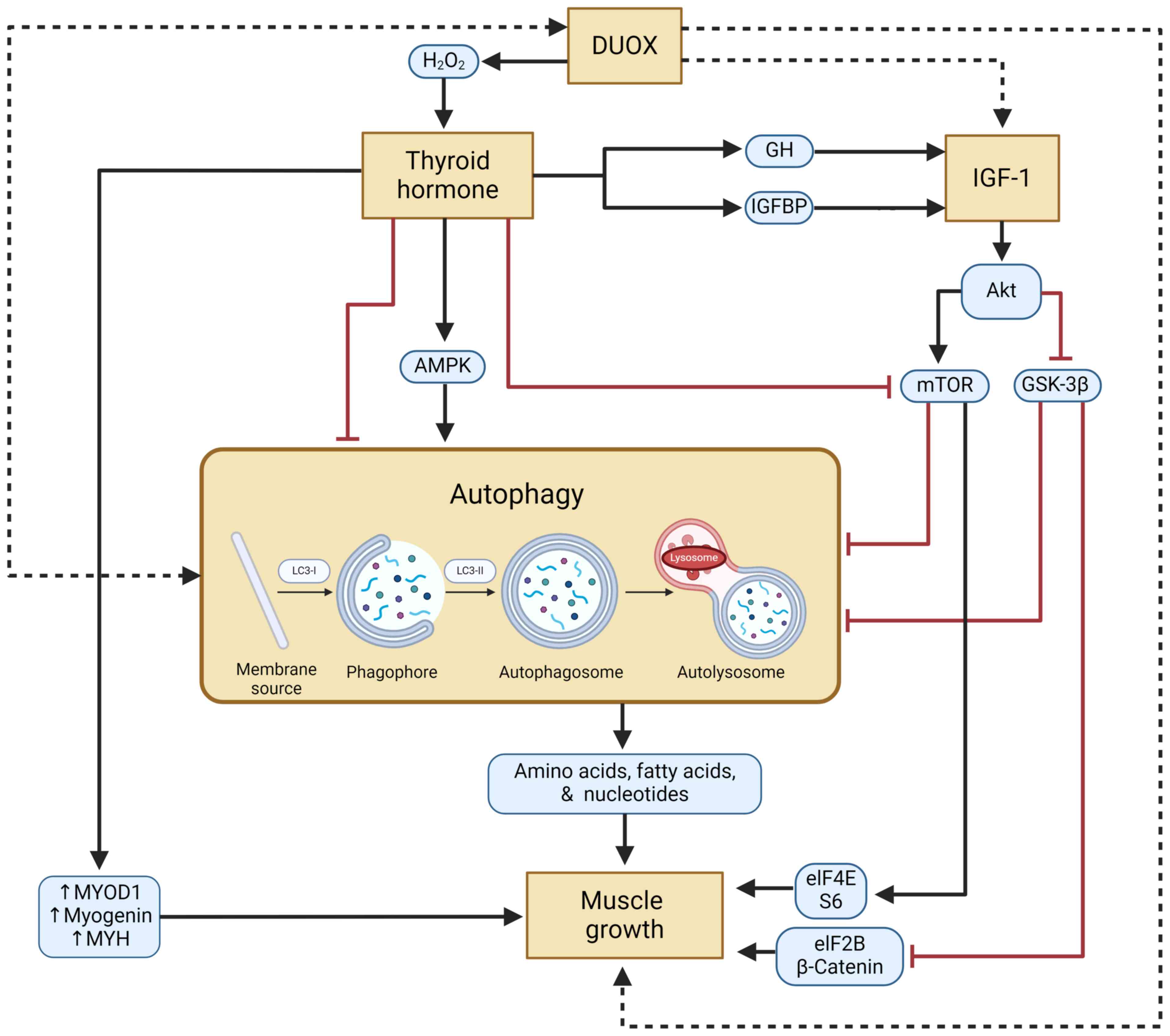
Skeletal muscle is an important body tissue with the
largest mass in the human body, accounting for ~40% of total body
weight and is the main source of protein reserves in the body
(1,2). Skeletal muscle is the most flexible
and plastic tissue in the human body and is responsible for
carrying out its functions in daily physical activities, including
movement, gestures and life activities (2,3).
Skeletal muscles also serve as the primary tissue involved in
energy metabolism, taking in, using and storing substrates,
including glucose, lipids, and amino acids (2,3).
In its development into the tissue with the largest
mass in the human body, the development of skeletal muscle is
influenced by a number of factors, such as nutritional status,
physical activity, exercise, injury or disease, autophagy processes
and hormones, one of which is thyroid hormone (4,5). Thus,
thyroid hormone deficiency has an effect on skeletal muscle and can
cause muscle atrophy if it remains at low levels (6,7). The
interaction of thyroid hormone with insulin-like growth factor 1
(IGF-1) and autophagy can also affect muscle development (8-11).
One form of thyroid hormone deficiency can be caused by
interference with thyroid hormone synthesis, such as a lack of
protein enzyme dual oxidases (DUOX) in the formation process
(12).
Consequently, DUOX and muscle development are
related via autophagy and IGF-1. To the best of the authors'
knowledge, no review or study has addressed this mechanism. The
regulatory relationships that will subsequently be connected to
muscle growth in relation to DUOX, thyroid hormone, IGF-1 and
autophagy are covered in the present review.
Dual oxidase is an enzyme that belongs to the Nox
family and performs a role in the oxidation of nicotinamide adenine
dinucleotide phosphate (NADPH) (13,14).
The DUOX enzyme has 2 types, DUOX1 and DUOX2, whose function is
mainly to produce reactive oxygen species (ROS) in various tissues
such as thyroid, colon, kidney, testis, salivary glands,
respiratory and lymphoid (13-15)
DUOX has a major contribution in the synthesis of
H2O2, a substance that has an important role
in the host defense system, fertilization, embryogenesis, signal
transduction, cell differentiation, cell death programs and hormone
synthesis, especially thyroid hormone (16-18).
During the maturation phase, additional proteins known as dual
oxidase maturation factors (DUOXA1 and DUOXA2) are needed to
support the production of H2O2 (19,20).
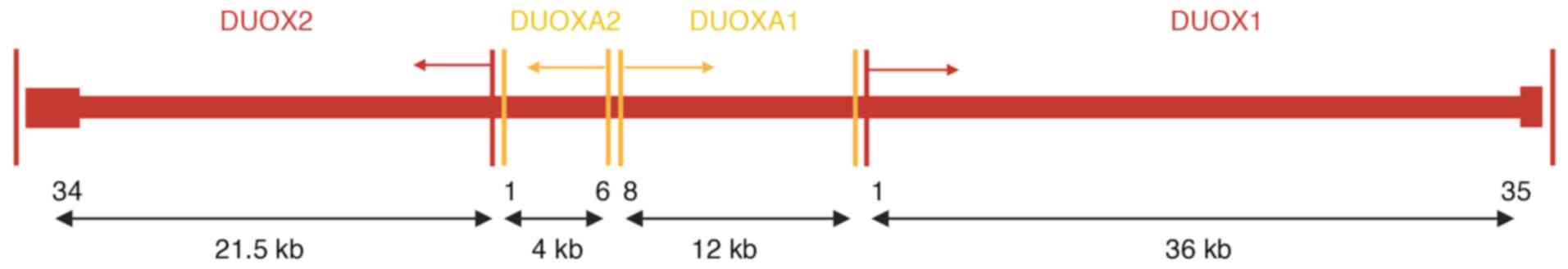
The DUOX and DUOXA genes are located next to each
other in an operon-like unit and are paired with each other on the
long arm of chromosome 15 (Fig. 1)
(21,22). In addition to thyroid cells, the
DUOXA1 and DUOXA2 genes are also expressed in human respiratory
epithelial cells (DUOXA1) and salivary glands (DUOXA2), although
the highest expression occurs in thyroid cells (23,24).
Research on mice and zebrafish demonstrates that DUOX expression
emerges only when the follicle structure is functioning optimally,
specifically at the final stage of cell differentiation during
thyroid embryogenesis (25,26).
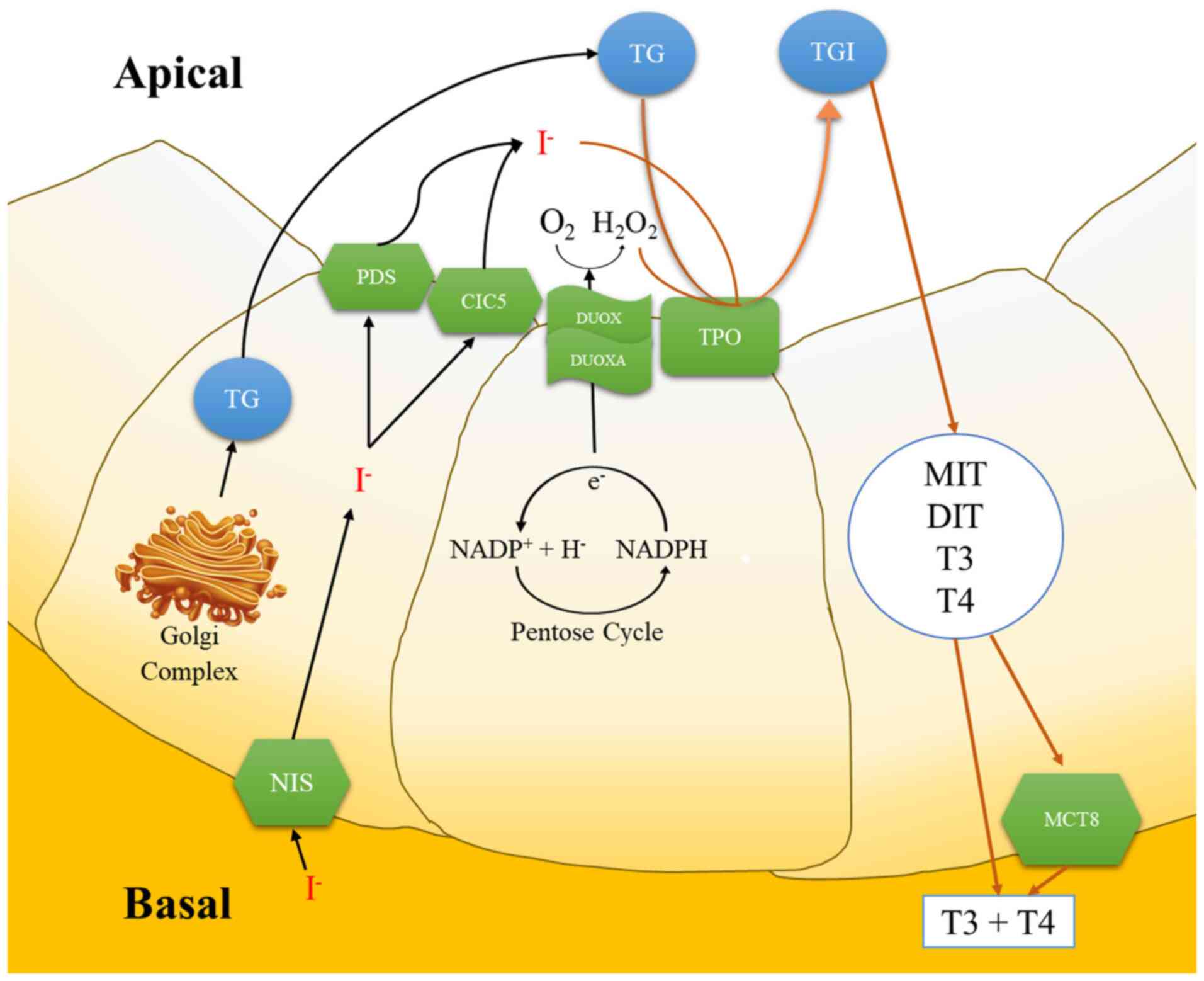
DUOX then travels toward the apex of the thyroid
cell and the N-linked glycosylation process occurs in the Golgi
apparatus, where it changes into an active form (27,28).
In the absence of DUOXA as a maturation factor for DUOX, the
oxidase process is arrested in the endoplasmic reticulum and only a
small amount of superoxide is detected (29). The activation of DUOX1 and DUOX2
isoform occurs through Ca2+ binding to the EF-hand motif
found in the N-terminal cytoplasmic segment (30,31).
Based on its sequence homology with NADPH oxidase 2 (NOX2), DUOX
should produce only superoxide. However, DUOX1 and DUOX2
co-expressed with DUOXA1 and DUOXA2 produced more
H2O2 (32).
This difference is caused by the presence of the seventh
transmembrane domain and the N-terminal peroxidase ectodomain,
which showed 40% homology to thyroid peroxidase (TPO), so that the
superoxide produced was directly converted into
H2O2 (33-35).
Thyroid stimulating hormone (TSH), via cAMP
transmission, significantly regulates DUOX2 mRNA transcription in
dog and pig thyrocytes (14,40). A
study conducted in mice showed an autoregulatory mechanism by
thyroglobulin (Tg) that suppresses DUOX2 and DUOXA2 mRNA to control
thyroid hormone synthesis (41). In
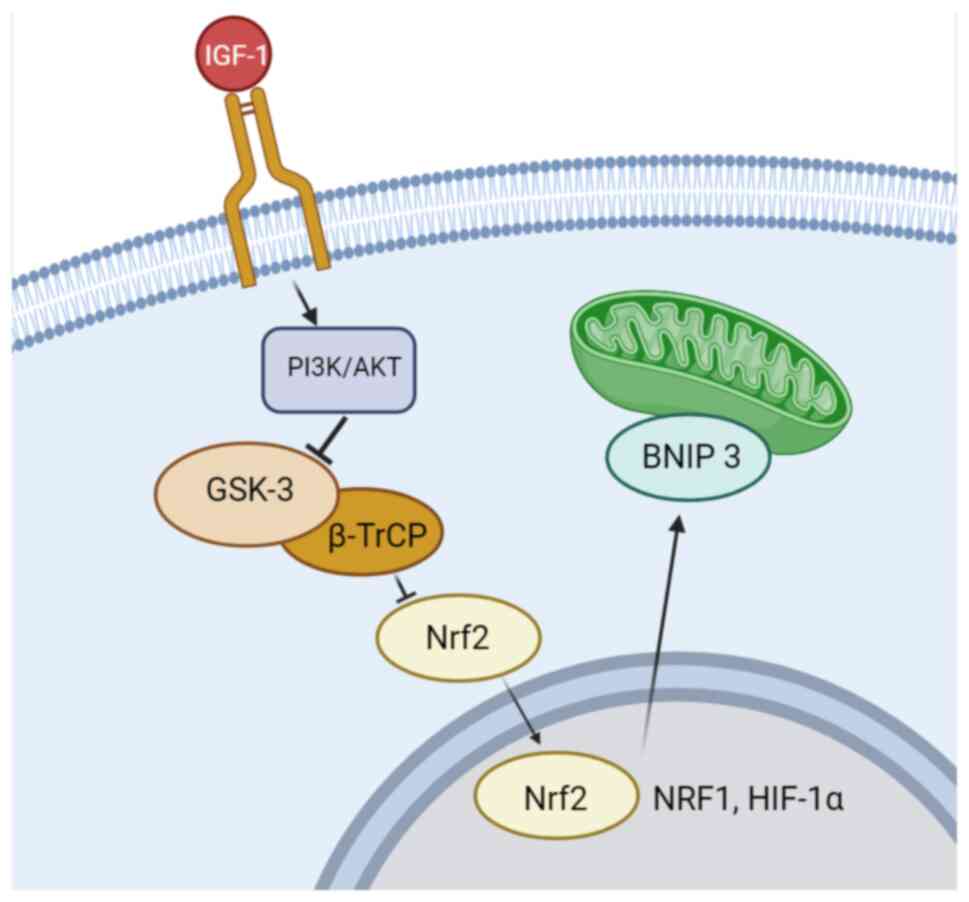
addition to TSH, increasing intracellular
H2O2 concentration may affect DUOX function
(27,33,42).
Excess amounts of iodide (I-) will inhibit the
production of H2O2, which causes a decrease
in TPO activity and reduced incorporation of I- into Tg.
This effect is called ‘Wolf-Chaikof effect’ (42-45).
Tyrosine in Tg produced in follicular cells by the
Golgi complex is transported into colloids through the process of
exocytosis and binds to the resulting iodotyrosine residue, which
ultimately forms iodinated Tg (TgI). TgI then forms a complex with
proteolytic cleavage to produce monoiodotyrosine, diiodotyrosine,
3,5,3'-triiodothyronine (T3); and 3,5,3',5'-tetraiodothyronine
(T4/thyroxin), all of which will be stored in colloids and released
when needed (17,47). When the stimulus to release hormones
is received, thyrosite will engulf some of the colloid, form an
endocytosis and proteolysis process assisted by lysosomes and then
separate Tg from T3 and T4, which will then be transported by
monocarboxylate transporters and diffuse into the blood (17,47,50).
It has been reported that mice with a double knockout of the
DUOXA1/DUOXA2 gene show that the loss of DUOX cells resulted in
hypothyroidism and a decrease in H2O2 levels
due to disruption of T4 production in the thyroid follicles
(51,52).
Thyroid hormones, particularly T3, have a direct
effect on the pituitary gland and regulate the secretion of growth
hormone (GH). This direct influence operates through thyroid
hormone receptors found in the somatotroph cells of the anterior
pituitary gland. Once T3 binds to these receptors, it can adjust
the transcription of the GH gene, thereby affecting the synthesis
and release of GH. Maintaining appropriate thyroid hormone levels
is crucial for normal GH production, as evidenced by decreased GH
mRNA levels in the pituitary gland and reduced GH secretion in
hypothyroidism cases. Conversely, administering thyroid hormone
under hypothyroid conditions can restore GH secretion to normal
levels, underscoring the necessity of adequate thyroid hormone
levels for regular GH synthesis and release (53,54).
Thyroid hormones also indirectly affect GH secretion
by affecting the hypothalamus. The hypothalamus synthesizes two key
hormones, growth hormone-releasing hormone (GHRH), which prompts GH
secretion and somatostatin, which inhibits GH secretion. Thyroid
hormones can regulate the release of hypothalamic hormones, thus
indirectly influencing GH secretion. For instance, thyroid hormones
can increase the production of GHRH in the hypothalamus, resulting
in increased GH release from the pituitary gland (54,55).
The amount of thyroid hormone circulating in the blood is related
to an increase in IGF-1, a polypeptide that shares structural
similarities with human pro-insulin and is an essential hormone for
the growth and development of the body. The primary source of GH
production is the liver and is triggered by GH secreted by the
anterior pituitary gland (56). The
bioavailability and physiological effects of IGF-1 are controlled
by a set of proteins called IGF-binding proteins (IGFBP) that are
secreted. These proteins have a strong affinity for IGF-1 and serve
as transporters of circulating IGF-1(57).
Thyroid hormone can modulate GH and subsequently
affect the production of IGF-1. Some studies have mentioned that
hypothyroidism leads to decreased GH levels and consequently lower
IGF-1 levels, whereas replacement therapy with thyroid hormone can
elevate IGF-1 levels (58,59). Another study found that patients
with hyperthyroidism have higher serum IGF-1 levels than those with
euthyroidism (60). On the other
hand, previous studies have shown that not all effects of thyroid
hormones on the IGF-1 pathway are mediated through GH. Thyroid
hormones directly affect the transcription of the IGF-1 gene.
Specifically, triiodothyronine can attach to thyroid hormone
receptors, which function as transcription factors. These thyroid
hormone receptors can subsequently bind to thyroid hormone response
elements situated in the promoter region of IGF-1, resulting in
altered IGF-1 mRNA. This direct regulation of transcription occurs
in different tissues, such as the liver and bone, where IGF-1 plays
a crucial role in growth and development (60-62).
Thyroid hormones can also adjust intracellular
signaling pathways of IGF-1 intracellularly. They can regulate the
expression of IGF-1 receptors (IGF-1R) on target cells, thus
influencing the sensitivity and responsiveness of these cells to
IGF-1. Furthermore, thyroid hormones can affect the production of
IGFBP, which controls the availability and function of IGF-1 by
binding to it in the bloodstream. Variations in IGFBP levels can
change the quantity of unbound or free IGF-1 that can be attached
to its receptor to produce its effects (58,60,61).
One study showed that T4 replacement therapy increased serum IGFBP1
levels. In hypothyroid animals, serum IGFBP3 and IGFBP4 levels are
reduced, and thyroid hormone replacement can correct these changes.
Patients who undergo thyroidectomy and have their thyroid hormone
replacement discontinued experience a decrease in the levels of
circulating IGFBP1. Treatment with thyroxine raises these levels
(58). These findings highlight the
complex relationship between thyroid hormone levels and the IGF-1
pathway, suggesting that thyroid hormones may influence the
activity of IGF-1 and related pathways in multiple ways.
Research has revealed that IGF-1 signaling controls
autophagy in a bidirectional manner (63). Autophagy is a widely occurring
recycling process in which cellular material, including organelles,
is taken up by membrane-bound vacuoles referred to as
autophagosomes and transported to lysosomes and are degraded by the
lysosomal compartment's store of proteolytic enzymes. This process
is essential for maintaining cell, tissue, and organism homeostasis
(64,65).
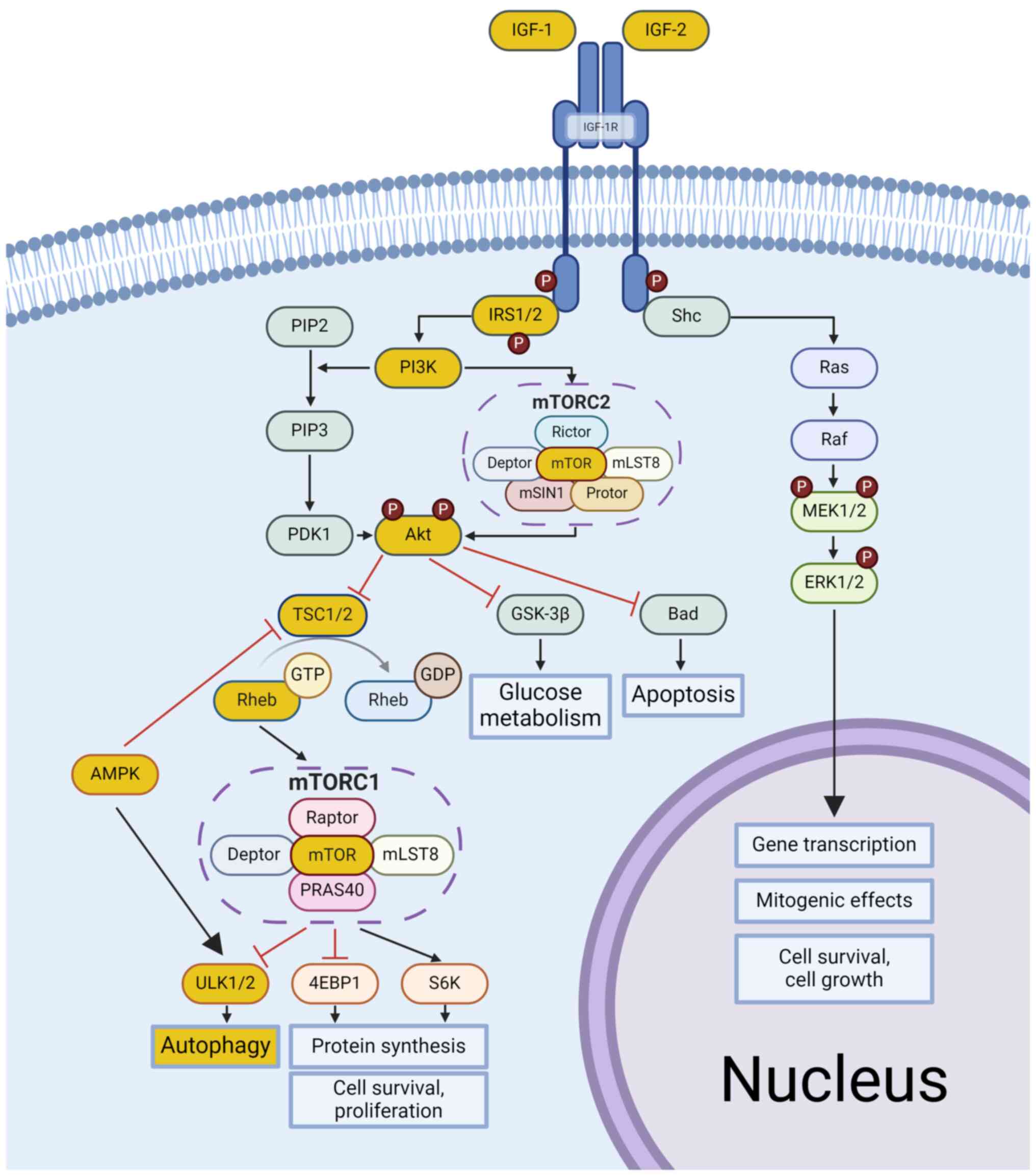
The PI3K/AKT/mTOR activation pathway involves the
phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2), a
lipid protein, to phosphatidylinositol 3,4,5-trisphosphate (PIP3)
by the PI3K kinase subunits p85 and p110. PIP3 signaling proteins,
such as phosphoinositide-dependent kinase-1 (PDK1), then activate
AKT, which in turn suppresses serine and threonine residues on its
targets, including glycogen synthase kinase-3 beta (GSK-3β) and
Tuberous sclerosis complex 1/Tuberous sclerosis complex 2
(TSC1/TSC2) (67,71). Inactive GSK-3β and TSC1/TSC2 prevent
their inhibition, activating the small G protein Ras homolog
enriched in the brain (Rheb) that binds with GTP. GTP-bound Rheb
activates mTOR-complex 1 (mTORC1) at the lysosomal surface by
binding to specific domains, including N-heat, M-heat, and the
focal adhesion targeting domain. This binding allosterically
modulates ATP binding at the active site, facilitating subsequent
phosphorylation events (67,72).
Through the MAPK pathway, Shc/Ras/Raf/MEK modulates
ERK1/2 and phosphorylates and inhibits the TSC complex, thus
activating mTORC1. Subsequently, this activation affects downstream
effectors such as ribosomal S6 kinase (S6K), Eukaryotic translation
initiation factor 4E-binding protein 1 (4EBP1) and Unc-51-like
kinase (ULK) 1/2, which in turn control processes such as autophagy
inhibition, delayed apoptosis, protein synthesis, cell survival and
proliferation (73). Several
studies have found that changes in IGF-1 levels alter autophagy
(74,75) Renna et al (76) found that IGF-1R knockdown reduced
LC3-II levels in HeLa cells grown in normal media. In addition,
IGF-1R knockdown reduced autophagosome formation in mouse embryonic
fibroblasts derived from hemizygous IGF-1R mice. In another study,
it was found that an increase in IGF-1 was accompanied by an
increase in Beclin1, ULK1, and autophagy-related 5 (Atg5), which
are markers of autophagy (77).
The bidirectional effects of IGF-1 are
context-dependent and are influenced by various factors, including
the cellular context and situations. Under normal conditions, where
cellular growth and proliferation are prioritized, IGF-1 signaling
inhibits autophagy (63,67,69,81).
Nonetheless, it stimulates autophagy and mitophagy in pathological
situations where cellular recycling processes are required, such as
energy deficiencies, starvation, hypoxia and cancer (78-80,82).
Muscle development can be directly influenced by
thyroid hormones through several pathways and one of them is the
autophagy pathway (83) thyroid
hormones require autophagy to regulate lipid homeostasis and
mitochondrial quality control in the liver (84,85).
To date, research data are insufficient to explain the role of
thyroid hormones in autophagy.
Thyroid hormone has a major role in the growth,
regeneration and differentiation of skeletal muscle through the
induction of autophagy, which involves the stimulation of ROS of
AMPK and mTOR-ULK1 signaling (86).
Thyroid hormone can also trigger changes in the muscle fiber
profile, such as the loss of embryonic and neonatal myosin and
increase in fast or slow myosin genes in certain muscles (88). In addition, rats with hypothyroidism
exhibit delayed transition to adult myosin in their fast muscles
but not in their slow muscles (89-91).
Weight-bearing exercise and electrical stimulation
are essential for the postnatal growth of slow fibers, whereas T3
signaling is critical for the development of fast fibers,
particularly for the conversion of neonatal fiber to fiber IIb
(89,91-93).
The typical pattern of fiber dispersion in every muscle is
determined in part by the physiological levels of thyroid hormone
(92,94). Thyroid hormone, especially T3,
induces muscle contractions to become faster in rats by increasing
the expression of myosin heavy chain (MYH)2, MYH 1, MYH 4, fibers
IIa, IIx, and IIb; and suppressing the expression of MYH7 and
myosin from fiber type I. Furthermore, T3 promotes the conversion
of muscle fiber types from slow to fast by causing changes from
MYH7 to MYH2, MYH2 to MYH1, and MYH1 to MYH4 (95,96).
Triiodothyronine induces miR-133a expression in fast-twitch muscles
and also induces slow-to-fast muscle fiber transition (97). Furthermore, mice with miR-133a
deletion exhibit a fast-to-slow muscle transition (98).
Muscle growth can be directly regulated by T3
hormone, which stimulates signals to myoblast determination protein
(MYOD)1, a protein that regulates the transcription process during
myogenesis (5). Moreover, MYOD1
stimulates muscle satellite cells to differentiate into myoblasts
and myotubes. Furthermore, myogenin in immature myotubes and myosin
heavy chain (MYH) in mature myotubes are two additional mechanisms
that directly affect muscle development and function caused by T3
(5,99).
Autophagy provides the fundamental components for
metabolism and cellular renewal (11,100,101). Additionally, autophagy controls
intracellular quality control, which aids in the breakdown of
defective proteins and basal protein turnover (102). Inhibition of autophagy causes the
aggregation of ubiquitin proteins and inclusion bodies in various
types of cells, and the abnormalities can also occur in
mitochondria, peroxisomes, the endoplasmic reticulum, and Golgi
bodies (103-105).
Studies using Atg7 knockout mice and focused on muscles that
presented abnormal concentric membranous formations, reticulum
distension, disordered sarcomere and aberrant mitochondria. The
Atg7 knockout mice displayed muscular phenotypes such myopathy's
morphological characteristics, muscle atrophy and degeneration
under catabolic environments, this showed how autophagy provides
benefits for preserving the integrity of myofiber and muscle mass
(104,105). Additionally, an Atg16L hypomorph
mouse model showed reduced autophagy flux but still present and
impaired muscle fiber development and generation (105,106).
Autophagy plays an important role in skeletal muscle
regeneration due to its ability to regenerate muscle stem
cells/satellite cells by maintaining a state of quiescence and
preventing aging (111,112). In senescent muscle stem cells, the
phosphorylation of AMPK and its downstream target P27Kip1 is
reduced and the accompanying stress of inhibited autophagy renders
muscle stem cells more susceptible to apoptosis (113). Autophagy helps prevent aging by
clearing the autophagosome and providing an energy source for
activation (111,112). Failure of autophagy in satellite
cells will cause aging, oxidative stress and mitochondrial
dysfunction, as well as accumulation of organelles and proteins,
but satellite cells are not the cause of muscle fiber hypertrophy
because satellite cells are only needed for the de novo
formation of new fibers (114-118).
These results show that the decrease in the number of satellite
cells may not be due to atrophy.
Previous studies have reported that the autophagy
process occurs throughout the entire myoblast differentiation
cycle, so it can be concluded that there is a two-way process
between autophagy and muscle cell differentiation (112,119-121).
There is a relationship with thyroid hormone, where T3 is needed
for the process of differentiation and fusion of myoblasts, which
will later trigger upregulation of autophagy (122). Disruption of the autophagy
process, such as knockdown of Atg5 and Atg7, affects myogenesis,
which is followed by mitochondrial dysfunction (121).
The differentiation of primitive myoblasts into
mature myotubes necessitates a metabolic change to meet the
increasing energy demand, which involves mitochondrial renewal,
which has been proven to be an essential step (123,124). In this situation, autophagy plays
an important role, as evidenced by an increase in mitophagy prior
to a rise in the amount of mitochondrial proteins at the beginning
of the mitochondrial renewal process during myogenic
differentiation (124).
Autophagy is required in the myoblast
differentiation process due to its relationship with signaling
preventing the apoptosis process (125,126). Inhibition of Atg7 enhanced
transient caspase 3 activation, DNA fragmentation and the
proportion of apoptotic nuclei (125). In addition, mitophagy also has the
function of removing damaged mitochondria prior to apoptotic
signaling, reducing cell stress and death (126). Notably, the increased ROS levels
induced by cellular stress are also required for skeletal muscle
development and inhibition of mitochondrial ROS production leads to
the failure of myoblast differentiation (127,128). Furthermore, DUOX, which is a
member of the NADPH family, may produce ROS, leading to the
hypothesis that there is a link between DUOX gene and skeletal
muscle growth.
To the best of the authors' knowledge, DUOX research
on autophagy and cell or tissue growth is currently limited to
smooth muscle organs such as the respiratory system. A study using
mice with inflammation in their lungs showed that autophagy
regulates the increase in superoxide levels by directing DUOX1 to
the apical surface of the airway epithelium (130). Another similar study in
Drosophila showed the activation of the DUOX gene downstream
of autophagy by activating the ATG1-dependent lipophagy pathway,
which is required for tumor necrosis factor receptor-associated
factor 3 (TRAF3)-AMPK/Warts gene (WTS)-pathway-induced DUOX
activation (131,132). Additionally, NOX, which is in the
same family as DUOX, produces ROS, which affects autophagy. The
exact methods by which ROS trigger autophagy remain unclear. For
example, by directly oxidizing parts of the autophagic machinery,
this activation serves as a compensatory and survival mechanism to
reduce cellular death caused by excess ROS (133,134). Another study reported that ROS
produced from NOX activates autophagy by stimulating the protein
kinase RNA-like endoplasmic reticulum kinase signaling pathway,
which in turn enhances the activation of autophagy and survival in
cardiomyocytes in response to food restriction and ischaemia
(135).
Hypothetically, ROS produced by the DUOX gene has a
similar effect to NOX in directly regulating autophagy, where
autophagy is triggered when the cell is under stress (136-142)
or, in the other words, DUOX and autophagy have a reciprocal
relationship (143). Furthermore,
ROS has an effect on muscle tissue development and it is
hypothesized that DUOX has a direct effect on muscle development
via ROS production. Skeletal muscle formation depends on elevated
ROS levels brought on by cellular stress, and inhibition of
mitochondrial ROS synthesis impairs myoblast differentiation
(127,128). It is also hypothesized that IGF-1
promotes muscle growth by upregulating DUOX. However, no studies
have specifically investigated this relationship, and the direct
interactions are largely unknown.
In conclusion, the DUOX gene has several benefits in
various life processes, including muscular development. By
understanding the function of DUOX, clinicians will be able to
diagnose and administer appropriate treatment if a disorder occurs
in this gene. DUOX is an enzyme that can produce
H2O2, which is needed for thyroid hormone
production. In addition, thyroid hormone can trigger muscle growth
directly and indirectly through the IGF-1 signaling pathway. IGF-1
will later have a bidirectional effect on the autophagy process.
Autophagy itself is a process necessary for muscle development. In
this pathway, the effect of DUOX on skeletal muscle growth is
unclear. It was hypothesized that there is a direct relationship
between DUOX and IGF-1, autophagy and muscle development.
Therefore, further studies are required to provide new insights
into the influence of DUOX on skeletal muscle growth through IGF-1
signaling.
The authors are grateful to Dr Aurelia Angelica
Erawan, Dr Ivana Iman Santosa, and Dr Gladys Danielle Novianto
(Universitas Padjadjaran, Bandung, Indonesia) for the insightful
suggestions and constructive feedback on the manuscript and to Dr
Audrey Averina Santoso, Dr Cici Pratama Dea Mantong, and Dr Jody
Garcia Hartanto Sitorus (Universitas Padjadjaran, Bandung,
Indonesia) who provided assistance in the editing of images of the
current paper.
Funding: The present study was supported by the Ministry of
Education, Culture, Research and Technology of Indonesia through
the Fundamental Research-Regular grant no. 3018/UN6.3.1/PT.00/2023
to RL.
Not applicable.
AAT completed the first draft of the manuscript. HG
and RL proposed ideas. HG, RL and AC reviewed and edited the
manuscript. NS and JWG analyzed the data and revised the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Liang JL, Xie JF, Wang CY and Chen N:
Regulatory roles of microRNAs in sarcopenia and exercise
intervention. Sheng Li Xue Bao. 72:667–676. 2020.PubMed/NCBI(In Chinese).
|
|
2
|
Evans WJ: Skeletal muscle loss: Cachexia,
sarcopenia, and inactivity. Am J Clin Nutr. 91:1123S–1127S.
2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Frontera WR and Ochala J: Skeletal muscle:
A brief review of structure and function. Calcif Tissue Int.
96:183–195. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xia Q, Huang X, Huang J, Zheng Y, March
ME, Li J and Wei Y: The role of autophagy in skeletal muscle
diseases. Front Physiol. 12(638983)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Muscat GE, Mynett-johnson L, Dowhan D,
Downes M and Griggs R: Activation of myoD gene transcription by
3,5,3'-triiodo-L-thyronine: A direct role for the thyroid hormone
and retinoid X receptors. Nucleic Acids Res. 22:583–591.
1994.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brent GA: Mechanisms of thyroid hormone
action. J Clin Invest. 122:3035–3043. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yu F, Göthe S, Wikström L, Forrest D,
Vennström B and Larsson L: Effects of thyroid hormone receptor gene
disruption on myosin isoform expression in mouse skeletal muscles.
Am J Physiol Regul Integr Comp Physiol. 278:R1545–R1554.
2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Grosvenor CE and Turner CW: Effect of
growth hormone upon thyroid secretion rate in the rat. Proc Soc Exp
Biol Med. 100:70–72. 1959.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Saji M, Tsushima T, Isozaki O, Murakami H,
Ohba Y, Sato K, Arai M, Mariko A and Shizume K: Interaction of
insulin-like growth factor I with porcine thyroid cells cultured in
monolayer. Endocrinology. 121:749–756. 1987.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Malaguarnera R, Frasca F, Garozzo A, Gianì
F, Pandini G, Vella V, Vigneri R and Belfiore A: Insulin receptor
isoforms and insulin-like growth factor receptor in human
follicular cell precursors from papillary thyroid cancer and normal
thyroid. J Clin Endocrinol Metab. 96:766–774. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kang C, You NJ and Avery L: Dual roles of
autophagy in the survival of Caenorhabditis elegans during
starvation. Genes Dev. 21:2161–2171. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Carvalho DP and Dupuy C: Role of the NADPH
oxidases DUOX and NOX4 in thyroid oxidative stress. Eur Thyroid J.
2:160–167. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Donkó Á, Péterfi Z, Sum A, Leto T and
Geiszt M: Dual oxidases. Philos Trans R Soc Lond B Biol Sci.
360:2301–2308. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dupuy C, Ohayon R, Valent A, Noël-Hudson
MS, Dème D and Virion A: Purification of a novel flavoprotein
involved in the thyroid NADPH oxidase. Cloning of the porcine and
human cdnas. J Biol Chem. 274:37265–37269. 1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Geiszt M and Leto TL: The Nox family of
NAD(P)H oxidases: Host defense and beyond. J Biol Chem.
279:51715–51718. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Conner GE: Regulation of dual oxidase
hydrogen peroxide synthesis results in an epithelial respiratory
burst. Redox Biol. 41(101931)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Szanto I, Pusztaszeri M and Mavromati M:
H2O2 metabolism in normal thyroid cells and
in thyroid tumorigenesis: Focus on NADPH oxidases. Antioxidants
(Basel). 8(126)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Korzeniowska A, Donkó ÁP, Morand S and
Leto TL: Functional characterization of DUOX enzymes in
reconstituted cell models. Methods Mol Biol. 1982:173–190.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Grasberger H and Refetoff S:
Identification of the maturation factor for dual oxidase. Evolution
of an eukaryotic operon equivalent. J Biol Chem. 281:18269–18272.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hulur I, Hermanns P, Nestoris C, Heger S,
Refetoff S, Pohlenz J and Grasberger H: A single copy of the
recently identified dual oxidase maturation factor (DUOXA) 1 gene
produces only mild transient hypothyroidism in a patient with a
novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J
Clin Endocrinol Metab. 96:E841–E851. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xu C, Linderholm A, Grasberger H and
Harper RW: Dual oxidase 2 bidirectional promoter polymorphisms
confer differential immune responses in airway epithelia. Am J
Respir Cell Mol Biol. 47:484–490. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Christophe-Hobertus C and Christophe D:
Delimitation and functional characterization of the bidirectional
THOX-DUOXA promoter regions in thyrocytes. Mol Cell Endocrinol.
317:161–167. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Luxen S, Belinsky SA and Knaus UG:
Silencing of DUOX NADPH oxidases by promoter hypermethylation in
lung cancer. Cancer Res. 68:1037–1045. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Grasberger H, De Deken X, Miot F, Pohlenz
J and Refetoff S: Missense mutations of dual oxidase 2 (DUOX2)
implicated in congenital hypothyroidism have impaired trafficking
in cells reconstituted with DUOX2 maturation factor. Mol
Endocrinol. 21:1408–1421. 2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Milenkovic M, De Deken X, Jin L, De Felice
M, Di Lauro R, Dumont JE, Corvilain B and Miot F: Duox expression
and related H2O2 measurement in mouse
thyroid: Onset in embryonic development and regulation by TSH in
adult. J Endocrinol. 192:615–626. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Opitz R, Maquet E, Zoenen M, Dadhich R and
Costagliola S: TSH receptor function is required for normal thyroid
differentiation in zebrafish. Mol Endocrinol. 25:1579–1599.
2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
De Deken X, Wang D, Dumont JE and Miot F:
Characterization of ThOX proteins as components of the thyroid
H(2)O(2)-generating system. Exp Cell Res. 273:187–196.
2002.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Raad H, Eskalli Z, Corvilain B, Miot F and
De Deken X: Thyroid hydrogen peroxide production is enhanced by the
Th2 cytokines, IL-4 and IL-13, through increased expression of the
dual oxidase 2 and its maturation factor DUOXA2. Free Radic Biol
Med. 56:216–225. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
El Hassani RA, Benfares N, Caillou B,
Talbot M, Sabourin JC, Belotte V, Morand S, Gnidehou S, Agnandji D,
Ohayon R, et al: Dual oxidase2 is expressed all along the digestive
tract. Am J Physiol Gastrointest Liver Physiol. 288:G933–G942.
2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rigutto S, Hoste C, Grasberger H,
Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F and De
Deken X: Activation of dual oxidases Duox1 and Duox2: differential
regulation mediated by camp-dependent protein kinase and protein
kinase C-dependent phosphorylation. J Biol Chem. 284:6725–6734.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ameziane-El-Hassani R, Schlumberger M and
Dupuy C: NADPH oxidases: New actors in thyroid cancer? Nat Rev
Endocrinol. 12:485–494. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lambeth JD: Nox enzymes, ROS, and chronic
disease: An example of antagonistic pleiotropy. Free Radic Biol
Med. 43:332–347. 2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Song Y, Ruf J, Lothaire P, Dequanter D,
Andry G, Willemse E, Dumont JE, Van Sande J and De Deken X:
Association of duoxes with thyroid peroxidase and its regulation in
thyrocytes. J Clin Endocrinol Metab. 95:375–382. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ameziane-El-Hassani R, Morand S, Boucher
JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R,
Noël-Hudson MS, Francon J, et al: Dual oxidase-2 has an intrinsic
Ca2+-dependent H2O2-generating
activity. J Biol Chem. 280:30046–30054. 2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Caillou B, Dupuy C, Lacroix L, Nocera M,
Talbot M, Ohayon R, Dème D, Bidart JM, Schlumberger M and Virion A:
Expression of reduced nicotinamide adenine dinucleotide phosphate
oxidase (ThoX, LNOX, Duox) genes and proteins in human thyroid
tissues. J Clin Endocrinol Metab. 86:3351–3358. 2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hoste C, Dumont JE, Miot F and De Deken X:
The type of DUOX-dependent ROS production is dictated by defined
sequences in DUOXA. Exp Cell Res. 318:2353–2364. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zamproni I, Grasberger H, Cortinovis F,
Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S,
Persani L and Weber G: Biallelic inactivation of the dual oxidase
maturation factor 2 (DUOXA2) gene as a novel cause of congenital
hypothyroidism. J Clin Endocrinol Metab. 93:605–610.
2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Morand S, Ueyama T, Tsujibe S, Saito N,
Korzeniowska A and Leto TL: Duox maturation factors form cell
surface complexes with Duox affecting the specificity of reactive
oxygen species generation. FASEB J. 23:1205–1218. 2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pachucki J, Wang D, Christophe D and Miot
F: Structural and functional characterization of the two human
ThOX/Duox genes and their 5'-flanking regions. Mol Cell Endocrinol.
214:53–62. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
De Deken X, Wang D, Many MC, Costagliola
S, Libert F, Vassart G, Dumont JE and Miot F: Cloning of two human
thyroid cDNAs encoding new members of the NADPH oxidase family. J
Biol Chem. 275:23227–23233. 2000.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yoshihara A, Hara T, Kawashima A, Akama T,
Tanigawa K, Wu H, Sue M, Ishido Y, Hiroi N, Ishii N, et al:
Regulation of dual oxidase expression and
H2O2 production by thyroglobulin. Thyroid.
22:1054–1062. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cardoso LC, Martins DC, Figueiredo MD,
Rosenthal D, Vaisman M, Violante AH and Carvalho DP:
Ca(2+)/nicotinamide adenine dinucleotide phosphate-dependent
H(2)O(2) generation is inhibited by iodide in human thyroids. J
Clin Endocrinol Metab. 86:4339–4343. 2001.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wolff J and Chaikoff IL: Plasma inorganic
iodide, a chemical regulator of normal thyroid function.
Endocrinology. 42:468–471. 1948.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Carvalho DP, Dupuy C, Gorin Y, Legue O,
Pommier J, Haye B and Virion HA: The Ca2+- and reduced
nicotinamide adenine dinucleotide phosphate-dependent hydrogen
peroxide generating system is induced by thyrotropin in porcine
thyroid cells. Endocrinology. 137:1007–1012. 1996.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Corvilain B, Van Sande J and Dumont JE:
Inhibition by iodide of iodide binding to proteins: The
‘Wolff-Chaikoff’ effect is caused by inhibition of
H2O2 generation. Biochem Biophys Res Commun.
154:1287–1292. 1988.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Pochin EE: Investigation of thyroid
function and disease with radioactive iodine. Lancet. 2:84–91.
1950.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Godlewska M, Góra M, Buckle AM, Porebski
BT, Kemp EH, Sutton BJ, Czarnocka B and Banga JP: A redundant role
of human thyroid peroxidase propeptide for cellular, enzymatic, and
immunological activity. Thyroid. 24:371–382. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Varela V, Rivolta CM, Esperante SA,
Gruñeiro-Papendieck L, Chiesa A and Targovnik HM: Three mutations
(p.Q36H, p.G418fsX482, and g.IVS19-2A>C) in the dual oxidase 2
gene responsible for congenital goiter and iodide organification
defect. Clin Chem. 52:182–191. 2006.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Di Candia S, Zamproni I, Cortinovis F,
Passoni A, Vigone MC, Fugazzola L, Persani L and Weber G:
Congenital hypothyroidism and partial iodide organification
defects: Two mutations in DUOX2 gene. Horm Res. 65(38)2006.
|
|
50
|
Thomas J, Sairoz Jose A, Poojari VG,
Shetty S, K SP, Prabhu R V K and Rao M: Role and clinical
significance of monocarboxylate transporter 8 (MCT8) during
pregnancy. Reprod Sci. 30:1758–1769. 2023.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Grasberger H, De Deken X, Mayo OB, Raad H,
Weiss M, Liao XH and Refetoff S: Mice deficient in dual oxidase
maturation factors are severely hypothyroid. Mol Endocrinol.
26:481–492. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Eskalli Z, Achouri Y, Hahn S, Many MC,
Craps J, Refetoff S, Liao XH, Dumont JE, Van Sande J, Corvilain B,
et al: Overexpression of interleukin-4 in the thyroid of transgenic
mice upregulates the expression of Duox1 and the anion transporter
pendrin. Thyroid. 26:1499–1512. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Pappa T and Refetoff S: Resistance to
thyroid hormone beta: A focused review. Front Endocrinol
(Lausanne). 12(656551)2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Giustina A and Wehrenberg WB: Influence of
thyroid hormones on the regulation of growth hormone secretion. Eur
J Endocrinol. 133:646–653. 1995.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kamegai J, Tamura H, Ishii S, Sugihara H
and Wakabayashi I: Thyroid hormones regulate pituitary growth
hormone secretagogue receptor gene expression. J Neuroendocrinol.
13:275–278. 2001.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Al-Samerria S and Radovick S: The role of
insulin-like growth factor-1 (IGF-1) in the control of
neuroendocrine regulation of growth. Cells. 10(2664)2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yakar S and Adamo ML: Insulin-like growth
factor-1 physiology: Lessons from mouse models. Endocrinol Metab
Clin North Am. 41:231–247, v. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Smith TJ: Insulin-like growth factor
pathway and the thyroid. Front Endocrinol (Lausanne).
12(653627)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chang YJ, Hwu CM, Yeh CC, Wang PS and Wang
SW: Effects of subacute hypothyroidism on metabolism and
growth-related molecules. Molecules. 19:11178–11195.
2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Tseng FY, Chen YT, Chi YC, Chen PL and
Yang WS: Serum levels of insulin-like growth factor 1 are
negatively associated with log transformation of
thyroid-stimulating hormone in Graves' disease patients with
hyperthyroidism or subjects with euthyroidism: A prospective
observational study. Medicine (Baltimore).
98(e14862)2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Smith TJ and Janssen JAMJL: Insulin-like
growth factor-i receptor and thyroid-associated ophthalmopathy.
Endocr Rev. 40:236–267. 2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Robson H, Siebler T, Shalet SM and
Williams GR: Interactions between GH, IGF-I, glucocorticoids, and
thyroid hormones during skeletal growth. Pediatr Res. 52:137–147.
2002.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Sipos F, Székely H, Kis ID, Tulassay Z and
Műzes G: Relation of the IGF/IGF1R system to autophagy in colitis
and colorectal cancer. World J Gastroenterol. 23:8109–8119.
2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Gómez-Virgilio L, Silva-Lucero MDC,
Flores-Morelos DS, Gallardo-Nieto J, Lopez-Toledo G,
Abarca-Fernandez AM, Zacapala-Gómez AE, Luna-Muñoz J, Montiel-Sosa
F, Soto-Rojas LO, et al: Autophagy: A key regulator of homeostasis
and disease: An overview of molecular mechanisms and modulators.
Cells. 11(2262)2022.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Levine B and Kroemer G: SnapShot:
Macroautophagy. Cell. 132:162.e1–162.e3. 2008.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu Q, Guan JZ, Sun Y, Le Z, Zhang P, Yu D
and Liu Y: Insulin-like growth factor 1 receptor-mediated cell
survival in hypoxia depends on the promotion of autophagy via
suppression of the PI3K/Akt/mTOR signaling pathway. Mol Med Rep.
15:2136–2142. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Kasprzak A: Autophagy and the insulin-like
growth factor (IGF) system in colonic cells: Implications for
colorectal neoplasia. Int J Mol Sci. 24(3665)2023.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Wang Z, Li W, Guo Q, Wang Y, Ma L and
Zhang X: Insulin-like growth factor-1 signaling in lung development
and inflammatory lung diseases. Biomed Res Int.
2018(6057589)2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Gonçalves DA, Silveira WA, Manfredi LH,
Graça FA, Armani A, Bertaggia E, O Neill BT, Lautherbach N, Machado
J, Nogara L, et al: Insulin/IGF1 signalling mediates the effects of
β2-adrenergic agonist on muscle proteostasis and growth.
J Cachexia Sarcopenia Muscle. 10:455–475. 2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Zhang B, Li H, Wang Y, Li Y, Zhou Z, Hou
X, Zhang X and Liu T: Mechanism of autophagy mediated by IGF-1
signaling pathway in the neurotoxicity of lead in pubertal rats.
Ecotoxicol Environ Saf. 251(114557)2023.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Mercurio L, Albanesi C and Madonna S:
Recent updates on the involvement of PI3K/AKT/mTOR molecular
cascade in the pathogenesis of hyperproliferative skin disorders.
Front Med (Lausanne). 8(665647)2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Shams R, Ito Y and Miyatake H: Evaluation
of the binding kinetics of RHEB with mTORC1 by in-cell and in vitro
assays. Int J Mol Sci. 22(8766)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Germano CA, Clemente G, Storniolo A, Romeo
MA, Ferretti E, Cirone M and Di Renzo L: mTORC1/ERK1/2 interplay
regulates protein synthesis and survival in acute myeloid leukemia
cell lines. Biology (Basel). 12(676)2023.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Ravikumar B, Vacher C, Berger Z, Davies
JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ and
Rubinsztein DC: Inhibition of mTOR induces autophagy and reduces
toxicity of polyglutamine expansions in fly and mouse models of
Huntington disease. Nat Genet. 36:585–595. 2004.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Renna M, Bento CF, Fleming A, Menzies FM,
Siddiqi FH, Ravikumar B, Puri C, Garcia-Arencibia M, Sadiq O,
Corrochano S, et al: IGF-1 receptor antagonism inhibits autophagy.
Hum Mol Genet. 22:4528–4544. 2013.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Yu Q, Zhao B, He Q, Zhang Y and Peng XB:
microRNA-206 is required for osteoarthritis development through its
effect on apoptosis and autophagy of articular chondrocytes via
modulating the phosphoinositide 3-kinase/protein kinase B-mTOR
pathway by targeting insulin-like growth factor-1. J Cell Biochem.
120:5287–5303. 2019.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Guan X, Yan Q, Wang D, Du G and Zhou J:
IGF-1 signaling regulates mitochondrial remodeling during myogenic
differentiation. Nutrients. 14(1249)2022.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Riis S, Murray JB and O'Connor R: IGF-1
signalling regulates mitochondria dynamics and turnover through a
conserved GSK-3β-Nrf2-BNIP3 pathway. Cells. 9(147)2020.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lyons A, Coleman M, Riis S, Favre C,
O'Flanagan CH, Zhdanov AV, Papkovsky DB, Hursting SD and O'Connor
R: Insulin-like growth factor 1 signaling is essential for
mitochondrial biogenesis and mitophagy in cancer cells. J Biol
Chem. 292:16983–16998. 2017.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Zecchini S, Giovarelli M, Perrotta C,
Morisi F, Touvier T, Di Renzo I, Moscheni C, Bassi MT, Cervia D,
Sandri M, et al: Autophagy controls neonatal myogenesis by
regulating the GH-IGF1 system through a NFE2L2- and DDIT3-mediated
mechanism. Autophagy. 15:58–77. 2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Shan Y, Lu C, Wang J, Li M, Ye S, Wu S,
Huang J, Bu S and Wang F: IGF-1 contributes to liver cancer
development in diabetes patients by promoting autophagy. Ann
Hepatol. 27(100697)2022.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Dentice M, Marsili A, Ambrosio R,
Guardiola O, Sibilio A, Paik JH, Minchiotti G, DePinho RA, Fenzi G,
Larsen PR and Salvatore D: The FoxO3/type 2 deiodinase pathway is
required for normal mouse myogenesis and muscle regeneration. J
Clin Invest. 120:4021–4030. 2010.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Sinha RA, Singh BK, Zhou J, Wu Y, Farah
BL, Ohba K, Lesmana R, Gooding J, Bay BH and Yen PM: Thyroid
hormone induction of mitochondrial activity is coupled to mitophagy
via ROS-AMPK-ULK1 signaling. Autophagy. 11:1341–1357.
2015.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Sinha RA, You SH, Zhou J, Siddique MM, Bay
BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al:
Thyroid hormone stimulates hepatic lipid catabolism via activation
of autophagy. J Clin Invest. 122:2428–2438. 2012.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Lesmana R, Sinha RA, Singh BK, Zhou J,
Ohba K, Wu Y, Yau WW, Bay BH and Yen PM: Thyroid hormone
stimulation of autophagy is essential for mitochondrial biogenesis
and activity in skeletal muscle. Endocrinology. 157:23–38.
2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Kurashige T, Nakajima Y, Shimamura M,
Yamada M and Nagayama Y: Hormonal regulation of autophagy in
thyroid PCCL3 cells and the thyroids of male mice. J Endocr Soc.
4(bvaa054)2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Schiaffino S, Rossi AC, Smerdu V, Leinwand
LA and Reggiani C: Developmental myosins: Expression patterns and
functional significance. Skelet Muscle. 5(22)2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Gambke B, Lyons GE, Haselgrove J, Kelly AM
and Rubinstein NA: Thyroidal and neural control of myosin
transitions during development of rat fast and slow muscles. FEBS
Lett. 156:335–339. 1983.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Butler-Browne GS, Herlicoviez D and Whalen
RG: Effects of hypothyroidism on myosin isozyme transitions in
developing rat muscle. FEBS Lett. 166:71–75. 1984.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Di Maso NA, Caiozzo VJ and Baldwin KM:
Single-fiber myosin heavy chain polymorphism during postnatal
development: Modulation by hypothyroidism. Am J Physiol Regul
Integr Comp Physiol. 278:R1099–R1106. 2000.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Baldwin KM and Haddad F: Effects of
different activity and inactivity paradigms on myosin heavy chain
gene expression in striated muscle. J Appl Physiol (1985).
90:345–357. 2001.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Adams GR, Haddad F and Baldwin KM: The
interaction of space flight and thyroid state on somatic and
skeletal muscle growth and myosin heavy chain expression on
neonatal rodents. J Gravit Physiol. 7:P15–P18. 2000.PubMed/NCBI
|
|
94
|
Mahdavi V, Izumo S and Nadal-Ginard B:
Developmental and hormonal regulation of sarcomeric myosin heavy
chain gene family. Circ Res. 60:804–814. 1987.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Simonides WS and Van Hardeveld C: Thyroid
hormone as a determinant of metabolic and contractile phenotype of
skeletal muscle. Thyroid. 18:205–216. 2008.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Larsson L, Li X, Teresi A and Salviati G:
Effects of thyroid hormone on fast- and slow-twitch skeletal
muscles in young and old rats. J Physiol. 481:149–161.
1994.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Zhang D, Wang X, Li Y, Zhao L, Lu M, Yao
X, Xia H, Wang YC, Liu MF, Jiang J, et al: Thyroid hormone
regulates muscle fiber type conversion via miR-133a1. J Cell Biol.
207:753–766. 2014.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Liu N, Bezprozvannaya S, Shelton JM,
Frisard MI, Hulver MW, McMillan RP, Wu Y, Voelker KA, Grange RW,
Richardson JA, et al: Mice lacking microRNA 133a develop dynamin
2-dependent centronuclear myopathy. J Clin Invest. 121:3258–3268.
2011.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Downes M, Griggs R, Atkins A, Olson EN and
Muscat GE: Identification of a thyroid hormone response element in
the mouse myogenin gene: Characterization of the thyroid hormone
and retinoid X receptor heterodimeric binding site. Cell Growth
Differ. 4:901–910. 1993.PubMed/NCBI
|
|
100
|
Ito K and Suda T: Metabolic requirements
for the maintenance of self-renewing stem cells. Nat Rev Mol Cell
Biol. 15:243–256. 2014.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Scott RC, Schuldiner O and Neufeld TP:
Role and regulation of starvation-induced autophagy in the
Drosophila fat body. Dev Cell. 7:167–178. 2004.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Mizushima N: The pleiotropic role of
autophagy: From protein metabolism to bactericide. Cell Death
Differ. 12 (Suppl 2):S1535–S1541. 2005.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Jung HS, Chung KW, Won Kim J, Kim J,
Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al:
Loss of autophagy diminishes pancreatic beta cell mass and function
with resultant hyperglycemia. Cell Metab. 8:318–224.
2008.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Komatsu M, Waguri S, Ueno T, Iwata J,
Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et
al: Impairment of starvation-induced and constitutive autophagy in
Atg7-deficient mice. J Cell Biol. 169:425–434. 2005.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Masiero E, Agatea L, Mammucari C, Blaauw
B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S and
Sandri M: Autophagy is required to maintain muscle mass. Cell
Metab. 10:507–515. 2009.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Paolini A, Omairi S, Mitchell R, Vaughan
D, Matsakas A, Vaiyapuri S, Ricketts T, Rubinsztein DC and Patel K:
Attenuation of autophagy impacts on muscle fibre development,
starvation induced stress and fibre regeneration following acute
injury. Sci Rep. 8(9062)2018.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Carnio S, LoVerso F, Baraibar MA, Longa E,
Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, et al:
Autophagy impairment in muscle induces neuromuscular junction
degeneration and precocious aging. Cell Rep. 8:1509–1521.
2014.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Vainshtein A, Grumati P, Sandri M and
Bonaldo P: Skeletal muscle, autophagy, and physical activity: The
ménage à trois of metabolic regulation in health and disease. J Mol
Med (Berl). 92:127–137. 2014.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Carmignac V, Svensson M, Körner Z,
Elowsson L, Matsumura C, Gawlik KI, Allamand V and Durbeej M:
Autophagy is increased in laminin α2 chain-deficient muscle and its
inhibition improves muscle morphology in a mouse model of MDC1A.
Hum Mol Genet. 20:4891–4902. 2011.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Grumati P, Coletto L, Sabatelli P, Cescon
M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini
L, et al: Autophagy is defective in collagen VI muscular
dystrophies, and its reactivation rescues myofiber degeneration.
Nat Med. 16:1313–1320. 2010.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Rayagiri SS, Ranaldi D, Raven A, Mohamad
Azhar NIF, Lefebvre O, Zammit PS and Borycki AG: Basal lamina
remodeling at the skeletal muscle stem cell niche mediates stem
cell self-renewal. Nat Commun. 9(1075)2018.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Tang AH and Rando TA: Induction of
autophagy supports the bioenergetic demands of quiescent muscle
stem cell activation. EMBO J. 33:2782–2797. 2014.PubMed/NCBI View Article : Google Scholar
|
|
113
|
White JP, Billin AN, Campbell ME, Russell
AJ, Huffman KM and Kraus WE: The AMPK/p27Kip1 axis
regulates autophagy/apoptosis decisions in aged skeletal muscle
stem cells. Stem Cell Reports. 11:425–439. 2018.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Fukada SI: The roles of muscle stem cells
in muscle injury, atrophy and hypertrophy. J Biochem. 163:353–358.
2018.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Mccarthy JJ, Mula J, Miyazaki M, Erfani R,
Garrison K, Farooqui AB, Srikuea R, Lawson BA, Grimes B, Keller C,
et al: Effective fiber hypertrophy in satellite cell-depleted
skeletal muscle. Development. 138:3657–3666. 2011.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Sousa-Victor P, Gutarra S, García-Prat L,
Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E,
González S, Serrano AL, et al: Geriatric muscle stem cells switch
reversible quiescence into senescence. Nature. 506:316–321.
2014.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Cosgrove BD, Gilbert PM, Porpiglia E,
Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL and Blau HM:
Rejuvenation of the muscle stem cell population restores strength
to injured aged muscles. Nat Med. 20:255–264. 2014.PubMed/NCBI View Article : Google Scholar
|
|
118
|
García-Prat L, Martínez-Vicente M,
Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla
V, Gutarra S, Ballestar E, Serrano AL, et al: Autophagy maintains
stemness by preventing senescence. Nature. 529:37–42.
2016.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Call JA, Wilson RJ, Laker RC, Zhang M,
Kundu M and Yan Z: Ulk1-mediated autophagy plays an essential role
in mitochondrial remodeling and functional regeneration of skeletal
muscle. Am J Physiol Cell Physiol. 312:C724–C732. 2017.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Fortini P, Ferretti C, Iorio E, Cagnin M,
Garribba L, Pietraforte D, Falchi M, Pascucci B, Baccarini S,
Morani F, et al: The fine tuning of metabolism, autophagy and
differentiation during in vitro myogenesis. Cell Death Dis.
7(e2168)2016.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Sin J, Andres AM, Taylor DJR, Weston T,
Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS and Gottlieb RA:
Mitophagy is required for mitochondrial biogenesis and myogenic
differentiation of C2C12 myoblasts. Autophagy. 12:369–80.
2016.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Chargé SBP and Rudnicki MA: Cellular and
molecular regulation of muscle regeneration. Physiol Rev.
84:209–238. 2004.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Horie T, Kawamata T, Matsunami M and
Ohsumi Y: Recycling of iron via autophagy is critical for the
transition from glycolytic to respiratory growth. J Biol Chem.
292:8533–8543. 2017.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Duguez S, Féasson L, Denis C and
Freyssenet D: Mitochondrial biogenesis during skeletal muscle
regeneration. Am J Physiol Endocrinol Metab. 282:E802–E809.
2002.PubMed/NCBI View Article : Google Scholar
|
|
125
|
McMillan EM and Quadrilatero J: Autophagy
is required and protects against apoptosis during myoblast
differentiation. Biochem J. 462:267–277. 2014.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Hoshino A, Matoba S, Iwai-Kanai E,
Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M,
Mita Y, et al: p53-TIGAR axis attenuates mitophagy to exacerbate
cardiac damage after ischemia. J Mol Cell Cardiol. 52:175–184.
2012.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Le Moal E, Pialoux V, Juban G, Groussard
C, Zouhal H, Chazaud B and Mounier R: Redox control of skeletal
muscle regeneration. Antioxid Redox Signal. 27:276–310.
2017.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Yin W, Yang L, Kong D, Nie Y, Liang Y and
Teng CB: Guanine-rich RNA binding protein GRSF1 inhibits myoblast
differentiation through repressing mitochondrial ROS production.
Exp Cell Res. 381:139–149. 2019.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Ornatowski W, Lu Q, Yegambaram M, Garcia
AE, Zemskov EA, Maltepe E, Fineman JR, Wang T and Black SM: Complex
interplay between autophagy and oxidative stress in the development
of pulmonary disease. Redox Biol. 36(101679)2020.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Dickinson JD, Sweeter JM, Warren KJ, Ahmad
IM, De Deken X, Zimmerman MC and Brody SL: Autophagy regulates
DUOX1 localization and superoxide production in airway epithelial
cells during chronic IL-13 stimulation. Redox Biol. 14:272–284.
2018.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Lee KA, Kim B, Bhin J, Kim DH, You H, Kim
EK, Kim SH, Ryu JH, Hwang D and Lee WJ: Bacterial uracil modulates
Drosophila DUOX-dependent gut immunity via Hedgehog-induced
signaling endosomes. Cell Host Microbe. 17:191–204. 2015.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Lee KA, Cho KC, Kim B, Jang IH, Nam K,
Kwon YE, Kim M, Hyeon DY, Hwang D, Seol JH and Lee WJ:
Inflammation-modulated metabolic reprogramming is required for
DUOX-dependent gut immunity in Drosophila. Cell Host
Microbe. 23:338–352.e5. 2018.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Tian Y, Kuo CF, Sir D, Wang L,
Govindarajan S, Petrovic LM and Ou JHJ: Autophagy inhibits
oxidative stress and tumor suppressors to exert its dual effect on
hepatocarcinogenesis. Cell Death Differ. 22:1025–1034.
2015.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Peng YF, Shi YH, Shen YH, Ding Bin Z, Ke
AW, Zhou J, Qiu SJ and Fan J: Promoting colonization in metastatic
HCC cells by modulation of autophagy. PLoS One.
8(e74407)2013.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Sciarretta S, Zhai P, Shao D, Zablocki D,
Nagarajan N, Terada LS, Volpe M and Sadoshima J: Activation of
NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte
autophagy and survival during energy stress through the protein
kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic
initiation factor 2α/activating transcription factor 4 pathway.
Circ Res. 113:1253–1264. 2013.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Chen Y, Azad MB and Gibson SB: Superoxide
is the major reactive oxygen species regulating autophagy. Cell
Death Differ. 16:1040–1052. 2009.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim
HP, Choi AMK and Kim YS: Carbon monoxide activates autophagy via
mitochondrial reactive oxygen species formation. Am J Respir Cell
Mol Biol. 45:867–873. 2011.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Cho IH, Choi YJ, Gong JH, Shin D, Kang MK
and Kang YH: Astragalin inhibits autophagy-associated airway
epithelial fibrosis. Respir Res. 16(51)2015.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Scherz-Shouval R and Elazar Z: ROS,
mitochondria and the regulation of autophagy. Trends Cell Biol.
17:422–427. 2007.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Recuero M, Munive VA, Sastre I, Aldudo J,
Valdivieso F and Bullido MJ: A free radical-generating system
regulates AβPP metabolism/processing: involvement of the
ubiquitin/proteasome and autophagy/lysosome pathways. J Alzheimers
Dis. 34:637–647. 2013.PubMed/NCBI View Article : Google Scholar
|
|
144
|
De Deken X and Miot F: DUOX defects and
their roles in congenital hypothyroidism. Methods Mol Biol.
1982:667–693. 2019.PubMed/NCBI View Article : Google Scholar
|