Introduction
Congenital disorders of kidney are a major disorder
that frequently affect children and contribute to prenatal and
perinatal deaths (1). The most
prevalent kidney disorders include polycystic kidney disease
(2), unilateral renal agenesis
(3), and bilateral renal agenesis
(4). Renal hypodysplasia is one of
the most lethal renal disorders resulting in fetal death in
utero. It is a highly phenotypically heterogeneous autosomal
recessive disease (5). Key
characteristics of renal hypodysplasia include oligohydramnios
in utero and Potter facies, which can both be detected using
ultrasonography (6). Other less
common clinical observations include facial dysmorphism, pulmonary
hypoplasia, congenital hip dislocation and clubfoot (7).
Kidney anomalies are estimated to account for 20-50%
of all congenital anomalies in developing fetus, with renal
hypodysplasia affecting 0.1-0.2% of the global population (4). Renal hypodysplasia can be inherited in
both autosomal recessive and autosomal dominant pattern, with a
with a male-to-female ratio of 2.7:1(8).
Studies have identified pathogenic variants in
several genes as causes of renal hypodysplasia, some of which are
syndromic (9), impacting other
organs such as the eyes and limbs; other gene mutation leads to
non-syndromic form of renal hypodysplasia (10). While heterozygous mutations in
several genes can cause renal hypodysplasia with varying severity,
only mutations in the RET and ITGA8 genes are associated
with bilateral renal hypodysplasia (5,11).
In the present clinical study, a 25 year-old woman
was reported with a history of multiple pregnancy losses presented
within 18-25 weeks of pregnancy with oligohydramnios, facial
dysmorphism, and spontaneous abortion at 25 weeks. Clinical
observations indicated that the fetus may have been affected by
renal hypodysplasia. To investigate further, whole exome sequencing
was performed using DNA from both parents and the current
pregnancy. Among several variants identified, evidence was provided
associating a novel missense variant, Ser654Leu in the ITGA8
gene, with renal hypodysplasia. This variant was confirmed in the
current pregnancy through Sanger sequencing.
Case report
A 25-year-old woman was admitted to Sanjeevani
Multi-speciality Hospital and Trauma Centre in December 2021. She
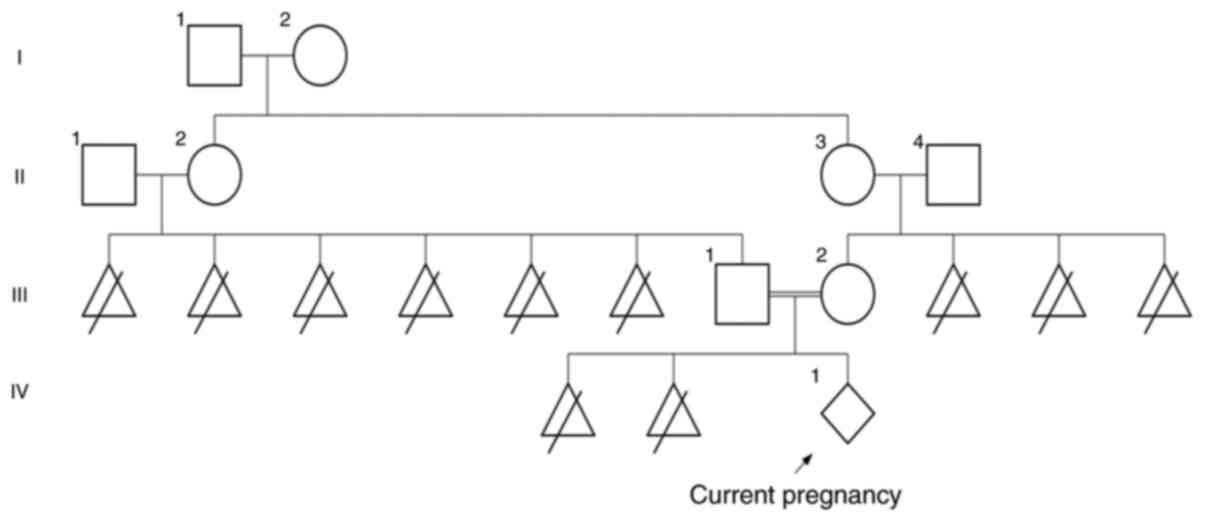
was born to second-degree consanguineous parents (Fig. 1) and had a history of two prior
pregnancy losses. Her first spontaneous abortion occurred at age 22
after 20 weeks of gestation, and her second at age of 23 after 18
weeks of gestation. Both instances were accompanied by mild
abdominal pain and severe vaginal bleeding, that was unresponsive
to medication. Genetic counselling revealed that her previous
pregnancies had involved facial dysmorphism and oligohydramnios in
the fetus.
During her third pregnancy, she reported similar
features of abdominal pain and blood spotting which were managed
with medication. Initial ultrasonography at 14 weeks revealed mild
oligohydramnios, with no congenital anomalies detected. However, at
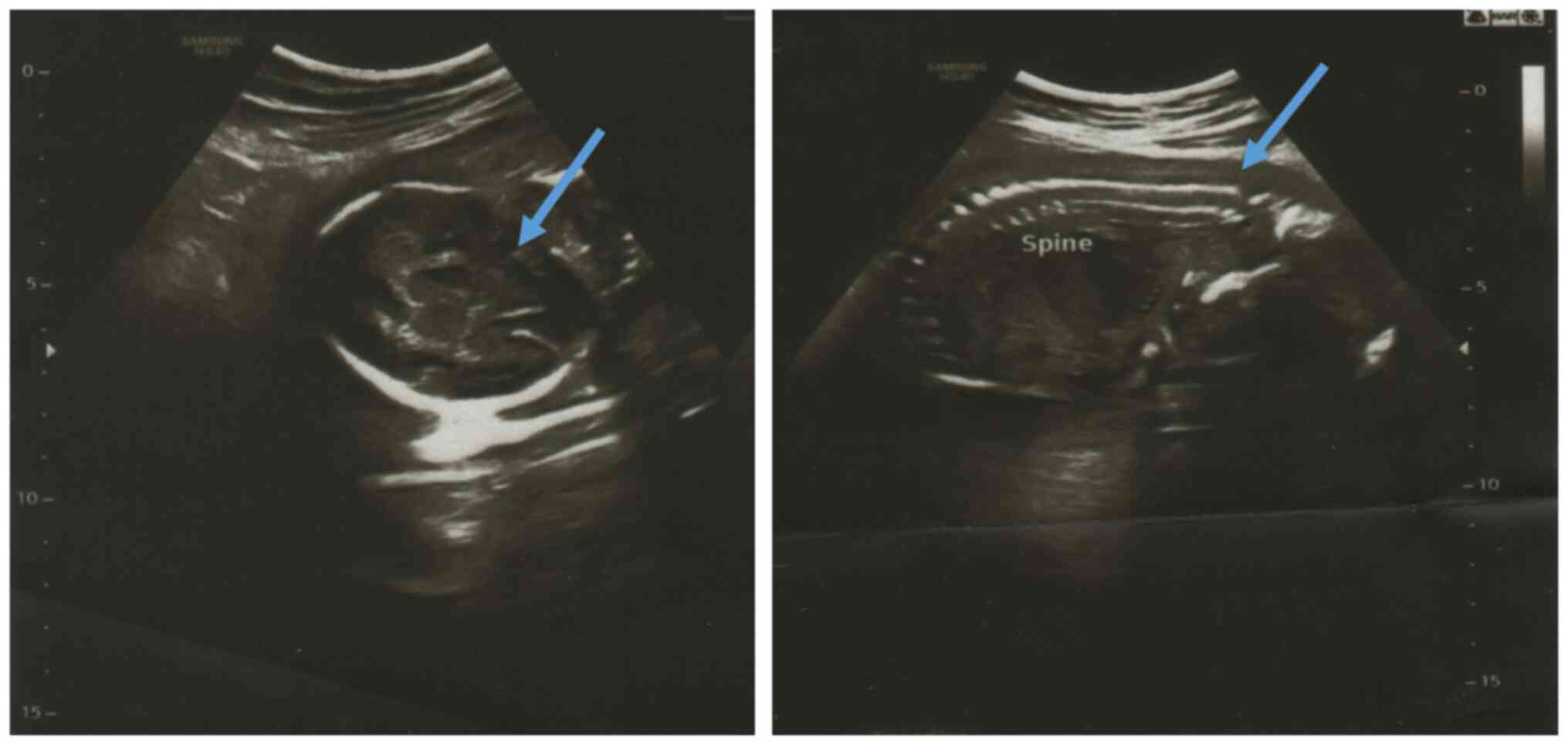
20 weeks, ultrasonography revealed facial abnormalities and severe
oligohydramnios (Fig. 2), with
normal placental attachment and development. Karyotyping confirmed
the absence of chromosomal anomalies in both parents and the
current pregnancy.
During early pregnancy, amniotic fluid is produced
by the mother's body; however, after 10 weeks, fetal urine becomes
the primary source of amniotic fluid (12). Usually, oligohydramnios in the
developing fetus is primarily caused by an undeveloped kidney or
abnormal placenta (13). In the
present case, since placenta development was completely normal, an
abnormal kidney development was considered to be the cause. Due to
a strong history of oligohydramnios associated with all pregnancies
in a span of 3 years, genetic testing was recommended by the
clinician, and trio whole exome sequencing was performed, followed
by Sanger sequencing.
There was a history of six pregnancy losses in the
paternal grandparents and three pregnancy losses in the maternal
grandparents. As per information collected in genetic counselling
sessions, there were no major other health condition present in the
family. All pathological tests were negative.
Genomic DNA was isolated from 5 ml of blood from
each parent using QIAamp DNA Blood Mini Kits (cat. no. 51104;
Thermo Fisher Scientific, Inc.) as per manufacturer's protocol.
Whole exome sequencing was performed with AmpliSeq Exome RDY kit
(cat. no. A38264; Thermo Fisher Scientific, Inc.) in an Ion
GeneStudio S5 Plus System (Thermo Fisher Scientific, Inc.),
covering all exonic regions with a depth of at least 120X. Raw data
were quality trimmed to remove low-quality data having Phred score
of <30, followed by mapping to the human genome (hg19), and
variant calling was performed with Torrent Suite v5.5.5 (Thermo
Fisher Scientific, Inc.). Variants were annotated using the gnomAD
population database (https://gnomad.broadinstitute.org/), ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/), mutation
impact prediction scores, and conservation databases. Finally
screening of the variants was performed manually by removing all
variants with an allelic frequency of >1% or those predicted to
be benign as determined by the mutation impact prediction tools
consensually; only variants present in the genes known to cause
renal dysplasia were eventually evaluated. Consequently, a single
variant in ITGA8 gene was linked to the clinical
condition.
To confirm the presence of same variant in the
current pregnancy, amniotic fluid was obtained from current
pregnancy and maternal cell contamination (MCC) followed by whole
exome sequencing and Sanger sequencing were performed. Using MCC
testing it was confirmed that the obtained amniotic fluid doesn't
contains any maternal DNA. Sanger sequencing amniotic fluid were
performed using primers (forward, 5'-CCAAACCACAGGCTAACCCA-3' and
reverse, 5'-GAGGAAAGCTCTGGTTCCGT-3') to amplify 332 bp region of
the ITGA8 gene.
Discussion
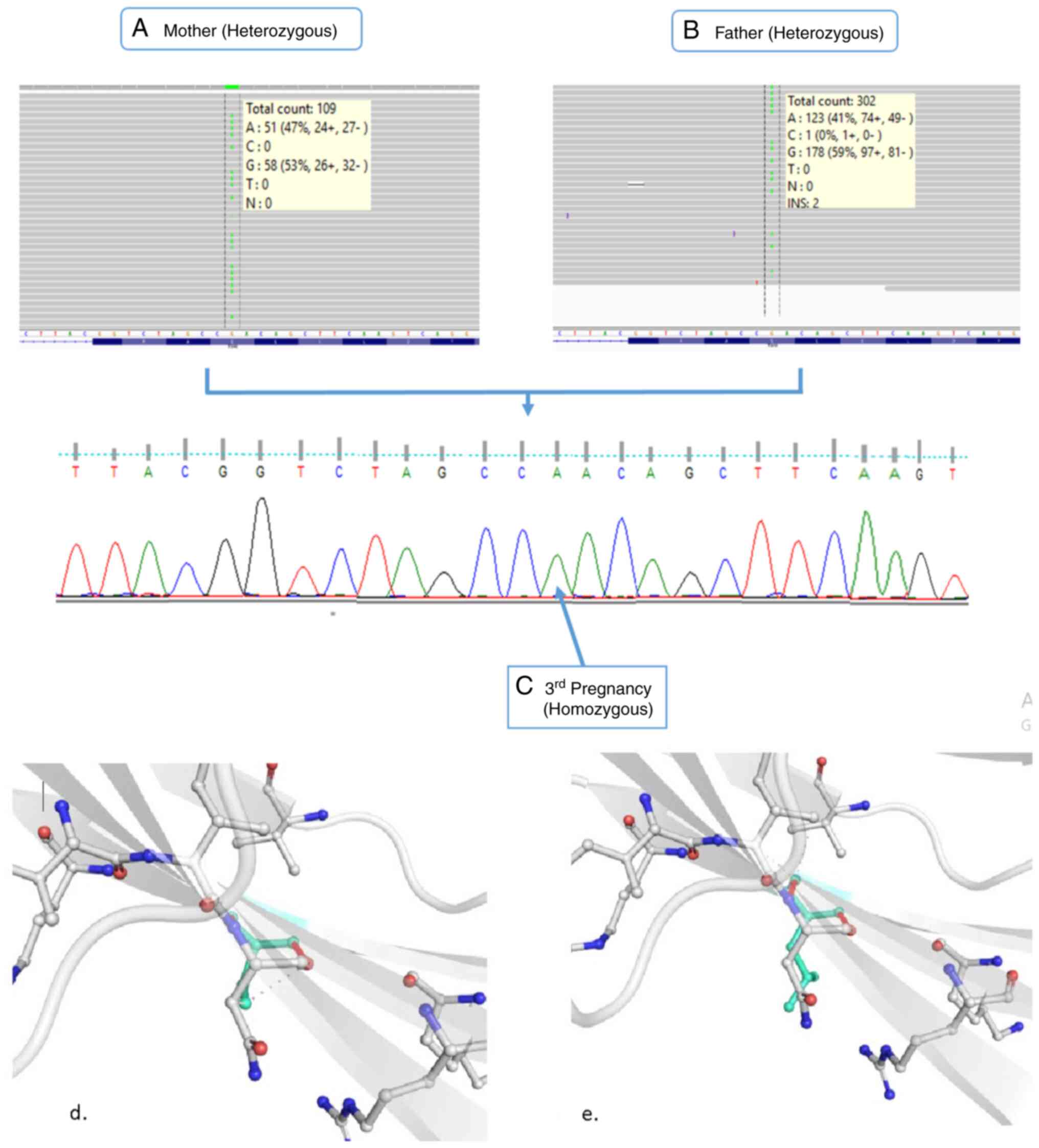
After following all the steps, a single carrier
missense variant (NM_003638.2:c.1961C>T; p.Ser654Leu) in
ITGA8 gene was identified, which was present in both
parents. Whole exome sequencing was also performed using the
amniotic fluid of the pregnant mother, confirming that no other
de novo variant was causative in the current pregnancy. This
carrier missense variant (NM_003638.2:c.1961C>T; p.Ser654Leu) in
the ITGA8 gene was found to be homozygous in the amniotic
fluid of the pregnant mother (Fig.
3A-C).
This variant was classified a variant of uncertain
significance (PM2, PP3 and PP4) based on ACMG guidelines (14) following these criteria: i) The
homozygous variant c.1961C>T in the ITGA8 gene is absent
in both the gnomAD genome and gnomAD exome databases, while the
heterozygous state of the variant has a frequency of 0.001209 in
South Asian population and 0.0001835 worldwide in the gnomAD exome
database (PM2); ii) As demonstrated in Table SI, various software predicted this
variant as damaging, and it is conserved across several species
tested, as indicated in Table SII
(PP3); and iii) the family history demonstrated a high specificity
to multiple pregnancy losses with oligohydramnios (PP4).
After confirming the presence of this variant using
whole exome sequencing and Sanger sequencing, its impact on the
protein was evaluated. Homology modelling was performed to predict
the structures of both the native ITGA8 protein and the
protein with the variant, using Robetta (15) tool. The generated structures were
compared using DynaMut (16) and
SDM (17) tools to assess changes
in stability (∆∆G). Both DynaMut and SDM predicted this change as
stabilizing with ∆∆G value of 0.980 and 1.570 kcal/mol,
respectively (Table SIII).
Vibrational entropy (ΔΔSVib) was estimated using the
ENCoM tool (18) between both
protein structures (wild-type and mutated) to evaluate the change
in molecular flexibility, which revealed that the Ser654Leu variant
decreases protein flexibility (ΔΔSVib=-0.0992 kcal/mol).
Structural analysis using PyMOL (19), revealed that substitution of the
polar uncharged amino acid serine with the hydrophobic amino acid
leucine altered the intermolecular interaction pattern, resulting
in the loss of carbonyl contact and ionic interaction, along with
the gain of a new carbonyl contact (Fig. 3D and E). These comparative observations
confirmed the damaging impact of this missense variant, leading to
the amino acid change from serine to leucine at position 654.
ITGA8 proteins are heterodimeric
transmembrane receptors composed of alpha and beta subunits. They
possess a functionally active domain known as ‘integrin alpha-2’,
which contains FG-GAP repeats, proven to be active participants in
ligand binding (20). These repeats
are also essential for cell-cell interaction (21), host pathogen recognition, and
regulation of neurite outgrowth in the sensory and motor neurons
and play a vital role in kidney organogenesis. Absence of ITGA8
gene product can potentially affect normal epithelial mesenchymal
transition that results in renal hypodysplasia (22). Improved understanding of mechanism
underlaying the impact of mutation in this gene will help in
increasing the prenatal diagnostic yield (23).
In the present study, both parents were heterozygous
for the ITGA8 (Ser654Leu) missense variant, while the fetus
was homozygous for the same variant. This suggests that the
identified variant in the ITGA8 gene has a significant role
in proper kidney development. The conservation score and variant
impact prediction indicated that the variant is deleterious, and
structural analysis further supported its damaging effect on
protein structure. The present study reports a novel variant of the
ITGA8 gene and provides clinical evidence for the role of
this genetic variant in renal development. In vitro or in
vivo functional study will add more strength to this finding
for use in prenatal diagnosis.
Prenatal genetic diagnosis offers wide variety of
information about the health of the fetus. The present case study
demonstrated the impact of missense mutation (Ser654Leu) in
ITGA8 gene and will help in risk assessment for the renal
hypodysplasia in further pregnancies thereby, enabling clinicians
to take proactive measures for surveillance and preventions. As
both parents were carrier for the same mutation, the women may be
suggested to conceive through donor sperm.
Supplementary Material
Impact of the variant predicted by
different mutation impact prediction tools.
Conservation score and conservation
prediction of the variant predicted based on different conservation
tools.
Change in Gibbs free energy and
vibrational entropy predicted by different tools while comparing
structure of wild type protein and mutated (Ser654Leu) protein of
ITGA8 gene.
Acknowledgements
The authors would like to thank Dr Archana Singh
from Sanjeevani Multi-Speciality Hospital and Trauma Centre
(Rampur, India) for facilitating the sample collection and
collecting patient information.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the Sequence Read Archive database under accession numbers
SRR31344619, SRR31344620 and SRR31344621 or at the following URL:
https://www.ncbi.nlm.nih.gov/sra/?term=SRR31344619,
https://www.ncbi.nlm.nih.gov/sra/?term=SRR31344620 and
https://www.ncbi.nlm.nih.gov/sra/?term=SRR31344621.
Authors' contributions
AM conceived the idea and designed the project. KGS
performed the experiments. Both authors read and approved the final
version of the manuscript. KGS and AM confirm the authenticity of
all the raw data.
Ethics approval and consent of
participation
The study design and protocol were conducted in
accordance with the guidelines of the ACMG, and were approved
(approval no. IECH/2022/SEP-014) by the ethical review committee of
Sanjeevani Multi-Speciality Hospital and Trauma Centre, (Rampur,
India).
Patient consent for publication
Written informed consent for the publication of her
data and associated images was obtained from the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Capone VP, Morello W, Taroni F and Montini
G: Genetics of congenital anomalies of the kidney and urinary
tract: The current state of play. Int J Mol Sci.
18(796)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Xue C and Mei CL: Polycystic KIDNEY
DISEASE AND RENAL FIBROSis. Adv Exp Med Biol. 1165:81–100.
2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pichler R, Oswald J, Glodny B, Skradski V,
Aigner F and Rehder P: Unilateral renal agenesis with absent ductus
deferens, epididymis and seminal vesicle: Incidental finding in a
22-year-old patient with maldevelopment of the mesonephric duct.
Urol Int. 86:365–369. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huber C, Shazly SA, Blumenfeld YJ, Jelin E
and Ruano R: Update on the prenatal diagnosis and outcomes of fetal
bilateral renal agenesis. Obstet Gynecol Surv. 74:298–302.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gómez-Conde S, Dunand O, Hummel A,
Morinière V, Gauthier M, Mesnard L and Heidet L: Bi-allelic
pathogenic variants in ITGA8 cause slowly progressive renal disease
of unknown etiology. Clin Genet. 103:114–118. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Schmidt W, Schroeder TM, Buchinger G and
Kubli F: Genetics, pathoanatomy and prenatal diagnosis of Potter I
syndrome and other urogenital tract diseases. Clin Genet.
22:105–127. 1982.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cain DR, Griggs D, Lackey DA and Kagan BM:
Familial renal agenesis and total dysplasia. Am J Dis Child.
128:377–380. 1974.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pashayan HM, Dowd T and Nigro AV:
Bilateral absence of the kidneys and ureters. Three cases reported
in one family. J Med Genet. 14:205–209. 1977.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sanna-Cherchi S, Caridi G, Weng PL,
Scolari F, Perfumo F, Gharavi AG and Ghiggeri GM: Genetic
approaches to human renal agenesis/hypoplasia and dysplasia.
Pediatr Nephrol. 22:1675–1684. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Weber S, Moriniere V, Knüppel T, Charbit
M, Dusek J, Ghiggeri GM, Jankauskiené A, Mir S, Montini G,
Peco-Antic A, et al: Prevalence of mutations in renal developmental
genes in children with renal hypodysplasia: Results of the ESCAPE
study. J Am Soc Nephrol. 17:2864–2870. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Humbert C, Silbermann F, Morar B, Parisot
M, Zarhrate M, Masson C, Tores F, Blanchet P, Perez MJ, Petrov Y,
et al: Integrin alpha 8 recessive mutations are responsible for
bilateral renal agenesis in humans. Am J Hum Genet. 94:288–294.
2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fitzsimmons ED and Bajaj T: Embryology,
amniotic fluid. StatPearls Publishing, pp1-4, 2019.
|
|
13
|
Zilberman Sharon N, Pekar-Zlotin M, Kugler
N, Accart Z, Nimrodi M, Melcer Y, Cuckle H and Maymon R:
Oligohydramnios: How severe is severe? J Matern Fetal Neonatal Med.
35:5754–5760. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kim DE, Chivian D and Baker D: Protein
structure prediction and analysis using the Robetta server. Nucleic
Acids Res. 32:W526–W531. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rodrigues CHM, Pires DEV and Ascher DB:
DynaMut: Predicting the impact of mutations on protein
conformation, flexibility and stability. Nucleic Acids Res.
46:W350–W355. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pandurangan AP, Ochoa-Montaño B, Ascher DB
and Blundell TL: SDM: A server for predicting effects of mutations
on protein stability. Nucleic Acids Res. 45:W229–W235.
2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Frappier V, Chartier M and Najmanovich RJ:
ENCoM server: Exploring protein conformational space and the effect
of mutations on protein function and stability. Nucleic Acids Res.
43:W395–W400. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Delano WL: The PyMOL molecular graphics
system. CCP4 Newsletter on protein crystallography. Computer
Science, Chemistry, 2002.
|
|
20
|
Springer TA: Folding of the N-terminal,
ligand-binding region of integrin alpha-subunits into a
beta-propeller domain. Proc Natl Acad Sci USA. 94:65–72.
1997.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Loftus JC, Smith JW and Ginsberg MH:
Integrin-mediated cell adhesion: The extracellular face. J Biol
Chem. 269:25235–25238. 1994.PubMed/NCBI
|
|
22
|
Pavlović N, Kelam N, Racetin A, Filipović
N, Pogorelić Z, Prusac IK and Vukojević K: Expression profiles of
ITGA8 and VANGL2 Are altered in congenital anomalies of the kidney
and urinary tract (CAKUT). Molecules. 29(3294)2024.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Marek I, Hilgers KF, Rascher W, Woelfle J
and Hartner A: A role for the alpha-8 integrin chain (itga8) in
glomerular homeostasis of the kidney. Mol Cell Pediatr.
7(13)2020.PubMed/NCBI View Article : Google Scholar
|