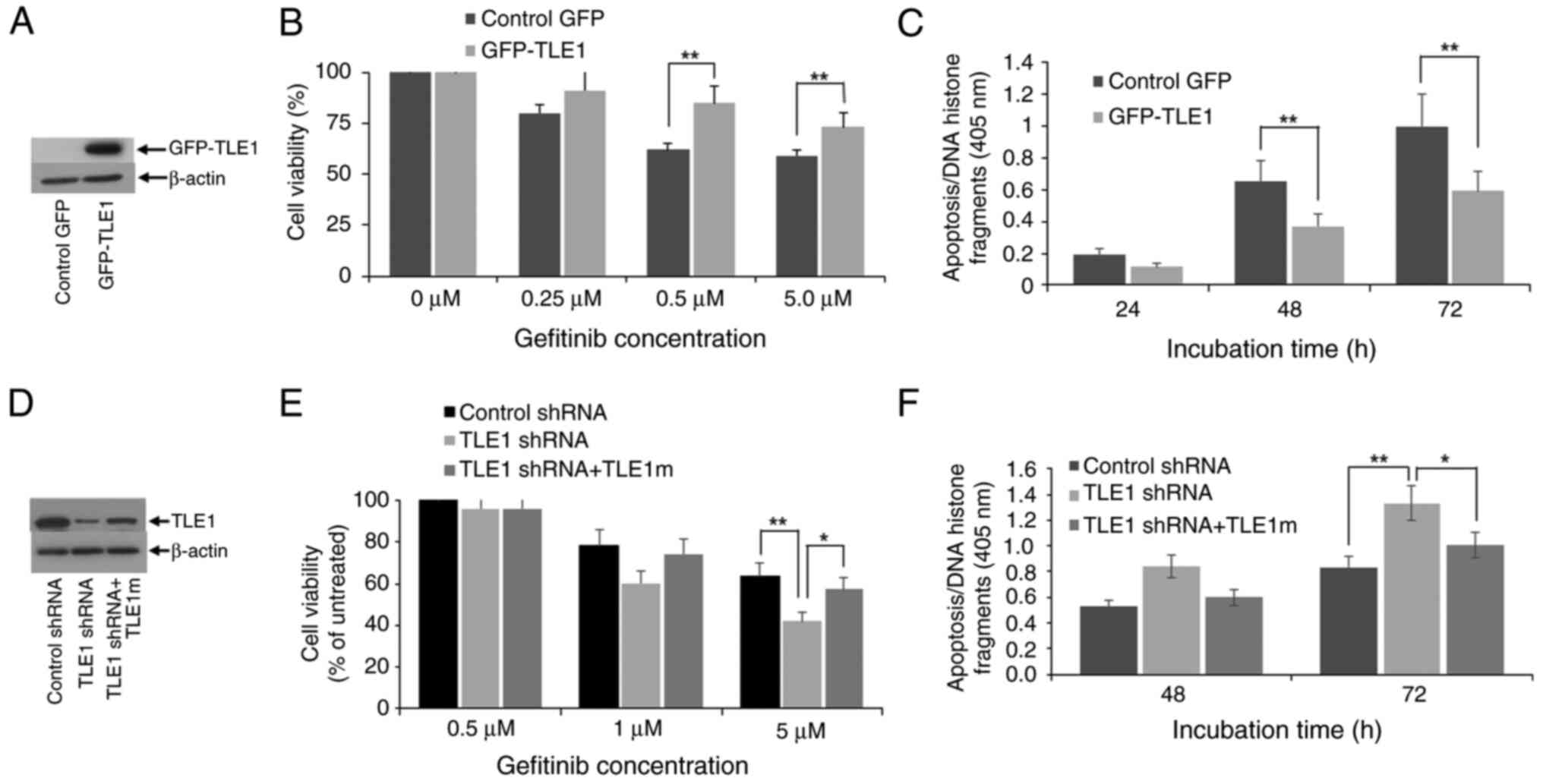
stable exogenous GFP-TLE1-expressing and control GFP A549 clonal
pool of cells (Fig. 2A) were
treated with the EGFR-TKI gefitinib and their proliferation
(Fig. 2B) and basal apoptosis
(Fig. 2C) were assessed. Treatment
of control GFP A549 cells with gefitinib resulted in proliferation
inhibition and apoptosis induction, indicating sensitivity of A549
cells to gefitinib as previously reported (10,15).
As compared with control GFP cells, the GFP-TLE1 A549 cells showed
enhanced proliferation and reduced apoptosis following gefitinib
treatment, signifying that exogenous TLE1 expression conferred
gefitinib resistance in A549 cells.
To confirm the specificity of ectopic TLE1 effects
on the antitumor activity of gefitinib, endogenous TLE1 expression
in A549 cells, which exhibit moderate levels of TLE1, was
downregulated via the shRNA technology (Fig. 2D). Following gefitinib treatment,
TLE1 shRNA cells exhibited greater proliferation inhibition and
apoptosis as compared with control shRNA cells (Fig. 2E and F). The enhanced gefitinib sensitivity of
TLE1 shRNA cells was lost upon restoration of TLE1 expression with
a TLE1 plasmid containing silent mutations in the shRNA target
sequence (TLE1m) (Fig. 2D-F).
Collectively, these findings indicated that TLE1 expression
attenuates the proliferation-inhibitory and apoptosis-inducing
effects of gefitinib in the A549 cell line, and its upregulation
may contribute to gefitinib resistance in these cells. Consistent
with these results, TLE1 expression in A549 cells also conferred
protection against the proliferation-inhibitory and apoptotic
effects of the third generation EGFR-TKI osimertinib (Fig. S2A-D) and attenuated the antitumor
effect of gefitinib in the EGFR-TKI sensitive, EGFR-mutant LUAD
HCC827 cell line (Fig. S3A-C).
TLE1 enhances gefitinib resistance in
A549 cells through E-cadherin repression
As a transcriptional corepressor, TLE1 promotes EMT
in LUAD cells via epigenetic silencing of E-cadherin expression
(8,9) (Fig.
S3A). Since E-cadherin expression correlates with gefitinib
sensitivity in lung cancer (16),
it was investigated if TLE1 confers resistance to gefitinib through
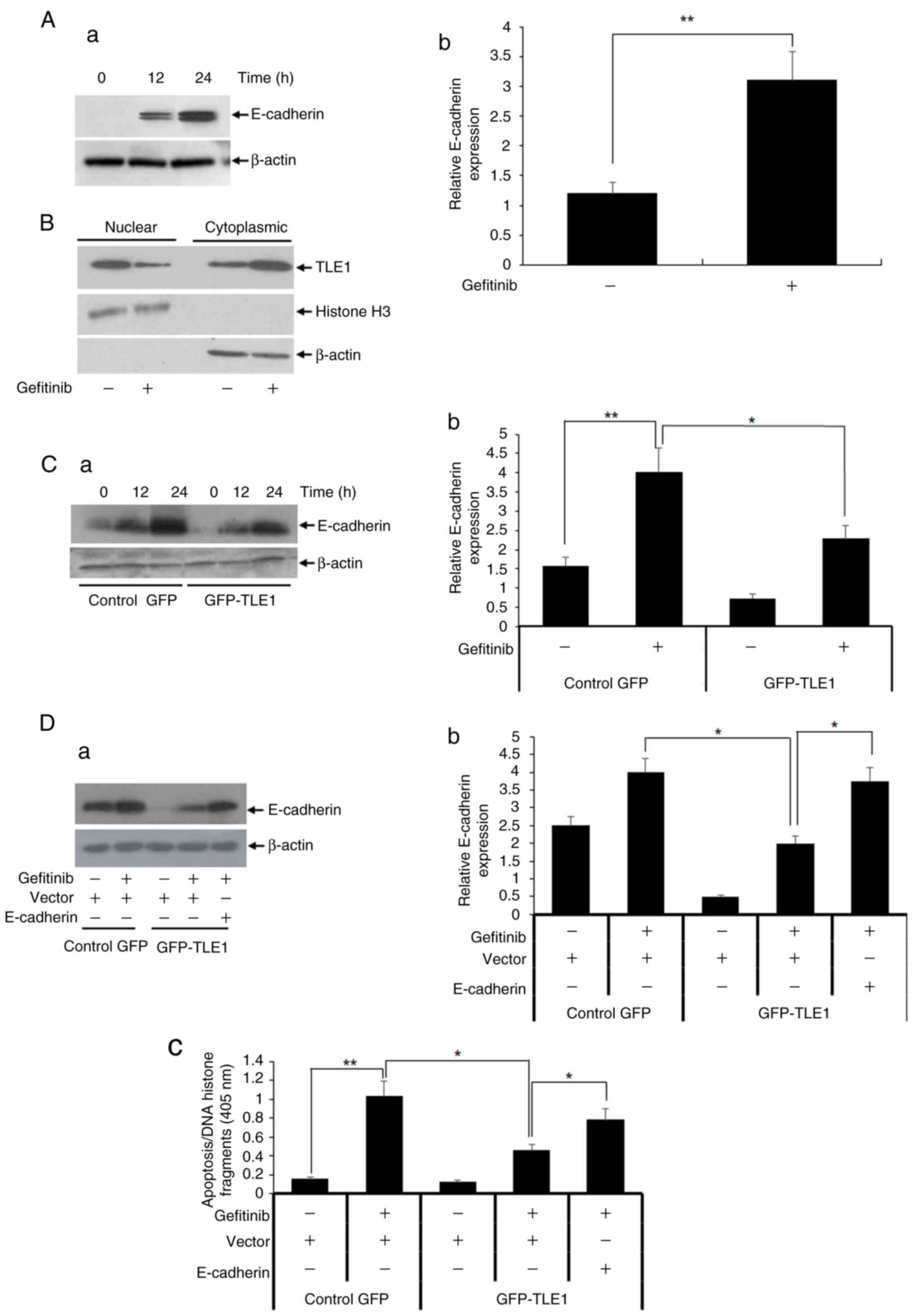
transcriptional silencing of E-cadherin. At first, the levels of
E-cadherin expression in A549 cells in the presence or absence of
gefitinib were examined. As demonstrated in Fig. 3A, gefitinib treatment upregulated
E-cadherin (a) protein and (b) mRNA expression levels. The
E-cadherin upregulation by gefitinib treatment coincided with a
significant re-localization of the nuclear TLE1 protein to the
cytoplasm as evidenced by subcellular fractionation and western
blotting assays (Fig. 3B),
consistent with the notion that the observed gefitinib-induced
E-cadherin expression is associated with inhibition of TLE1 nuclear
function. Importantly, exogenous TLE1 expression significantly
inhibited the upregulation of E-cadherin by gefitinib (Fig. 3Ca and Cb). These findings indicated that TLE1
serves as a downstream target of gefitinib, and its upregulation
antagonizes the gefitinib-induced E-cadherin expression in A549
cells.
The aforementioned findings raise the possibility
that TLE1 may confer EGFR-TKI resistance through silencing of
E-cadherin. To test directly whether the TLE1-mediated gefitinib
resistance can be attributed to regulation of E-cadherin, forced
expression of E-cadherin in TLE1 expressing A549 cells was
performed followed by gefitinib treatment. The upregulation of
E-cadherin expression in GFP-TLE1 A549 cells was confirmed by
immunoblotting (Fig. 3Da) and
RT-qPCR (Fig. 3Db) assays. As shown
in Fig. 3Dc, ectopic E-cadherin
expression significantly increased the level of gefitinib-induced
apoptosis in exogenous TLE1 expressing A549 cells, indicating
partial restoration of their gefitinib sensitivity. Consistent with
this finding, forced upregulation of E-cadherin in exogenous TLE1
expressing HCC827 cell line which exhibited decreased E-cadherin
expression similarly attenuated the TLE1-mediated gefitinib
resistance (Fig. S4A and B). These data indicated that E-cadherin
repression in part underlies the TLE1-mediated resistance to
gefitinib in lung cancer cells.
Increased TLE1 expression in
gefitinib-resistant A549 (A549GR) cells confers EMT features and
gefitinib resistance
To explore the role of TLE1 in acquired EGFR-TKI
resistance in lung cancer cells, gefitinib-resistant A549 cells
(A549GR) were established from the parental A549 cell line through
a continuous low dose exposure to gefitinib. In line with a
previous study (10), the A549GR
cells exhibited EMT features such as acquisition of increased cell
size, flattened phenotype (Fig.
4Aa), and enhanced cell migration capacity (Fig. 4Ab and Ac) as compared with parental A549 cells.
Molecular changes associated with EMT were also observed in A549GR
cells including decreased E-cadherin and increased vimentin
expression (Fig. 4Ad). Importantly,
the A549GR cells displayed reduced sensitivity to gefitinib-induced
apoptosis relative to parental A549 cells (Fig. 4B).
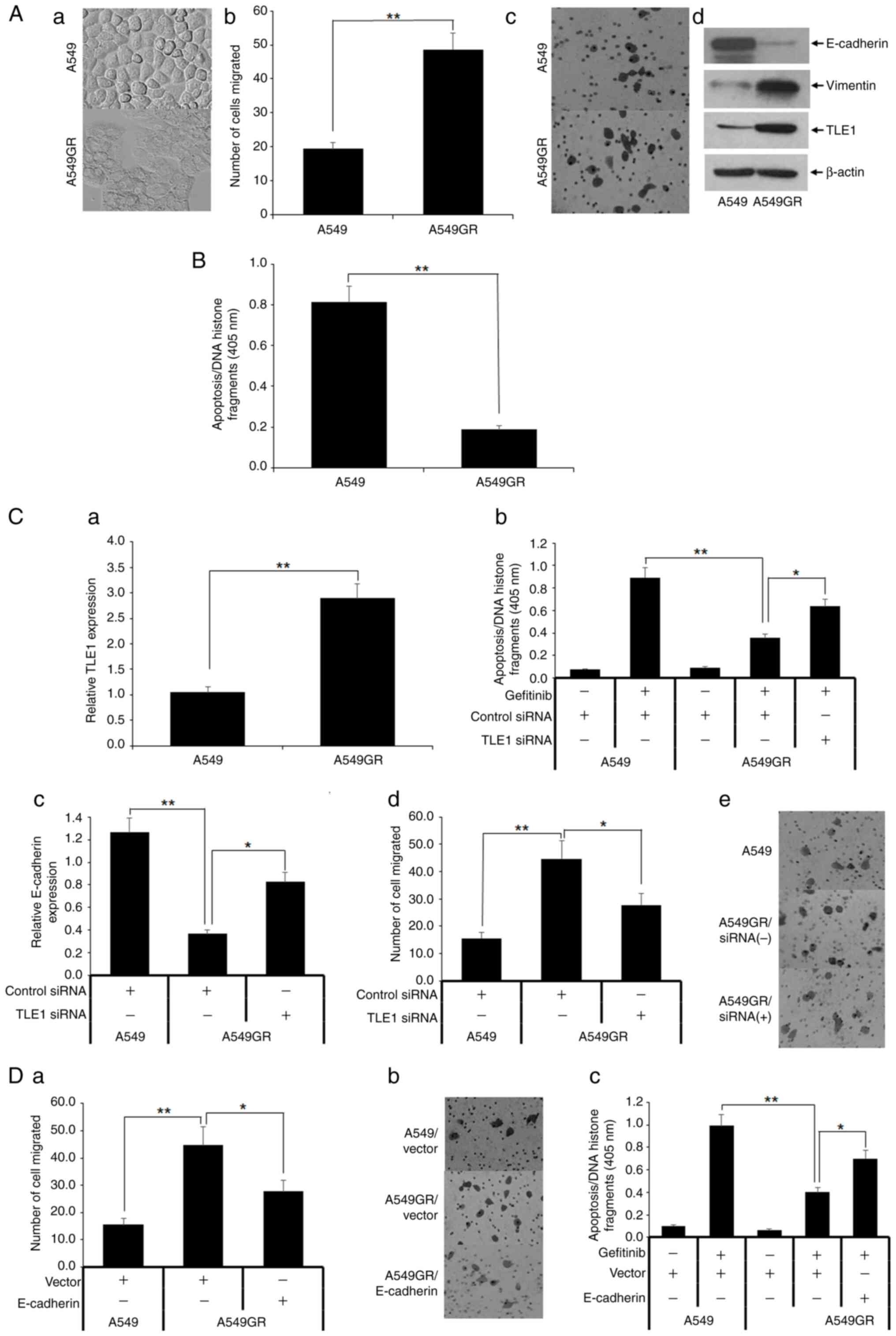 | Figure 4Gefitinib-resistant A549 (A549GR)
cells display EMT and increased TLE1 expression, and knockdown of
TLE1 attenuates the EMT phenotype and gefitinib resistance of
A549GR cells. (A) The parental A549 and gefitinib-resistant A549GR
cells were subjected to (Aa) phase contrast light microscopy to
assess their morphology, (Ab and Ac) Borden chamber assay to
evaluate migration potential, and (Ad) western blotting to measure
protein expression of different EMT markers including TLE1 with
specific antibodies. (B) A549 and A549GR cells were cultured in the
presence or absence of 10 mmol/l gefitinib for 48 h followed by
cell death ELISA apoptosis assay. (C) A549 and A549GR cells were
subjected to (Ca) RT-qPCR analysis to measure TLE1 mRNA expression
level; (Cb) A549GR cells transfected with a pool of TLE1 specific,
or control siRNAs were cultured in the presence of absence of 10
mmol/l gefitinib for 48 h followed by cell death ELISA apoptosis
assay. The control siRNA or TLE1 siRNA transfected A549GR cells
were also subjected to (Cc) RT-qPCR analysis to assess E-cadherin
mRNA level and (Cd and Ce) Boyden chamber migration assay. (D)
A549GR cells transfected with a E-cadherin expressing or vector
construct were subjected to (Da and Db) a Boyden chamber migration
or cultured in the presence of absence of 10 mmol/l gefitinib for
48 h followed by (Dc) cell death ELISA apoptosis assay. In Fig. 4Ab, Ac, Cd,
Ce, Da and Dc,
cells were added to the upper compartment of the Boyden chamber,
and after 12 h, cells attached on the underside of the membrane
were stained with 0.1% crystal violet, counted (4Ab, Cd and Da),
and images were captured (4Ac, Ce, and Db). In the aforementioned
experiments, the results are representative of three independent
experiments. *P<0.05 and **P<0.01
[Student's t test (Ca) and one-way ANOVA with post hoc Tukey's test
(Cb, Cc, Cd, Da and Dc)]. Error bars indicate SD. EMT,
epithelial-mesenchymal transition; TLE1, transducin-like enhancer
of split 1; RT-qPCR, reverse transcription-quantitative PCR; siRNA,
small interfering RNA. |
To investigate the role of TLE1 in acquired
gefitinib resistance of A549 cells, it was first examined if TLE1
expression is altered between A549 and A549 GR cells. Both RT-qPCR
(Fig. 4Ca) and western blotting
(Fig. 4Ac) assays demonstrated
induction of TLE1 expression at both the mRNA and protein levels,
respectively. To gain mechanistic insights on the observed
upregulation of TLE1 in gefitinib resistant cells, the TLE1
promoter region (-1,000 to 100 base pair (bp) relative to TSS,
cut-off P=0.001) was examined for TF binding motifs using the EPD
eukaryotic promoter database (13),
with emphasis on TFs that are associated with EGFR-TKI resistance
in human lung cancer cells. It was found that the TLE1 promoter
region is enriched for binding motif for transcription factors
STAT3 and ZNF263. The transcriptional activator STAT3 has been
shown to be activated upon acquisition of EGFR-TKI resistance
(17,18), while loss of expression of the
transcriptional repressor ZNF263 has been observed in EGFR-TKI
resistant LUAD cells (19). Thus,
the activation of STAT3 and/or downregulation of ZNF263 may
underlie the observed transcriptional upregulation of TLE1
expression in gefitinib-resistant A549GR cells.
TLE1 expression was then downregulated in these
cells via a previously validated pool of TLE1-specific or control
siRNAs (8,9) (Fig.
S5), followed by gefitinib treatment and apoptosis assay. As
demonstrated in Fig. 4Cb, acute
ablation of TLE1 expression in A549GR cells partially restored
their sensitivity to gefitinib-induced apoptosis. Concurrent with
the attenuation of gefitinib resistance, loss of TLE1 expression in
A549GR cells also resulted in induced expression of the epithelial
marker E-cadherin (Fig. 4Cc) with
concomitant inhibition of cell migration (Fig. 4Cd and Ce), indicating that increased TLE1
promotes EMT in A549GR cells.
Since EMT is a known determinant of acquired
EGFR-TKI resistance in lung cancer cells (14,20)
and considering the present findings that E-cadherin repression, a
hallmark of EMT, underlies the TLE1-mediated gefitinib resistance
in A549 cells, a possibility remains that TLE1 may contribute to
gefitinib resistance in A549GR cells via EMT. Hence, it was
examined whether reversing induced EMT in A549GR cells by ectopic
expression of the TLE1 target E-cadherin gene restores their
sensitivity to gefitinib. Forced expression of E-cadherin in A549GR
cells attenuated not only their increased motility (Fig. 4Da and Db) but also gefitinib resistance (Fig. 4Dc). Taken together, these results
indicated that the TLE1-E-cadherin transcriptional axis plays a
role in acquired gefitinib resistance of A549 cells.
The cell death effector Bit1
potentiates gefitinib-induced apoptosis by inhibiting the TLE1
nuclear function in A549 cells
To induce cell death or apoptosis, the mitochondrial
Bit1 protein is released to the cytoplasm and complexes with the
transcriptional regulator Amino Enhancer Split (AES) protein to
turn off the TLE1-mediated survival gene transcriptional program
(21,22). While the mechanistic details
underlying the Bit1 apoptosis function remain to be fully
delineated, the formation of the pro-apoptotic Bit1-AES complex may
channel pre-existing nuclear AES-TLE1 hetero-oligomers to the
cytoplasm and lower nuclear TLE1 level, thus turning off the
survival promoting TLE1 gene transcriptional program (21,22).
To further address the role of TLE1 as an inhibitor of
EGFR-TKI-mediated apoptosis and a determinant of EGFR-TKI
resistance, it was investigated whether Bit1 can potentiate
EGFR-TKI-mediated apoptosis in A549 cells by targeting the TLE1
nuclear function. To test this possibility, Bit1 expression in the
mitochondria of A549 cells was targeted via transfection with a
C-terminally myc-tagged Bit1 (Bit1 mito) or empty vector plasmid
followed by treatment with or without gefitinib. In untreated
conditions, the exogenous C-terminally myc tagged Bit1 protein is
localized primarily in the mitochondria, and gefitinib treatment
resulted in a significant shuttling of the Bit1 mito protein to the
cytoplasm (Fig. 5A). The
cytoplasmic relocalization of Bit1 following gefitinib treatment is
associated with significantly greater gefitinib-mediated
proliferation inhibition and apoptosis (Fig. 5B). Consistent with the role of Bit1
as a caspase-independent apoptotic effector (21), the Bit1 induction of
gefitinib-mediated apoptosis in A549 cells is unresponsive to
pan-caspase inhibitors pretreatment (Fig. 5Bb). Importantly, the observed
potentiation of gefitinib-induced apoptosis in Bit1 transfected
cells was significantly attenuated by knocking down the expression
of AES (the Bit1 pro-apoptotic partner) with the use of a pool of
AES-specific siRNA (Figs. 5Bb and
S5), indicating specificity of
Bit1 effect on gefitinib-induced apoptosis.
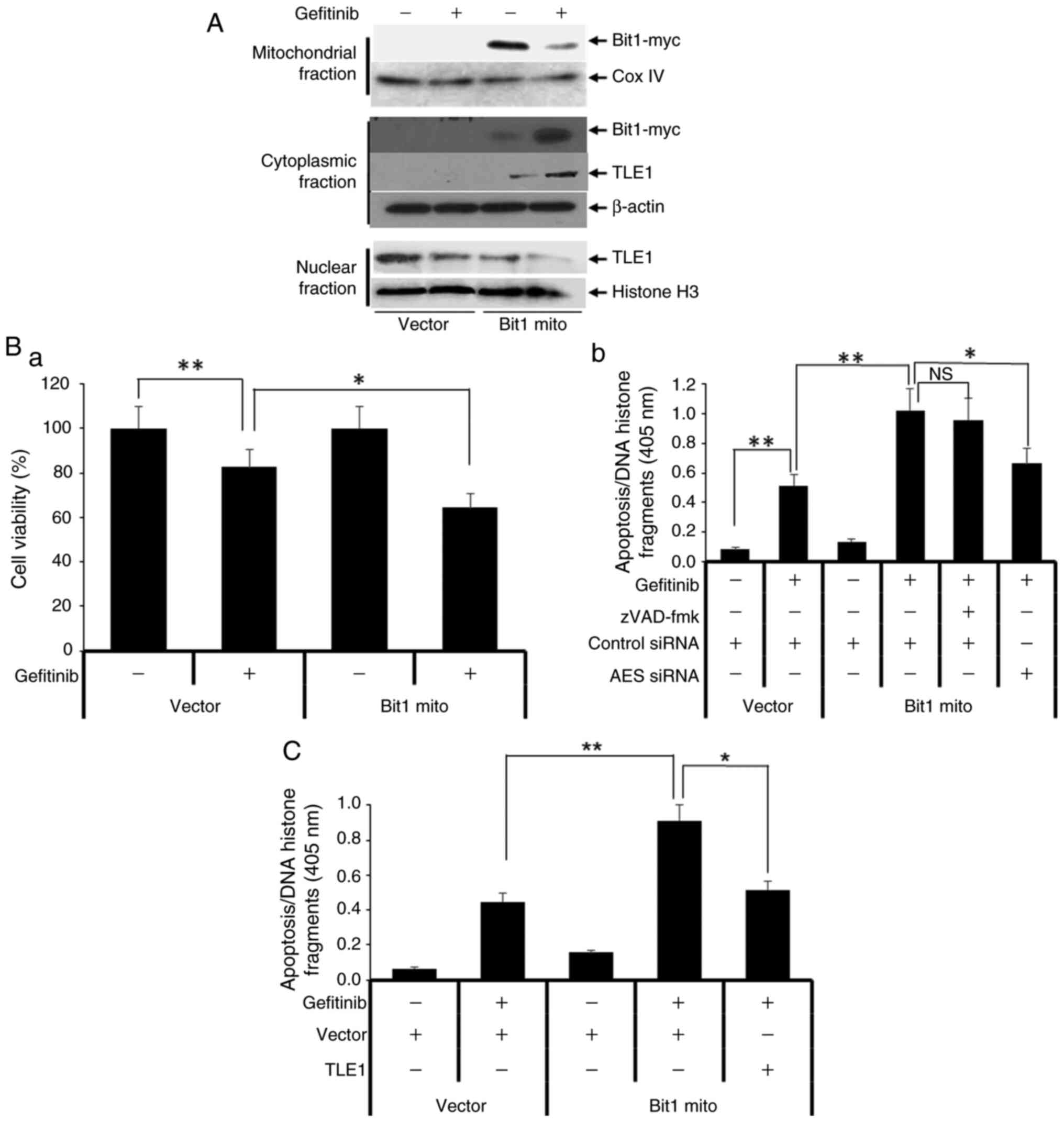 | Figure 5Cell death effector Bit1 enhances
gefitinib-induced apoptosis in A549 cells by targeting nuclear TLE1
protein to the cytoplasm. (A) A549 cells transfected with a
C-terminally myc-tagged mitochondrial Bit1 expressing or vector
construct were cultured in the presence or absence of 10 µmol/l
gefitinib for 16 h followed by Cell Fractionation assay. The
resulting mitochondrial, nuclear, and cytoplasmic fractions were
subjected to western blotting with the indicated antibodies. (B)
A549 cells transfected with a mitochondrial Bit1 expressing or
vector construct were cultured in the presence of absence of 10
mmol/l gefitinib for 48 h followed by (Ba) Alamar blue or (Bb) cell
death ELISA apoptosis assays. In (Bb), mitochondrial Bit1
expressing cells were pretreated with or without 20 mmol/l
Z-VAD-fmk or transfected with a pool of AES specific or control
siRNAs followed by gefitinib treatment and cell death Elisa
apoptosis assay. (C) The mitochondrial Bit1 expressing A549 cells
were transfected with TLE1 expressing or vector construct, and 24 h
post-transfection cells were cultured in the presence or absence of
10 mmol/l gefitinib for 48 h followed by cell death Elisa apoptosis
assay. The results are representative of three independent
experiments. *P<0.05 and **P<0.01
[one-way ANOVA with post hoc Tukey's test (Ba, Bb, C)]. Error bars
indicate SD. Bit1, Bcl-2-inhibitor of transcription 1; TLE1,
transducin-like enhancer of split 1; AES, Amino Enhancer Split;
siRNA, small interfering RNA; ns, not significant. |
To test if TLE1 is a downstream target in the Bit1
regulation of gefitinib-mediated apoptosis, it was examined if Bit1
impinges on the nuclear localization of TLE1. As shown in Fig. 5A, the gefitinib-induced apoptosis in
Bit1 expressing cells was associated with cytoplasmic translocation
of nuclear TLE1, consistent with our previous findings that Bit1
triggers apoptosis by inhibiting TLE1 nuclear function (21,22).
To directly examine the role of TLE1 as a downstream target of Bit1
regulation of gefitinib apoptosis, exogenous TLE1 was expressed in
Bit1 overexpressing cells followed by gefitinib treatment and
apoptosis assay. As revealed in Fig.
5C, forced expression of nuclear TLE1 significantly inhibited
the Bit1 induction of gefitinib-mediated apoptosis in A549 cells.
These collective data indicated that Bit1 augments
gefitinib-induced apoptosis in part by inhibiting the pro-survival
nuclear function of TLE1, thus highlighting TLE1 as a negative
regulator of EGFR-TKI sensitivity.
Discussion
Previous studies have provided evidence in support
of the TLE1 corepressor as an oncogenic driver of NSCLC through
induction of anoikis resistance and EMT in vitro and
tumorigenicity in vivo (7-9).
In the present study, it was demonstrated that TLE1 expression is
upregulated in both human LUAD and LUSC tumors but exerts poor
prognostic function only in LUAD tumors. This finding is consistent
with the in vivo transgenic mice data demonstrating that
overexpression of Grg1 (mouse homologue of TLE1) resulted in lung
tumors that resemble human LUAD (7). These findings highlight the lung
specific oncogenic function of TLE1 in LUAD and underscore the
different biological and genetics signatures of LUAD and LUSC.
In the present study, a novel TLE1 function in
inhibiting gefitinib's antiproliferative and apoptotic effects in
A549 cells was uncovered. Exhibiting moderate sensitivity to
gefitinib, the EGFR wild-type A549 cell line serves as a model
system to investigate molecular factors other than EGFR-TK
mutations that regulate sensitivity of EGFR-TKIs. Importantly, such
cells develop EGFR-TKI resistance via acquisition of EMT phenotype
(10). To the best of our
knowledge, this is the first study to implicate the TLE1
corepressor in drug sensitivity and resistance in human lung
cancer. As illustrated in Fig. 6, a
model is proposed by which inhibition of TLE1 nuclear function and
upregulation of E-cadherin and other TLE1 target genes' expression
contributes to EGFR-TKI-mediated apoptosis, and potentiation of
TLE1/E-cadherin transcriptional silencing axis is a determinant of
EGFR-TKI resistance.
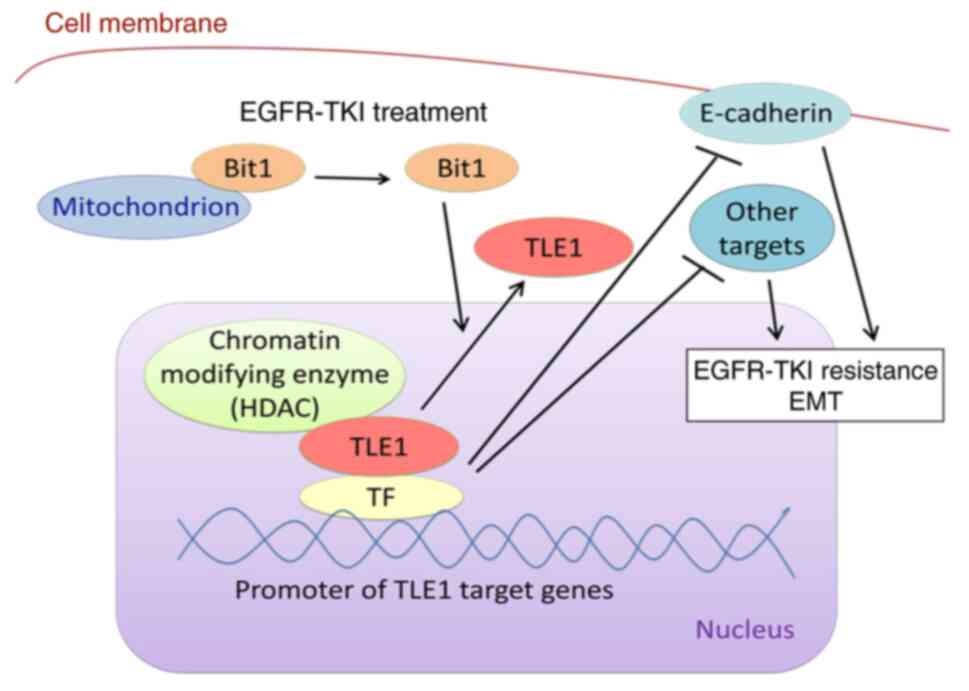 | Figure 6Working model illustrating how TLE1
may drive EGFR-TKI resistance in lung cancer cells via its
transcriptional and epigenetic program. As a transcriptional
corepressor, TLE1 in conjunction with a chromatin remodelling
enzyme (such as HDAC) binds to a TF (such as Zeb1) and is recruited
to its target gene promoters, repressing transcription of
E-cadherin and other target genes. The silencing of E-cadherin and
other target genes functions to block EGFR-TKI-induced apoptosis
and drive EGFR-TKI resistance as well as EMT. To initiate
apoptosis, EGFR-TKI induces cytoplasmic release of the cell death
effector mitochondrial Bit1 protein followed into the cytoplasm to
trigger translocation of nuclear TLE1 to the cytoplasm, thus
inhibiting TLE1 transcriptional silencing function. The ability of
cytoplasmic Bit1 to trigger TLE1 nuclear to cytoplasmic
redistribution is in part dependent on the Groucho transcriptional
regulator Amino Enhancer Split protein (21,22).
The resulting upregulation of E-cadherin and other TLE1 target
genes promotes EGFR-mediated cell death. In the development of
acquired EGFR-TKI resistance, the observed induction TLE1
expression and activation of the TLE1 transcriptional pathway
ensures complete blockade of the cell death and anti-EMT genetic
program. TLE1, transducin-like enhancer of split 1; EGFR-TKI,
epidermal growth factor receptor tyrosine kinase inhibitor; HDAC,
histone deacetylase; TF, transcription factor; EMT,
epithelial-mesenchymal transition; Bit1, Bcl-2-inhibitor of
transcription 1. |
A major antitumor effect of EGFR-TKIs is to induce
apoptosis. To date, the mechanisms by which EGFR-TKIs trigger
apoptosis are yet to be fully delineated. Detailed understanding of
apoptotic pathways targeted by EGFR-TKIs is imperative to generate
strategies to circumvent EGFR-TKI resistance, which develops in
part due to lack of cell death induction following EGFR inhibition.
In the last decade, while significant effort has been made to
determine the cell death pathways and apoptotic effectors impacted
by EGFR-TKIs, knowledge on the transcriptional and epigenetic
machineries downstream of EGFR remains limited. In the present
study, the findings indicated that the corepressor TLE1 serves a
cellular target of gefitinib to trigger apoptosis and whose
activation may contribute to EGFR-TKI resistance. First, gefitinib
treatment induced cytoplasmic relocalization of nuclear TLE1 in
part via activation of the cell death Bit1 pathway resulting in
inhibition of its gene transcriptional silencing function. Second,
forced expression of nuclear TLE1 was sufficient to attenuate the
level of gefitinib-mediated cell death and downregulation of
endogenous TLE1 potentiated gefitinib-induced apoptosis. Third, the
development of acquired gefitinib resistance in A549 cells resulted
in upregulation of TLE1 expression which functions to safeguard
cells from EGFR-mediated apoptosis. As described in our model, TLE1
may drive lung cancer resistance to EGFR-TKI via suppression of a
cell death and/or anti-EMT genetic program (Fig. 6).
The current data indicates that the transcriptional
repression of E-cadherin contributes to TLE1-mediated attenuation
of EGFR-TKI apoptosis. In addition to promoting cell-cell adhesion,
the tumor suppressor E-cadherin may also induce apoptosis in
several cellular models (16,23,24).
Consistent with the present results, previous studies demonstrated
that restoration of E-cadherin expression is sufficient to induce
EGFR-TKI-mediated apoptosis and EGFR-TKI sensitivity in lung cancer
cells (16,25). The exact mechanisms underlying the
apoptosis function of E-cadherin however remain to be determined
but may involve alteration of signaling pathways downstream of and
regulated by E-cadherin. To this end, forced E-cadherin expression
in breast carcinoma cells promoted etoposide-induced apoptosis via
inhibition of the anti-apoptotic Bcl-2 expression (24). The authors are currently exploring
which E-cadherin regulated pathways are involved in
EGFR-TKI-mediated apoptosis and their potential regulation by TLE1.
As a transcriptional corepressor that has ability to interact with
several DNA-binding TFs and regulate distinct gene regulatory or
signaling pathways, TLE1 corepressor likely regulates a survival
promoting transcriptional program and not just a single gene (for
example, E-cadherin). The authors' future direction is to identify
novel TLE1 target genes by performing an integrated RNA-sequencing
and ChIP-sequencing study and molecularly characterize their
function in regulating EGFR-TKI apoptosis.
While our current data supports the hypothesis that
the TLE1 antagonistic effect on EGFR-TKIs involves turning off the
Bit1 apoptosis pathway, TLE1 may inhibit alternative cell death
mechanisms such as the BIM pathway, a key effector of
EGFR-TKI-mediated apoptosis. It is noteworthy that TLE1 has been
shown to transcriptionally upregulate Bcl-2 expression (21). Thus, an exciting possibility exists
that the TLE1-mediated induction of Bcl-2 may lead to sequestration
and inactivation of BIM through direct Bcl-2-BIM interaction,
thereby inhibiting BIM apoptosis function. Whether the activation
of Bit1 cell death pathway impacts BIM dependent apoptosis in LUAD
cells in the context of EGFR-TKIs remains to be determined.
At this time, the possibility that TLE1-mediated
gefitinib resistance maybe related to its ability to promote EMT
cannot be excluded. Numerous studies have shown that acquisition of
EMT is associated EGFR-TKI resistance in EGFR-mutant and wild-type
NSCLC, and restoration of the epithelial marker E-cadherin
expression and reversal of EMT potentiate sensitivity of lung
cancer cells to EGFR-TKI (16,25).
While the exact mechanisms of how EMT contributes to EGFR-TKI
resistance remain to be defined, our data raises a possibility that
the EMT-mediated EGFR-TKI resistance may channel through TLE1.
Indeed, the gefitinib resistant A549 resistant cell line used in
the present study exhibits high levels of TLE1 expression and
pronounced EMT. While the presence of other gefitinib-resistant
promoting mutations cannot be excluded, sole downregulation of TLE1
in these cells is sufficient to reverse EMT and attenuate gefitinib
resistance, indicating that TLE1 may drive EGFR-TKI acquired
resistance via EMT. Importantly, inhibiting the pro-EMT function of
TLE1 via ectopic expression of E-cadherin was associated with
increased gefitinib sensitivity. To further examine the role of
TLE1 in EMT as a mechanism of EGFR-TKI resistance, it will be
interesting to determine if TLE1 regulates acquired resistance to
EGFR-TKIs driven by known EMT effectors such as the TF
Zeb1(26) and if TLE1
downregulation and/or inactivation is sufficient to restore
EGFR-TKI sensitivity in these contexts. It is noteworthy that it
has been previously shown that Zeb1 is a critical TF in mediating
TLE1-induced silencing of E-cadherin expression and TLE1-mediated
EMT in LUAD cells (9). In addition
to Zeb1, the authors are exploring other TFs that are associated
with LUAD EGFR-TKI resistance as potential mediators of TLE1
resistance. Via proteomics and genetic approaches, these
TLE1-binding TFs will be identified and their role in TLE1 gene
regulatory and EGFR-TKI resistance function will be characterized.
It is noteworthy that HES1, a known TLE1 interacting TF, is a
determinant of EGFR-TKI resistance in LUAD.
To generate mechanistic insights on the upregulation
of TLE1 expression during the development of EGFR-TKI resistance,
the promoter region of TLE1 was examined for binding sites for TFs
that are associated with EGFR-TKI resistance in human lung cancer
cells. Strikingly, using the EPD Eukaryotic promoter database, it
was found that the TLE1 promoter region is enriched for TFs STAT3
and ZNF263 binding motif. While numerous data in literature
demonstrate that the transcriptional activator STAT3 is activated
upon acquisition of EGFR-TKI resistance (17,18),
there are evidence supporting loss of expression of the
transcriptional repressor ZNF263 in EGFR-TKI resistant LUAD cells
(19). Thus, the activation of
STAT3 and/or downregulation of ZNF263 may result in transcriptional
upregulation of TLE1 expression. The authors are currently
performing molecular genetic studies to determine which of these
TFs contributes to the elevated TLE1 expression in EGFR-TKI
resistant cells.
Detailed knowledge and understanding of the
molecular components of the TLE1-mediated transcriptional and
epigenetic machinery may yield new therapeutic strategies to
overcome TLE1-mediated oncogenic and EGFR-TKI resistance. For
example, inhibiting TLE1 nuclear function via small molecules and
chemicals that target the individual components of the TLE1
transcriptional machinery would be a viable approach. As a
transcription corepressor, the authors previously showed that TLE1
recruits histone deacetylase (HDAC) to the E-cadherin gene promoter
to promote histone deacetylation and gene silencing. Thus, it will
be of interest to investigate if HDAC inhibitors could alleviate
the TLE1-mediated EGFR-TKI resistance.
In summary, it was demonstrated that the
transcriptional corepressor TLE1 suppresses gefitinib-induced
proliferation inhibition and apoptosis in part by silencing the
E-cadherin expression. Consistent with its role as a determinant of
EGFR-TKI resistance, TLE1 expression is upregulated in the
experimentally derived gefitinib-resistant cell line and its
downregulation partially restores gefitinib sensitivity. While the
detailed mechanism by which TLE1 inhibits gefitinib-induced
apoptosis is yet to be elucidated, our collective data indicate
that TLE1 may serve as a negative predictive marker of EGFR-TKI
sensitivity in LUAD with invasive EMT phenotype.
Supplementary Material
(A) The GEO DataSets (http://www.ncbi.nlm.nih.gov/geo/gds), hosted by
the National Center for Biotechnology Information, was employed as
the database for dataset retrieval. The search query used was
‘[(lung adenocarcinoma) AND (EGFR-TKI resistance) OR (gefitinib
resistance) OR (osimertinib resistance)]’, with additional filters
set for the organism as ‘Homo sapiens’ and entry type as either
‘DataSets’ or ‘Series’. The resulting datasets were then screened
to include only samples derived directly from patients, excluding
those from cell lines. Ultimately, dataset GSE231938 was selected,
comprising samples from one EGFR-TKI-sensitive patient and two
EGFR-TKI-resistant patients. The levels of TLE1 mRNA in these
samples were subsequently analyzed using Geo2R, with expression
levels reported in Transcripts Per Million (TPM). (B) Reanalysis of
Kaplan-Meier curve of overall survival (OS) up to six years after
the initial diagnosis for high and low expression levels of TLE1 in
patients with lung squamous cell carcinoma. TLE1, transducin-like
enhancer of split 1; EGFR-TKI, epidermal growth factor receptor
tyrosine kinase inhibitor.
TLE1 expression regulates sensitivity
of A549 cells to osimertinib. (A and B) Control GFP and GFP-TLE1
A549 cells were treated with the indicated concentration of
osimertinib. A total of 48 h post-treatments, cells were subjected
to (A) Alamar Blue cell viability and (B) apoptosis assays. (C and
D) A549 were transfected with a pool of TLE1 specific or control
siRNAs, and 6 h post-transfection, cells were treated with
osimertinib for 48 h followed by (C) cell viability and (D)
apoptosis assays. The results are representative of three
independent experiments. *P<0.05 and
**P<0.01 (Student’s t-test). Error bars indicate SD.
TLE1, transducin-like enhancer of split 1; siRNA, small interfering
RNA.
Exogenous TLE1 expression confers
gefitinib resistance in the human lung adenocarcinoma epidermal
growth factor receptor-mutant HCC827 cell line. (A-C) HCC827
expressing control GFP or GFP-TLE1 construct were subjected to (A)
western blotting against specific antibodies to GFP, E-cadherin and
β-actin and treated with the indicated concentration of
gefitinib for 48 h followed by (B) Alamar blue assay and (C)
apoptosis assay. The results are representative of three
independent experiments. *P<0.05 and
**P<0.01 (Student’s t-test). Error bars indicate SD.
TLE1, transducin-like enhancer of split 1.
Exogenous expression of E-cadherin
attenuates the TLE1-mediated resistance to gefitinib in HCC827 cell
line. (A and B) GFP-TLE1 HCC827 cells were transfected with the
vector or E-cadherin expressing construct and subjected to (A)
western blotting with the indicated antibodies or further cultured
in the indicated concentration of gefitinib for 48 h followed by
(B) Alamar Blue viability assay. The results are representative of
three independent experiments. *P<0.05 and
**P<0.01 (One-way ANOVA with post hoc Tukey’s test).
Error bars indicate SD. TLE1, transducin-like enhancer of split
1.
A549 cells are transfected with a pool
of TLE1-specific, AES-specific siRNAs or a non-targeting control
siRNA with the Lipofectamine RNAiMAX Transfection Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer’s protocol, and 24 h later cells are subjected to
western blotting with specific antibodies against TLE1, AES, or
β-actin. TLE1, transducin-like enhancer of split 1; AES,
Amino Enhancer Split; siRNA, small interfering RNA.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the (grant no.
NIH-1R16GM145484-01), the NIH RCMI (grant no. 8G12MD007595; Xavier
University of Louisiana), the NIH BUILD Student Training Core
(grant no. 1TL4MD009637; Xavier University of Louisiana), and the
NIH (grant no. R25GM060926; Xavier University of Louisiana).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XY, NR, PI, AC and MCDC performed molecular and
cell-based experiments and analysed the data. RC and HB wrote the
manuscript. RC and HB designed the experiments, wrote and edited
the manuscript. XY, NR, PI, AC, RC, CDC and HB confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Uribe ML, Marrocco I and Yarden Y: EGFR in
cancer: Signaling mechanisms, drugs, and acquired resistance.
Cancers (Basel). 13(2748)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Koulouris A, Tsagkaris C, Corriero AC,
Metro G and Mountzios G: Resistance to TKIs in EGFR-mutated
non-small cell lung cancer: From mechanisms to new therapeutic
strategies. Cancers (Basel). 14(3337)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Li A, Cao W, Liu X, Zhang Y, Ma Y, Xu R,
Zhang R, Liu X, Zhou S, Wang R, et al: Gefitinib sensitization of
cisplatin-resistant wild-type EGFR non-small cell lung cancer
cells. J Cancer Res Clin Oncol. 146:1737–1749. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nishihara S, Yamaoka T, Ishikawa F,
Higuchi K, Hasebe Y, Manabe R, Kishino Y, Kusumoto S, Ando K,
Kuroda Y, et al: Mechanisms of EGFR-TKI-induced apoptosis and
strategies targeting apoptosis in EGFR-mutated non-small cell lung
cancer. Genes (Basel). 13(2183)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tanaka K, Yu HA, Yang S, Han S, Selcuklu
SD, Kim K, Ramani S, Ganesan YT, Moyer A, Sinha S, et al: Targeting
Aurora B kinase prevents and overcomes resistance to EGFR
inhibitors in lung cancer by enhancing BIM- and PUMA-mediated
apoptosis. Cancer Cell. 39:1245–1261.e6. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shimizu T, Nishio K, Sakai K, Okamoto I,
Okamoto K, Takeda M, Morishita M and Nakagawa K: Phase I safety and
pharmacokinetic study of YM155, a potent selective survivin
inhibitor, in combination with erlotinib in patients with EGFR TKI
refractory advanced non-small cell lung cancer. Cancer Chemother
Pharmacol. 86:211–219. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Allen T, van Tuyl M, Iyengar P, Jothy S,
Post M, Tsao MS and Lobe CG: Grg1 acts as a lung-specific oncogene
in a transgenic mouse model. Cancer Res. 66:1294–1301.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yao X, Ireland SK, Pham T, Temple B, Chen
R, Raj MH and Biliran H: TLE1 promotes EMT in A549 lung cancer
cells through suppression of E-cadherin. Biochem Biophys Res
Commun. 455:277–284. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yao X, Pham T, Temple B, Gray S, Cannon C,
Hardy C, Fletcher K, Ireland SK, Hossain A, Chen R, et al: TLE1
inhibits anoikis and promotes tumorigenicity in human lung cancer
cells through ZEB1-mediated E-cadherin repression. Oncotarget.
8:72235–72249. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lin Z, Li J, Zhang J, Feng W, Lu J, Ma X,
Ding W, Ouyang S, Lu J, Yue P, et al: Metabolic reprogramming
driven by IGF2BP3 promotes acquired resistance to EGFR inhibitors
in non-small cell lung cancer. Cancer Res. 83:2187–2207.
2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Périer RC, Praz V, Junier T, Bonnard C and
Bucher P: The eukaryotic promoter database (EPD). Nucleic Acids
Res. 28:302–303. 2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhu X, Chen L, Liu L and Niu X:
EMT-mediated acquired EGFR-TKI resistance in NSCLC: Mechanisms and
strategies. Front Oncol. 9(1044)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rho JK, Choi YJ, Ryoo BY, Na II, Yang SH,
Kim CH and Lee JC: p53 enhances gefitinib-induced growth inhibition
and apoptosis by regulation of Fas in non-small cell lung cancer.
Cancer Res. 67:1163–1169. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Witta SE, Gemmill RM, Hirsch FR, Coldren
CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita
M, et al: Restoring E-cadherin expression increases sensitivity to
epidermal growth factor receptor inhibitors in lung cancer cell
lines. Cancer Res. 66:944–950. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zheng Q, Dong H, Mo J, Zhang Y, Huang J,
Ouyang S, Shi S, Zhu K, Qu X, Hu W, et al: A novel STAT3 inhibitor
W2014-S regresses human non-small cell lung cancer xenografts and
sensitizes EGFR-TKI acquired resistance. Theranostics. 11:824–840.
2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Si J, Ma Y, Lv C, Hong Y, Tan H and Yang
Y: HIF1A-AS2 induces osimertinib resistance in lung adenocarcinoma
patients by regulating the miR-146b-5p/IL-6/STAT3 axis. Mol Ther
Nucleic Acids. 26:613–624. 2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liang J, Bi G, Sui Q, Zhao G, Zhang H,
Bian Y, Chen Z, Huang Y, Xi J, Shi Y, et al: Transcription factor
ZNF263 enhances EGFR-targeted therapeutic response and reduces
residual disease in lung adenocarcinoma. Cell Rep.
43(113771)2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Qin Q, Li X, Liang X, Zeng L, Wang J, Sun
L and Zhong D: Targeting the EMT transcription factor Snail
overcomes resistance to osimertinib in EGFR-mutant non-small cell
lung cancer. Thorac Cancer. 12:1708–1715. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Brunquell C, Biliran H, Jennings S,
Ireland SK, Chen R and Ruoslahti E: TLE1 is an anoikis regulator
and is downregulated by Bit1 in breast cancer cells. Mol Cancer
Res. 10:1482–1495. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yao X, Pham T, Temple B, Gray S, Cannon C,
Chen R, Abdel-Mageed AB and Biliran H: The anoikis effector bit1
inhibits emt through attenuation of TLE1-mediated repression of
E-cadherin in lung cancer cells. PLoS One.
11(e0163228)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bovan D, Krajnović T, Vuković NL, Vukić
MD, Mijatović S, Tanić N, Arsenijević N and Maksimović-Ivanić D:
Anoikis and cancer cell differentiation: Novel modes of shikonin
derivatives anticancer action in vitro. Mol Biol Rep.
51(218)2024.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sasaki CY, Lin HC and Passaniti A:
Expression of E-cadherin reduces bcl-2 expression and increases
sensitivity to etoposide-induced apoptosis. Int J Cancer.
86:660–666. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lee AF, Chen MC, Chen CJ, Yang CJ, Huang
MS and Liu YP: Reverse epithelial-mesenchymal transition
contributes to the regain of drug sensitivity in tyrosine kinase
inhibitor-resistant non-small cell lung cancer cells. PLoS One.
12(e0180383)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gohlke L, Alahdab A, Oberhofer A, Worf K,
Holdenrieder S, Michaelis M, Cinatl J Jr and Ritter CA: Loss of key
EMT-regulating miRNAs highlight the role of ZEB1 in EGFR tyrosine
kinase inhibitor-resistant NSCLC. Int J Mol Sci.
24(14742)2023.PubMed/NCBI View Article : Google Scholar
|