Introduction
Osteosarcoma (OS) is the most common primary
malignant bone tumor affecting adolescents and young adults and it
predominantly occurs in the long bones of the extremities, notably
in the femur (1,2). The lungs are the most common
metastatic site for OS and indicate a poor prognosis. OS incidence
is rare, accounting for <1% of all human cancers and ~2% of
childhood and adolescent cancers (3). OS clinical evaluation includes medical
history and physical examination followed by radiologic imaging and
tissue biopsy for histological diagnosis (4). The treatment modality generally
includes surgery and combination of chemotherapy (5). However, some patients do not respond
effectively to therapy; therefore, new treatment modalities are
needed. Several other oncological treatments showing promising
results in a number of cancers are being investigated for sarcomas,
including the use of poly(ADP-ribose) polymerase (PARP) inhibitors
(PARPi), new adjuvant therapies, immunotherapy with immune
checkpoint inhibitors (blockage of the programmed cell death
protein 1/programmed death-ligand 1 axis) and epigenetic therapies
(6-14).
Despite these efforts, the efficacy or clinical benefits of these
treatment strategies for sarcomas remain controversial.
Advances in understanding cancer-related genetic
alterations have had substantial effects on precision oncology
(15,16). Next-generation sequencing (NGS) is a
high-throughput sequencing offering robust genomic data for tumor
genotyping that is able to drive diagnosis and treatment decisions
(16,17). Despite this, little is known about
the molecular aspects involved in OS etiology and progression;
thus, genomic testing and targeted therapy to improve treatment are
still rare scenarios for patients with OS. Mutations in the
TP53 and RB1 tumor suppressor genes are commonly
described for OS (18,19). However, the clinical complexity of
OS suggests additional genetic drivers of this neoplasm.
The present study described the clinical course and
the genomic profiling of an adolescent patient who was diagnosed
with OS and treated with a standard protocol. It was approved by
the Ethics and Research Committee of the National Institute of
Traumatology and Orthopedics (approval no. CAAE:
60632822.4.0000.5273).
Case presentation
A 16-year-old female was referred to the Specialized
Care Center for Orthopedic Oncology (National Institute of
Traumatology and Orthopedics, Rio de Janeiro, Brazil) in November
2022 due to a mass, swelling and increasing pain in the right knee
which began two months earlier. Patient had no known family history
of cancer. The physical exam revealed a palpable mass in the right
knee and restricted range of motion (10-90˚) associated with pain,
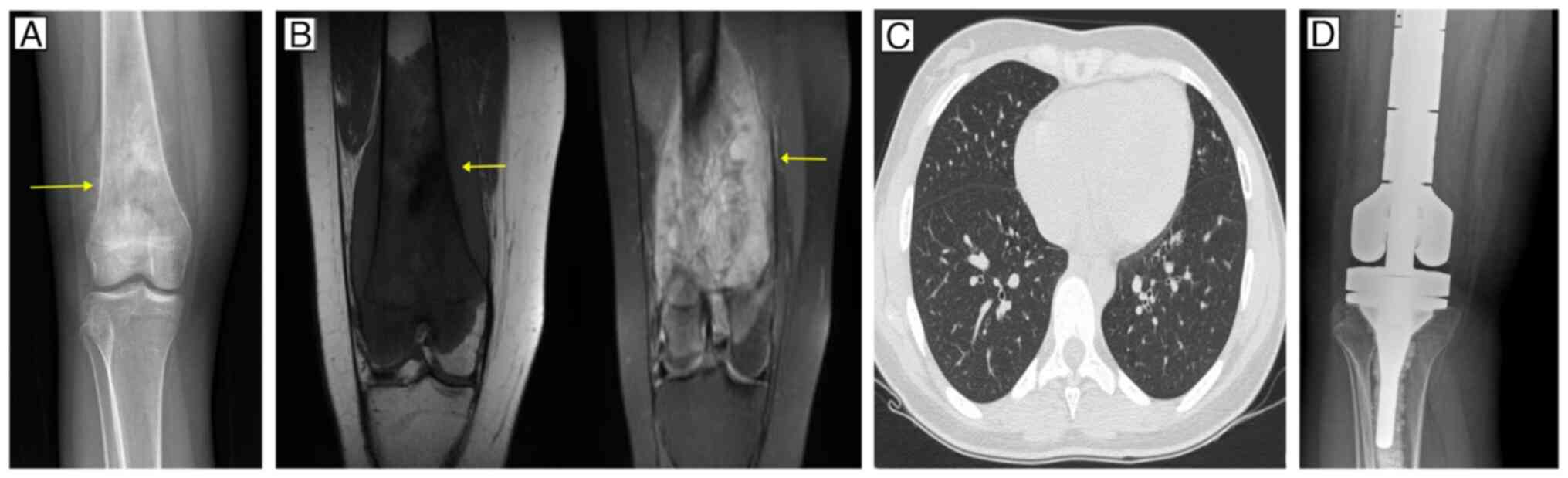
as major findings. Radiography revealed a blastic lesion affecting
the metadiaphyseal region of the right distal femur with a sunburst
periosteal reaction (Fig. 1A).
Magnetic resonance imaging revealed infiltration of the lesion
through the distal third of the femur and mild involvement of soft
tissues, resulting in a lesion that appeared hypointense on T1 and
hyperintense on T2 (Fig. 1B).
Computed tomography of the chest did not reveal evidence of distant
metastases (Fig. 1C). A needle
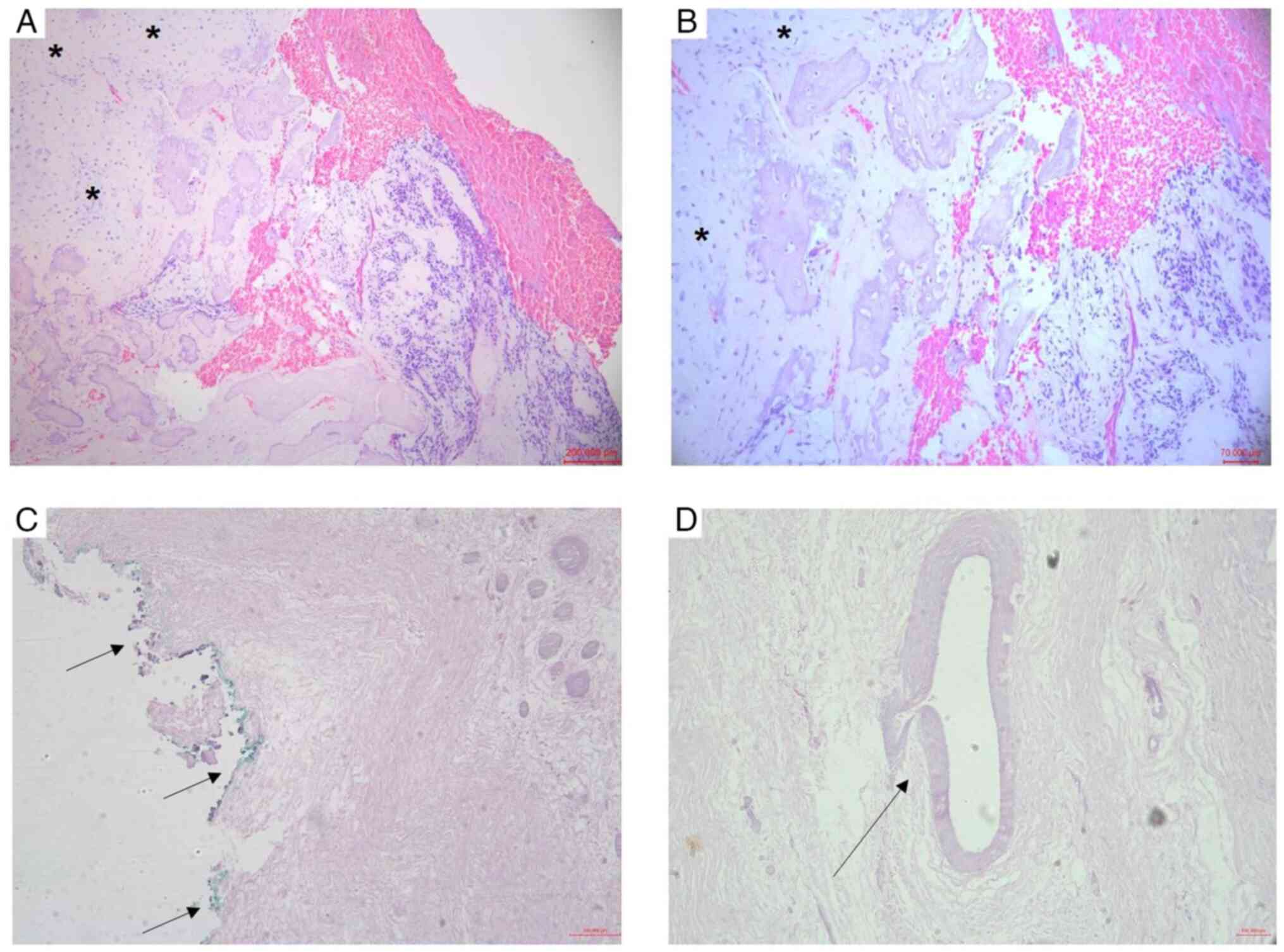
biopsy was performed and the diagnosis of conventional central OS,
grade III, with a chondroblastic area was confirmed by histological
examination (Fig. 2A and B). Tissue preparation for histological
examination was performed by hematoxylin and eosin (H&E)
staining. Briefly, the deparaffinization procedure of the tissue
section was performed with xylene followed by alcohol rinses and
then by rinsing in tap water to hydrate the section. Next, the
Harris hematoxylin solution was applied for 2 min to stain the
nuclear elements, followed by rinsing in tap water. After, a
treatment with 5% acid alcohol was carried out followed by rinsing
in tap water and subsequent addition of a 70% alcohol solution for
1 min. Finally, the eosin solution was applied for 15 sec to stain
nonnuclear elements, followed by 100% ethanol rinses for
dehydration and xylene treatment. For microscopy images, the
stained tissues were cut into 7-µm slices using a microtome.
In January 2023, the patient started treatment with
a neoadjuvant chemotherapy regimen of six cycles of cisplatin 60
mg/m2, doxorubicin 75 mg/m2, cardioxane 375
mg/m2 and high doses of methotrexate (12
g/m2). In May 2023, the patient underwent a wide
resection and endoprosthetic reconstruction (Fig. 1D). Histopathological analysis of the
surgical specimen revealed tumor necrosis of 60% (Huvos grade II),
tumor-free resection margin and absence of angiolymphatic or
perineural invasion (Fig. 2C and
D). In June 2023, the patient
started adjuvant chemotherapy with twelve cycles of cisplatin 60
mg/m2, doxorubicin 75 mg/m2, cardioxane 375
mg/m2 and high doses of methotrexate (12
g/m2). Until the last follow-up in April 2024, the
patient still had good clinical signs, with no evidence of
recurrence or pulmonary metastases.
The tumor tissue sample for DNA sequencing analysis
was obtained from the surgical resection, 4 months after
neoadjuvant chemotherapy. A peripheral blood sample was obtained at
the same time. NGS analysis of tumor tissue was performed using the
AmpliSeq for Illumina Focus Panel (Illumina, Inc.). Briefly, tumor
tissue was fragmented using the L-Beader 24 tissue disruptor
(Loccus do Brasil Ltda). Then, genomic DNA was extracted using
Quick-DNA Miniprep Kit (Zymo Research Corp.). For NGS, the library
preparation process was performed with 10 ng of DNA input, in which
DNA targets were amplified by PCR using the AmpliSeq Focus DNA
Panel and the AmpliSeq for Illumina Library PLUS (cat. no.
20019101; Illumina, Inc.). The AmpliSeq Focus DNA Panel contains 29
kb and the DNA amplicon size contains an average size of 107 bp in
length. Afterwards, the amplicons were partially digested and
subsequently ligated with index combination for dual-index
sequencing using the AmpliSeq for Illumina CD Indexes Set A (cat.
no. 20019105; Illumina, Inc.). All DNA library preparation assay
was performed according to the manufacturer's protocol, available
on the Illumina website (https://support.illumina.com/downloads/ampliseq-for-illumina-focus-panel-reference-guide-1000000039456.html).
The quality of DNA library was visualized on 2% agarose gel
electrophoresis using a DNA ladder 100 bp (Ludwig Biotecnologia
ltda) and stained with ethidium bromide, revealing a size
distribution corresponding to ~300 bp. The quantification of DNA
library was assessed by Qubit dsDNA HS Assay Kit (Thermo Fisher
Scientific, Inc.). Finally, after denaturing and diluting steps,
the library was loaded onto the reagent cartridge and transferred
automatically to a flow cell (NextSeq 500/550 Mid-Output v2.5 Kit;
cat. no. 20024905; Illumina, Inc.) for paired-end sequencing reads
based on sequencing by synthesis (SBS) technology on the NextSeq
550 Sequencing System. For bioinformatics analysis, genomics data
were analyzed using the DNA Amplicon App (Version 2.1.1) on
BaseSpace Sequence Hub (Illumina, Inc.) (https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps/dna-amplicon.html),
resulting in genomic variants identified in the following
databases: Single Nucleotide Polymorphism Database (dbSNP)
(https://www.ncbi.nlm.nih.gov/snp/?cmd=search),
Catalogue of Somatic Mutations in Cancer (COSMIC) (https://cancer.sanger.ac.uk/cosmic) and ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/). In the present
study, the single nucleotide variants (SNVs) in tumor tissue were
identified in coding and intronic regions described in the
following genes: JAK1, ALK, FGFR3,
PDGFRA, FGFR4, EGFR, RET and
KRAS.
The AmpliSeq for Illumina Focus Panel (Illumina,
Inc.) includes genes with known relevance to solid tumors. However,
the TP53 gene is not part of this panel. Since TP53
mutations are commonly described for OS, mutational hotspot exons
of TP53 (exons 4-8) were investigated in the present study
by Sanger sequencing. To investigate somatic and germline
TP53 mutations, DNA samples were obtained from tumor and
peripheral blood. Briefly, genomic DNA was extracted from blood
using QIAamp Blood Mini Kit (cat. no. 51104; Qiagen GmbH). The PCR
products of TP53 (exons 4-8) were purified by using the
PureLink Quick Gel Extraction and PCR Purification Combo Kit (cat.
no. K220001; Invitrogen; Thermo Fisher Scientific, Inc.), followed
by Sanger sequencing reaction using the BigDye Terminator v3.1
Cycle Sequencing kit on an ABI 3730XL DNA Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.), as previously
described (20). DNA sequencing
results were analyzed with ChromasPro software (Technelysium Pty
Ltd), version 2.1.10.1, using the reference sequence from NCBI
(NM_000546.6).
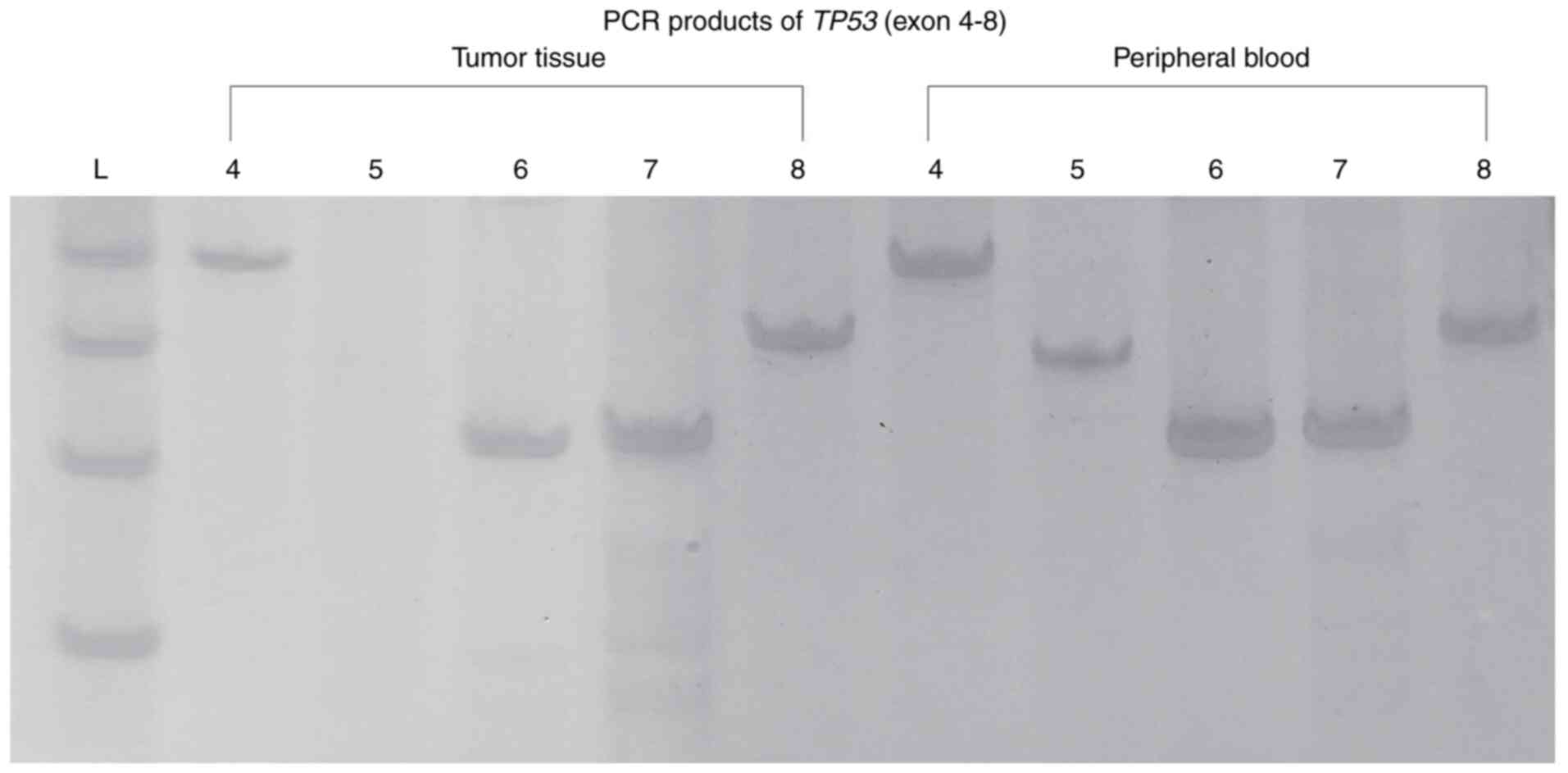
As shown in Fig. 3,
the PCR products of TP53 were initially visualized on
polyacrylamide gels. Only TP53 exon 5 was not amplified from
the tumor tissue sample, which suggested a significant somatic
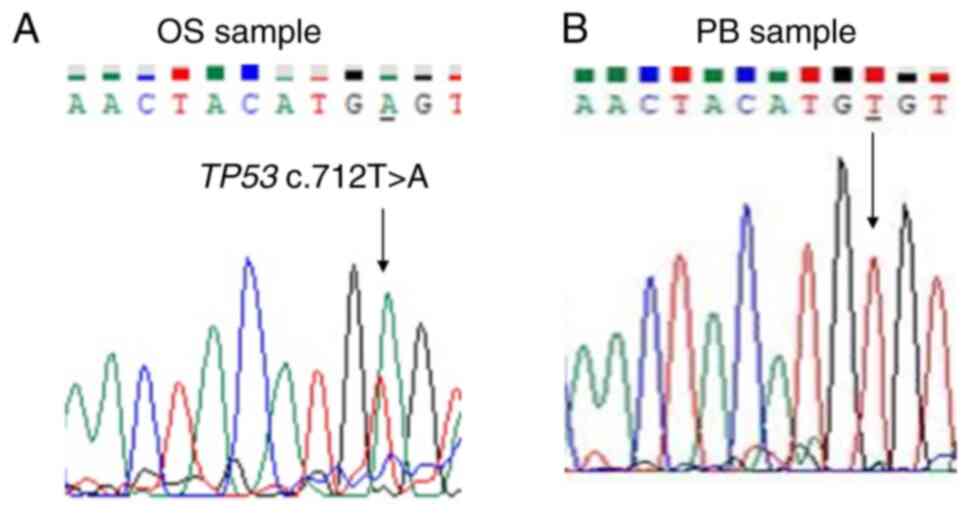
exonic deletion of TP53 in this OS tumor. Sanger sequencing
analysis revealed the missense variant TP53 c.712T>A
(p.Cys238Ser) in the tumor sample (Fig.
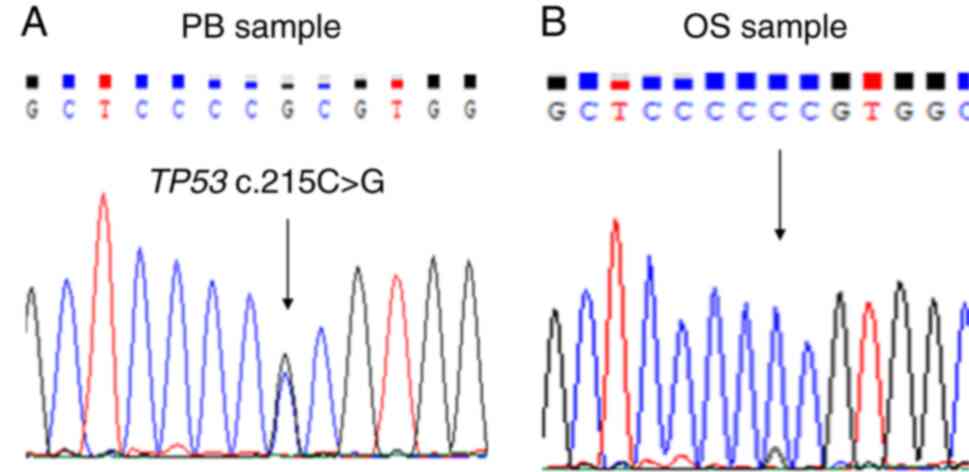
4) and the missense variant TP53 c.215C>G
(p.Pro72Arg) in the peripheral blood sample (Fig. 5).
All genetic variants identified in the present study
are described in Table I.
 | Table ISummary of the genetic variants
identified in the present study by NGS and Sanger sequencing
analyses. |
Table I
Summary of the genetic variants
identified in the present study by NGS and Sanger sequencing
analyses.
| A, NGS analysis-OS
tumor tissue |
|---|
| Gene ID | Chr | Pos | Ref | Alt | Type | Context | Consequence | dbSNP | COSMIC | ClinVar | Qual |
|---|
| JAK1 | chr1 | 65310489 | T | C | SNV | Coding |
synonymous_variant | rs2230588 | COSM3751351 | | 100 |
| ALK | chr2 | 29416572 | T | C | SNV | Coding |
missense_variant | rs1670283 | | benign | 100 |
| ALK | chr2 | 29445458 | G | T | SNV | Coding |
synonymous_variant | rs3795850 | COSM5351767 | benign | 100 |
| FGFR3 | chr4 | 1805799 | T | C | SNV | Intron | intron_variant | rs3135888 | | | 100 |
| FGFR3 | chr4 | 1807894 | G | A | SNV | Coding |
synonymous_variant | rs7688609 | | | 100 |
| PDGFRA | chr4 | 55097835 | G | C | SNV | Intron | intron_variant | rs4864504 | | | 100 |
| FGFR4 | chr5 | 176517326 | T | C | SNV | Intron | intron_variant | rs422421 | | | 100 |
| FGFR4 | chr5 | 176517797 | C | T | SNV | Coding |
missense_variant | rs376618 | | | 100 |
| FGFR4 | chr5 | 176519516 | A | G | SNV | Intron |
splice_region_variant, intron_variant | rs3135925 | | | 100 |
| FGFR4 | chr5 | 176523562 | C | A | SNV | Intron | intron_variant | rs31777 | | | 100 |
| FGFR4 | chr5 | 176523597 | A | G | SNV | Intron |
splice_region_variant, intron_variant | rs31776 | | | 100 |
| EGFR | chr7 | 55219909 | G | T | SNV | Intron | intron_variant | rs41364648 | | | 100 |
| EGFR | chr7 | 55228053 | A | T | SNV | Intron | intron_variant | rs1558544 | | | 100 |
| RET | chr10 | 43613843 | G | T | SNV | Coding |
synonymous_variant | rs1800861 | COSM4418405,
COSM4418406 | benign | 100 |
| KRAS | chr12 | 25400206 | G | T | SNV | Intron | intron_variant | rs10842518 | | | 100 |
| B, Sanger
sequencing (TP53 gene)-tumor tissue and peripheral blood |
| TP53
region | Sample | Type | Consequence | |
| TP53 exon 5
deletion | OS tumor | Deletion | deletion | |
| TP53
c.712T>A (p.Cys238Ser) | OS tumor | SNV | missense
variant | |
| TP53
c.215C>G (p.Pro72Arg) | peripheral
blood | SNV | missense
variant | |
Discussion
OS is the most common primary malignant bone tumor
and it is characterized by its rare incidence and occurrence in the
long bones of the extremities, notably in the femur. Adolescents
and young adults are usually affected by the disease and improved
5-year overall survival rates are observed for younger patients
(1). Molecular tests based on
potential cancer-related genomic drivers could improve precision
oncology for OS (21). However,
knowledge about the genomic hallmarks related to OS etiology and
progression is insufficient; thus, treatment options are still
limited. In the present study, NGS analysis and Sanger sequencing
were applied to investigate the genomic landscape of an adolescent
patient diagnosed with OS and treated with a standard protocol. NGS
analysis revealed that OS tumor sample harbored an intratumor
heterogeneity signature and identified somatic variants in the
following genes: JAK1, ALK, FGFR3,
PDGFRA, FGFR4, EGFR, RET and
KRAS.
Janus kinase (JAK) is a family of non-receptor
tyrosine kinase proteins involved in the signal transduction of
multiple cellular events, such as proliferation and differentiation
(22,23). Some JAK mutations have been
identified in different types of cancer. It has been reported that
tumors with JAK1 mutations may exhibit high mutation burden
and microsatellite instability, which may result in immune response
alterations and contribute to tumor immune evasion (24). In the present study, a coding
synonymous variant in the JAK1 gene (rs2230588; COSM3751351)
was identified. Carvalho et al (25) reported the same JAK1 genetic
variant in 25% (5/20) of tumor samples from patients diagnosed with
head and neck squamous cell carcinoma.
Anaplastic lymphoma kinase (ALK) gene encodes a
receptor tyrosine kinase and ALK mutations,
rearrangements/fusions, or amplifications have been identified in
several human cancers (26). ALK
inhibitors represent an effective treatment strategy for patients
with malignancies exhibiting ALK rearrangement and their
effectiveness in patients with OS should be further explored.
Ordulu et al (27) described
a 73-year-old male with high-grade OS and lung metastases who
showed sensitivity to ALK-targeted therapy harboring the
EML4::ALK fusion and the ALKL1196M mutation. By
contrast, Takeyasu et al (28) identified the ITSN2::ALK
fusion in a 17-year-old male patient with OS showing poor
sensitivity to alectinib therapy and progressive disease. In the
present study, only DNA targets were investigated; therefore, gene
fusions in RNA samples were not assessed. NGS analysis revealed a
coding missense variant (rs1670283) and a synonymous variant
(rs3795850; COSM5351767) in the ALK gene and both variants
were classified as benign in the ClinVar database: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000133472.35
and https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000335694.20,
respectively. The ALK variant rs1670283 has also been
associated with hereditary cancer-predisposing syndrome, breast
cancer (29), anaplastic large cell
lymphoma (30), gastric cancer
(31) and benign tumor of the
central nervous system (32). The
ALK variant rs3795850 (COSM5351767) identified in the
present study has also been associated with neuroblastoma
susceptibility and detected in other cancer studies, including
breast cancer (29), lung cancer
(33,34) and Wilms tumor (35).
The fibroblast growth factor receptors (FGFRs) are a
family of receptor tyrosine kinases involved in signaling pathways
of different biological processes, in which the FGF/FGFR pathway
plays a role in bone development and homeostasis (36,37).
FGFR genomic alterations, including gene amplifications and
gene mutations, have been described in OS (38,39).
Additionally, clinical trials and case studies have evaluated the
efficacy of FGFR inhibitors as a treatment option for patients with
OS and reported improvements in progression-free survival (40,41).
FGFR3 gene is a FGFR family member and FGFR3
mutations have been reported in several skeletal dysplasias
(42-44).
In the present study, the FGFR3 variants rs3135888 (intron
variant) and rs7688609 (synonymous variant) were identified. By
using NGS technology, Mansour et al also identified the
FGFR3 rs7688609 variant in a patient with lung carcinoma
(45). Kassem et al
identified the FGFR3 rs7688609 variant in seven patients
with brain tumors through liquid biopsy using NGS (46). The FGFR3 rs7688609 variant
was also identified by panel-based NGS in a case of glioblastoma
(47). FGFR4 also belongs to the
FGFR family and the present study identified five FGFR4
variants: rs422421 (intron variant), rs376618 (missense variant),
rs3135925 (splice region variant, intron variant), rs31777 (intron
variant) and rs31776 (splice region variant, intron variant). In a
meta-analysis study conducted by Moazeni-Roodi et al, the
FGFR4 rs376618 variant was described in three studies and no
association with overall cancer risk was found (48).
The platelet-derived growth factor receptor-alpha
(PDGFRA) gene encodes a receptor tyrosine kinase involved in
multiple cellular events. PDGFRA-mutated tumors can be found
in several cancers and targeted inhibitors have been described as a
treatment option to improve overall clinical outcomes (49-51).
PDGFRA mutations commonly occur in the exons 12/14/18,
notably in the exon 18 (D842V). Armstrong et al described a
refractory metastatic OS case (7-year-old) with partial response to
sorafenib (52). NGS technology
revealed a PDGFRA D846V mutation in the initial tumor sample
but not in the relapse sample, suggesting that this specific
PDGFRA mutation is a sorafenib target. Using a custom NGS
panel, a Brazilian study also identified molecular heterogeneity in
OS tumor tissues, in which copy number variations (CNVs) were
identified in the PDGFRA gene (53). In the present study, the intronic
variant rs4864504 in the PDGFRA gene was identified.
The epidermal growth factor receptor (EGFR) is a
receptor tyrosine kinase involved in downstream signaling cascades
resulting in cellular growth and proliferation. EGFR
deregulation has been identified in a number of cancers, especially
lung cancer and frequently includes activating mutations in
functional domains and amplifications (54,55).
Treatment with EGFR tyrosine kinase inhibitors (TKIs) has emerged
as an important strategy in cancer therapy to benefit patients
harboring EGFR mutations (56). Two EGFR intronic variants
(rs41364648 and rs1558544) were identified in the present study.
Geißler et al (57) reported
the EGFR rs1558544 variant in 7 of 25 patients with
colorectal cancer. This EGFR rs1558544 variant was also
found in a patient with cutaneous squamous cell carcinoma resistant
to conventional treatments but successfully treated with anti-EGFR
targeted therapy (58).
Rearranged during transfection (RET) is a
proto-oncogene that encodes a transmembrane receptor tyrosine
kinase involved in several cellular signaling pathways. RET
gene mutations are commonly found in medullary thyroid carcinoma
but can also be found in other cancers (59). Germline activating mutations and
somatic amplifications in the RET gene have been described
in patients with OS (60). The
RET rs1800861 (COSM4418405) synonymous variant was
identified in the present study. This RET variant was also
found by NGS analysis in a patient with chronic myeloid leukemia
(61).
Kirsten rat sarcoma viral oncogene homologue
(KRAS) is a well-known proto-oncogene playing a central role
as a signal transducer. KRAS mutations are commonly found in
numerous human cancers and targeted therapies with KRAS inhibitors
have emerged as promising treatment strategies (62). The uncommon KRAS intronic
variant rs10842518 (g.25400206 G>T) was identified in the
present study.
Most of the somatic SNVs described in the present
study by NGS analysis were notably identified in genes encoding
tyrosine kinase proteins, revealing an important intratumor
heterogeneity signature that may contribute as an additional event
for OS development. The application of TKIs in patients with OS has
been evaluated as a strategy to achieve improved therapeutic
efficacy, suggesting the simultaneous inhibition of several
relevant receptor tyrosine kinases in OS (63,64).
TP53 is a tumor suppressor gene that encodes
the p53 protein, which plays a key role in cell cycle control and
genome integrity; thus, it is referred to as the guardian of the
genome (65). Therefore, genetic
alterations in the TP53 gene leading to its malfunction are
hallmarks of several human cancers. Regarding OS, mutations in
TP53 are commonly described (18,19,66).
Additionally, OS is commonly diagnosed in Li-Fraumeni syndrome
(LFS), a cancer predisposition syndrome characterized by inherited
pathogenic germline variants in the TP53 gene (66,67).
Mutational hotspots in the TP53 gene notably occur by
nucleotide substitution in the coding sequence within exons
4-8(68). Wunder et al
(69) investigated TP53
mutations (exons 4-10) in 196 OS tumors by single-strand
conformation polymorphism and sequencing, identifying 38 mutations
(19.4%) described as 23 missense mutations, 11 nonsense mutations,
three splice site changes and one in-frame insertion. Chen et
al (18) investigated somatic
mutations in pediatric osteosarcoma samples by whole-genome
sequencing and identified multiple somatic chromosomal alterations
(notably structural variations) and SNVs. High rates of TP53
mutations, including TP53 rearrangements (50%; 16/32),
missense mutations (22%; 7/32), nonsense mutations (16%; 5/32) and
TP53 deletions (6%; 2/32), have been reported. By
whole-genome sequencing, Ribi et al (70) identified patients with OS with a
deletion in intron 1 of TP53, including exon 1, and a
patient harboring a deletion, including the entire TP53
gene. By whole-exome sequencing, Bousquet et al (71) identified TP53 mutations in OS
samples from three young patients, being described as STOP gained
mutation, nonsynonymous mutation, deletions and splice site
mutation. In the present study, no PCR products for TP53
exon 5 were detected in the tumor tissue sample, suggesting that a
deletion event in exon 5 can lead to somatic TP53
inactivation and contribute to OS development. Sanger sequencing
analysis revealed that the patient harbored the missense variant
TP53 c.712T>A (p.Cys238Ser) in tumor tissue sample and
the TP53 c.215C>G (p.Pro72Arg; rs1042522) germline
missense variant in the peripheral blood sample. The variant
TP53 c.712T>A (p.Cys238Ser) is reported to be pathogenic
in the ClinVar database and was also identified in a family with
LFS (72). The TP53
c.215C>G (p.Pro72Arg) is a common single nucleotide polymorphism
and has been reported to be benign for LFS in the ClinVar database.
Taken together, these findings reinforce the idea that TP53
mutations represent key oncogenic drivers in patients with OS.
The present study reported a case of an adolescent
patient with OS harboring an intratumor heterogeneity signature. OS
is a rare and challenging malignant tumor. In Brazil, patients
diagnosed with OS are still treated with a standard protocol,
reinforcing the need for genetic testing to guide diagnosis and
treatment. The present study has several limitations. First,
although the National Institute of Traumatology and Orthopedics is
a reference center for musculoskeletal sarcomas care, DNA
sequencing was not performed in all patients, which could improve
the investigation of the association between the genetic findings
and the development of pediatric OS or even variations in therapy
response. Second, the present study faced the lack of diagnostic
molecular genetic tests to help select patients at highest risk of
disease progression for this NGS-based study. It was hypothesized
that patients with OS at higher risk could harbor genetic
alterations that contribute to both disease development and
progression. Third, only DNA targets were investigated in the
present study; therefore, the detection of clinically actionable
gene fusions that have been described in OS was not assessed.
Despite this, the findings of the present study had the potential
to help to unravel part of the genetic landscape that constitutes
the biologic heterogeneity of OS, providing extensive genomic
information through a refined and advanced NGS-based technology.
Additionally, all genetic variants identified in the present study
had not previously been described in OS cases and might have
contributed to disease development.
Notably, the number of genome-targeted therapies and
the percentage of patients with cancer responding to these
therapies have increased over the years (73). By contrast, patients with OS
continue to be ineligible for targeted therapy due to lack of
genetic testing in clinical routine, being a consequence of little
investment and visibility, which notably represents a knowledge
gap. As researchers, it was hypothesized that the present study
facilitates understanding on the need for OS genetic investigation.
In the next 5 years, it is hoped that more efforts with global
collaboration initiatives in the scientific and medical communities
will include genetic testing in clinical routine for OS and
contribute to clinical trials for new treatment modalities with
targeted therapies to achieve improved clinical responses. In
conclusion, the present study added new information on genetic
aspects contributing for OS development, especially in pediatric
patients. Genomic testing for OS will represent the opportunity to
identify potential cancer driver genes to aid clinical decision
making. Therefore, genetic profiling of OS needs further
investigation.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The NGS datasets generated and/or analyzed during
the current study are available in Figshare (https://doi.org/10.6084/m9.figshare.26015227.v1).
Sanger sequencing datasets for TP53 (exons 4-8) in the
current study are available in Figshare (https://doi.org/10.6084/m9.figshare.27115351.v1).
Authors' contributions
MJ contributed to the conception of the present
study, performed the experiments, analyzed the molecular data and
wrote the original and final draft of the manuscript. RS and TC
contributed significantly to the NGS experiments and analysis. EL
and RP contributed to the acquisition and writing of the clinical
data. AC contributed to the acquisition and writing of the
histopathological data. AL and WM analyzed all the clinical data.
GA and MO reviewed the study and confirmed the authenticity of all
the raw data. All the authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics and
Research Committee of the National Institute of Traumatology and
Orthopedics (approval no. CAAE: 60632822.4.0000.5273). Written
informed consent was obtained from the patient and from the
patient's legal guardian.
Patient consent for publication
Written informed consent was obtained from the
patient and from her legal guardian.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
MJ: https://orcid.org/0000-0002-7351-5588
RS: https://orcid.org/0000-0003-2009-537X
TC: https://orcid.org/0009-0005-9493-1352
EL: https://orcid.org/0000-0002-1111-4906
RP: https://orcid.org/0000-0001-7837-5134
GA: https://orcid.org/0000-0003-2246-719X
MO: https://orcid.org/0000-0002-2983-9593
References
|
1
|
Lee JA, Lim J, Jin HY, Park M, Park HJ,
Park JW, Kim JH, Kang HG and Won YJ: Osteosarcoma in adolescents
and young adults. Cells. 10(2684)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48.
2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Balach T, Stacy GS and Peabody TD: The
clinical evaluation of bone tumors. Radiol Clin North Am.
49:1079–1093. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Carrle D and Bielack SS: Current
strategies of chemotherapy in osteosarcoma. Int Orthop. 30:445–451.
2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zoumpoulidou G, Alvarez-Mendoza C, Mancusi
C, Ahmed RM, Denman M, Steele CD, Tarabichi M, Roy E, Davies LR,
Manji J, et al: Therapeutic vulnerability to PARP1,2 inhibition in
RB1-mutant osteosarcoma. Nat Commun. 12(7064)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Astolfi A, Nannini M, Indio V, Schipani A,
Rizzo A, Perrone AM, De Iaco P, Pirini MG, De Leo A, Urbini M, et
al: Genomic database analysis of uterine leiomyosarcoma mutational
profile. Cancers (Basel). 12(2126)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rizzo A, Pantaleo MA, Saponara M and
Nannini M: Current status of the adjuvant therapy in uterine
sarcoma: A literature review. World J Clin Cases. 7:1753–1763.
2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rizzo A, Nannini M, Astolfi A, Indio V, De
Iaco P, Perrone AM, De Leo A, Incorvaia L, Di Scioscio V and
Pantaleo MA: Impact of chemotherapy in the adjuvant setting of
early stage uterine leiomyosarcoma: A systematic review and updated
meta-analysis. Cancers (Basel). 12(1899)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Boye K, Longhi A, Guren T, Lorenz S, Næss
S, Pierini M, Taksdal I, Lobmaier I, Cesari M, Paioli A, et al:
Pembrolizumab in advanced osteosarcoma: Results of a single-arm,
open-label, phase 2 trial. Cancer Immunol Immunother. 70:2617–2624.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Davis KL, Fox E, Merchant MS, Reid JM,
Kudgus RA, Liu X, Minard CG, Voss S, Berg SL, Weigel BJ and Mackall
CL: Nivolumab in children and young adults with relapsed or
refractory solid tumours or lymphoma (ADVL1412): A multicentre,
open-label, single-arm, phase 1-2 trial. Lancet Oncol. 21:541–550.
2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Monga V, Dodd R, Scherer A, Gutierrez WR,
Tanas M, Mott SL and Milhem M: Phase Ib study of decitabine in
combination with gemcitabine in treatment of advanced soft tissue
and bone sarcomas. J Clin Oncol. 38:11550. 2020.
|
|
13
|
Krishnadas DK, Shusterman S, Bai F, Diller
L, Sullivan JE, Cheerva AC, George RE and Lucas KG: A phase I trial
combining decitabine/dendritic cell vaccine targeting MAGE-A1,
MAGE-A3 and NY-ESO-1 for children with relapsed or
therapy-refractory neuroblastoma and sarcoma. Cancer Immunol
Immunother. 64:1251–1260. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chi SN, Yi JS, Williams PM, Roy-Chowdhuri
S, Patton DR, Coffey BD, Reid JM, Piao J, Saguilig L, Alonzo TA, et
al: Tazemetostat in patients with tumors with alterations in EZH2
or the SWI/SNF complex: Results from NCI-COG pediatric MATCH trial
Arm C (APEC1621C). J Clin Oncol. 40 (16 Suppl)(10009)2022.
|
|
15
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Schwartzberg L, Kim ES, Liu D and Schrag
D: Precision oncology: Who, how, what, when, and when not? Am Soc
Clin Oncol Educ Book. 37:160–169. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gagan J and Van Allen EM: Next-generation
sequencing to guide cancer therapy. Genome Med.
7(80)2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen X, Bahrami A, Pappo A, Easton J,
Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al:
Recurrent somatic structural variations contribute to tumorigenesis
in pediatric osteosarcoma. Cell Rep. 7:104–112. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Czarnecka AM, Synoradzki K, Firlej W,
Bartnik E, Sobczuk P, Fiedorowicz M, Grieb P and Rutkowski P:
Molecular biology of osteosarcoma. Cancers (Basel).
12(2130)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Leite C, Delmonico L, Alves G, Gomes RJ,
Martino MR, Silva AR, Moreira ADS, Maioli MC, Scherrer LR, Bastos
EF, et al: Screening of mutations in the additional sex combs like
1, transcriptional regulator, tumor protein p53, and KRAS
proto-oncogene, GTPase/NRAS proto-oncogene, GTPase genes of
patients with myelodysplastic syndrome. Biomed Rep. 7:343–348.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pestana RC, Groisberg R, Roszik J and
Subbiah V: Precision oncology in sarcomas: Divide and conquer. JCO
Precis Oncol. 3(PO.18.00247)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bousoik E and Aliabadi HM: ‘Do We Know
Jack’ about JAK? A closer look at JAK/STAT signaling pathway. Front
Oncol. 8(287)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hu X, Li J, Fu M, Zhao X and Wang W: The
JAK/STAT signaling pathway: From bench to clinic. Signal Transduct
Target Ther. 6(402)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Albacker LA, Wu J, Smith P, Warmuth M,
Stephens PJ, Zhu P, Yu L and Chmielecki J: Loss of function JAK1
mutations occur at high frequency in cancers with microsatellite
instability and are suggestive of immune evasion. PLoS One.
12(e0176181)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Carvalho TG, Carvalho AC, Maia DCC, Ogawa
JK, Carvalho AL and Vettore AL: Search for mutations in signaling
pathways in head and neck squamous cell carcinoma. Oncol Rep.
30:334–340. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Corte CMD, Viscardi G, Liello R, Fasano M,
Martinelli E, Troiani T, Ciardiello F and Morgillo F: Role and
targeting of anaplastic lymphoma kinase in cancer. Mol Cancer.
17(30)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ordulu Z, Giunta P, Hung WT, Hung YP,
Simon J, Fintelmann FJ, Lennerz JK, Naxerova K and Cote GM:
Sensitivity to ALK-directed therapy in osteosarcoma with an
acquired ALK rearrangement. JCO Precis Oncol.
7(e2300287)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Takeyasu Y, Okuma HS, Kojima Y, Nishikawa
T, Tanioka M, Sudo K, Shimoi T, Noguchi E, Arakawa A, Mori T, et
al: Impact of ALK inhibitors in patients with ALK-rearranged
nonlung solid tumors. JCO Precis Oncol.
5(PO.20.00383)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pan ZW, Wang XJ, Chen T, Ding XW, Jiang X,
Gao Y, Mo WJ, Huang Y, Lou CJ and Cao WM: Deleterious mutations in
DNA repair gene FANCC exist in BRCA1/2-negative Chinese familial
breast and/or ovarian cancer patients. Front Oncol.
9(169)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Noguchi K, Ikawa Y, Takenaka M, Sakai Y,
Fujiki T, Kuroda R, Ikeda H, Abe T, Sakai S and Wada T: Acquired
L1196M ALK mutation in anaplastic lymphoma kinase-positive
anaplastic large cell lymphoma during alectinib administration.
EJHaem. 4:305–308. 2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Machlowska J, Kapusta P, Baj J, Morsink
FHM, Wołkow P, Maciejewski R, Offerhaus GJA and Sitarz R:
High-throughput sequencing of gastric cancer patients: Unravelling
genetic predispositions towards an early-onset subtype. Cancers
(Basel). 12(1981)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Taher MM, Hassan AA, Saeed M, Jastania RA,
Nageeti TH, Alkhalidi H, Dairi G, Abduljaleel Z, Athar M,
Bouazzaoui A, et al: Next generation DNA sequencing of atypical
choroid plexus papilloma of brain: Identification of novel
mutations in a female patient by Ion Proton. Oncol Lett.
18:5063–5076. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Choi YL, Soda M, Yamashita Y, Ueno T,
Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H,
et al: EML4-ALK mutations in lung cancer that confer resistance to
ALK inhibitors. N Engl J Med. 363:1734–1739. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Doebele RC, Pilling AB, Aisner DL,
Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ,
Heasley LE, Franklin WA, et al: Mechanisms of resistance to
crizotinib in patients with ALK gene rearranged non-small cell lung
cancer. Clin Cancer Res. 18:1472–1482. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wegert J, Ishaque N, Vardapour R, Geörg C,
Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, et al:
Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA
microprocessor complex underlie high-risk blastemal type Wilms
tumors. Cancer Cell. 27:298–311. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Dai S, Zhou Z, Chen Z, Xu G and Chen Y:
Fibroblast growth factor receptors (FGFRs): Structures and small
molecule inhibitors. Cells. 8(614)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Su N, Jin M and Chen L: Role of FGF/FGFR
signaling in skeletal development and homeostasis: Learning from
mouse models. Bone Res. 2(14003)2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Barøy T, Chilamakuri CSR, Lorenz S, Sun J,
Bruland ØS, Myklebost O and Meza-Zepeda LA: Genome analysis of
osteosarcoma progression samples identifies FGFR1 overexpression as
a potential treatment target and CHM as a candidate tumor
suppressor gene. PLoS One. 11(e0163859)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Brown LM, Ekert PG and Fleuren EDG:
Biological and clinical implications of FGFR aberrations in
paediatric and young adult cancers. Oncogene. 42:1875–1888.
2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Davis LE, Bolejack V, Ryan CW, Ganjoo KN,
Loggers ET, Chawla S, Agulnik M, Livingston MB, Reed D, Keedy V, et
al: Randomized double-blind phase II study of Regorafenib in
patients with metastatic osteosarcoma. J Clin Oncol. 37:1424–1431.
2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Duffaud F, Mir O, Boudou-Rouquette P,
Piperno-Neumann S, Penel N, Bompas E, Delcambre C, Kalbacher E,
Italiano A, Collard O, et al: Efficacy and safety of regorafenib in
adult patients with metastatic osteosarcoma: A non-comparative,
randomised, double-blind, placebo-controlled, phase 2 study. Lancet
Oncol. 20:120–133. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Du X, Xie Y, Xian CJ and Chen L: Role of
FGFs/FGFRs in skeletal development and bone regeneration. J Cell
Physiol. 227:3731–3743. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xue Y, Sun A, Mekikian PB, Martin J,
Rimoin DL, Lachman RS and Wilcox WR: FGFR3 mutation frequency in
324 cases from the international skeletal dysplasia registry. Mol
Genet Genomic Med. 2:497–503. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wen X, Li X, Tang Y, Tang J, Zhou S, Xie
Y, Guo J, Yang J, Du X, Su N and Chen L: Chondrocyte FGFR3
regulates bone mass by inhibiting osteogenesis. J Biol Chem.
291:24912–24921. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Mansour H, Ouhajjou A, Bajic VB and
Incitti R: Next-Generation sequencing at high sequencing depth as a
tool to study the evolution of metastasis driven by genetic change
events of lung squamous cell carcinoma. Front Oncol.
10(1215)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kassem NM, Kassem HA, Selim H and Hafez M:
Targeted next generation sequencing provides insight for the
genetic alterations in liquid biopsy of Egyptian brain tumor
patients. Egypt J Med Hum Genet. 23(23)2022.
|
|
47
|
Loriguet L, Morisse MC, Dremaux J, Collet
L, Attencourt C, Coutte A, Boone M, Sevestre H, Galmiche A, Gubler
B, et al: Combining genomic analyses with tumour-derived slice
cultures for the characterization of an EGFR-activating kinase
mutation in a case of glioblastoma. BMC Cancer.
18(964)2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Moazeni-Roodi A, Sarabandi S, Karami S,
Hashemi M and Ghavami S: An updated meta-analysis of the
association between fibroblast growth factor receptor 4
polymorphisms and susceptibility to cancer. Biosci Rep.
40(BSR20192051)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sun Y, Yue L, Xu P and Hu W: An overview
of agents and treatments for PDGFRA-mutated gastrointestinal
stromal tumors. Front Oncol. 12(927587)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Velghe AI, Cauwenberghe SV, Polyansky AA,
Chand D, Montano-Almendras CP, Charni S, Hallberg B, Essaghir A and
Demoulin JB: PDGFRA alterations in cancer: Characterization of a
gain-of-function V536E transmembrane mutant as well as
loss-of-function and passenger mutations. Oncogene. 33:2568–2576.
2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wagner AJ, Kindler H, Gelderblom H,
Schöffski P, Bauer S, Hohenberger P, Kopp HG, Lopez-Martin JA,
Peeters M, Reichardt P, et al: A phase II study of a human
anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously
treated patients with metastatic gastrointestinal stromal tumors.
Ann Oncol. 28:541–546. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Armstrong AE, Walterhouse DO, Leavey PJ,
Reichek J and Walz AL: Prolonged response to sorafenib in a patient
with refractory metastatic osteosarcoma and a somatic PDGFRA D846V
mutation. Pediatr Blood Cancer. 66(e27493)2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Guimarães GM, Tesser-Gamba F, Petrilli AS,
Donato-Macedo CRP, Alves MTS, de Lima FT, Garcia-Filho RJ, Oliveira
R and Toledo SRC: Molecular profiling of osteosarcoma in children
and adolescents from different age groups using a next-generation
sequencing panel. Cancer Genet. 258-259:85–92. 2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jin R, Peng L, Shou J, Wang J, Jin Y,
Liang F, Zhao J, Wu M, Li Q, Zhang B, et al: EGFR-Mutated squamous
cell lung cancer and its association with outcomes. Front Oncol.
11(680804)2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Liu H, Zhang B and Sun Z: Spectrum of EGFR
aberrations and potential clinical implications: Insights from
integrative pan-cancer analysis. Cancer Commun (Lond). 40:43–59.
2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nan X, Xie C, Yu X and Liu J: EGFR TKI as
first-line treatment for patients with advanced EGFR
mutation-positive non-small-cell lung cancer. Oncotarget.
8:75712–75726. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Geißler AL, Geißler M, Kottmann D, Lutz L,
Fichter CD, Fritsch R, Weddeling B, Makowiec F, Werner M and
Lassmann S: ATM mutations and E-cadherin expression define
sensitivity to EGFR-targeted therapy in colorectal cancer.
Oncotarget. 8:17164–17190. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Capalbo C, Belardinilli F, Filetti M,
Parisi C, Petroni M, Colicchia V, Tessitore A, Santoni M, Coppa A,
Giannini G and Marchetti P: Effective treatment of a
platinum-resistant cutaneous squamous cell carcinoma case by EGFR
pathway inhibition. Mol Clin Oncol. 9:30–34. 2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Verrienti A, Grani G, Sponziello M, Pecce
V, Damante G, Durante C, Russo D and Filetti S: Precision oncology
for RET-related tumors. Front Oncol. 12(992636)2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kovac M, Woolley C, Ribi S, Blattmann C,
Roth E, Morini M, Kovacova M, Ameline B, Kulozik A, Bielack S, et
al: Germline RET variants underlie a subset of paediatric
osteosarcoma. J Med Genet. 58:20–24. 2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Sklarz LM, Wittke C, Krohn S, GROßE-Thie
C, Junghanss C, Escobar HM and Glaeser H: Genetic mutations in a
patient with chronic myeloid leukemia showing blast crisis 10 years
after presentation. Anticancer Res. 38:3961–3966. 2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Huang L, Guo Z, Wang F and Fu L: KRAS
mutation: From undruggable to druggable in cancer. Signal Transduct
Target Ther. 6(386)2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Chen C, Shi Q, Xu J, Ren T, Huang Y and
Guo W: Current progress and open challenges for applying tyrosine
kinase inhibitors in osteosarcoma. Cell Death Discov.
8(488)2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Tian Z, Niu X and Yao W: Receptor tyrosine
kinases in osteosarcoma treatment: Which is the key target? Front
Oncol. 10(1642)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Borrero LJH and El-Deiry WS: Tumor
suppressor p53: Biology, signaling pathways, and therapeutic
targeting. Biochim Biophys Acta Rev Cancer.
1876(188556)2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Mirabello L, Yeager M, Mai PL,
Gastier-Foster JM, Gorlick R, Khanna C, Patiño-Garcia A,
Sierrasesúmaga L, Lecanda F, Andrulis IL, et al: Germline TP53
variants and susceptibility to osteosarcoma. J Natl Cancer Inst.
107(djv101)2015.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hameed M and Mandelker D: Tumor syndromes
predisposing to osteosarcoma. Adv Anat Pathol. 25:217–222.
2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wunder JS, Gokgoz N, Parkes R, Bull SB,
Eskandarian S, Davis AM, Beauchamp CP, Conrad EU, Grimer RJ, Healey
JH, et al: TP53 mutations and outcome in osteosarcoma: a
prospective, multicenter study. J Clin Oncol. 23:1483–1490.
2005.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Ribi S, Baumhoer D, Lee K, Edison Teo AS,
Madan B, Zhang K, Kohlmann WK, Yao F, Lee WH, et al: TP53 intron 1
hotspot rearrangements are specific to sporadic osteosarcoma and
can cause Li-Fraumeni syndrome. Oncotarget. 6:7727–7740.
2015.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Bousquet M, Noirot C, Accadbled F, de
Gauzy JS, Castex MP, Brousset P and Gomez-Brouchet A: Whole-exome
sequencing in osteosarcoma reveals important heterogeneity of
genetic alterations. Ann Oncol. 27:738–744. 2016.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Balmaña J, Nomdedéu J, Díez O, Sabaté JM,
Balil A, Pericay C, López JJ, Brunet J, Baiget M and Alonso C:
Description of a new TP53 gene germline mutation in a family with
the Li-Fraumeni syndrome. Genetic counselling to healthy mutation
carriers. Med Clin (Barc). 119:497–499. 2002.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|
|
73
|
Haslam A, Kim MS and Prasad V: Updated
estimates of eligibility for and response to genome-targeted
oncology drugs among US cancer patients, 2006-2020. Ann Oncol.
32:926–932. 2021.PubMed/NCBI View Article : Google Scholar
|