Introduction
Identifying the impact of cholesterol reduction
agents and cardiovascular morbidity in the scientific world was
crucial. Since then, statins, the cholesterol-lowering class of
drugs, are considered to have achieved remarkable results in this
field and are the most prescribed drugs worldwide (1,2).
Statins offer a considerable health benefit for both primary and
secondary prevention of cardiovascular diseases (3). Conversely, concerns have been raised
about the negative consequences of long-term statin usage. In a
significant percentage of individuals, statin medications appear to
be safe to utilize. However, individuals with various other disease
co-morbidities are more likely to experience undesirable side
effects from long-term statin treatment. Though the advantages of
statins exceed their heightened risks, the FDA has recently added
diabetes as a black box label warning for statins (4). Statins have been proven to double an
individual's chance of developing diabetes mellitus due to how they
may interfere with insulin signaling pathways, impair the function
of pancreatic beta cells, and possibly raise insulin resistance
(5). The risk of developing a new
onset of diabetes rose by 46% after statin therapy in a METSIM
cohort study, and numerous studies point towards the
diabetogenicity of statins (6-8).
It is concerning to realize that individuals with cardiovascular
disease consuming statins are also vulnerable to other fatal
conditions such as diabetes, which necessitates severe
consideration and further research in this area. Globally and
markedly more quickly, the burden of diabetes mellitus is
increasing. Diabetes disease comprises 6.28% of the global
population (9).
The specific mechanism(s) of diabetogenesis with
statins are unresolved. However, they may include decreased insulin
sensitivity, mitigated beta-cell function and elevated
intracellular cholesterol absorption (10,11).
Insulin resistance was found in a study with 10-week statin
administration in non-diabetic individuals, indicating the
propensity of the risk of developing diabetes (12). An interventional RCT study testing
rosuvastatin (JUPITER) revealed a slight but substantial rise in
diabetes incidence rates in individuals who received statins over a
median of 1.9 years (13).
Individuals who are at high-risk categories for developing diabetes
had a greater incidence of the disease with statin medication in a
cohort study indicating its likelihood of developing diabetes at a
faster rate (14). Statins produced
insulin resistance by diminishing the phosphorylation of insulin
receptor, insulin receptor substrate-1, AKT, glycogen synthase 3β
and downregulated Glut-4 expression; and it was demonstrated that
statin-induced insulin resistance is independent of cholesterol
biosynthesis inhibition by a fatty acid-mediated effect on the
insulin signaling pathway (15).
Statins cause increased IL-1β secretion from macrophages. Long-term
fluvastatin treatment of obese mice led to impaired
insulin-stimulated glucose uptake in adipose tissue and disrupted
insulin signaling in lipopolysaccharide (LPS)-primed 3T3-L1
adipocytes. This effect was associated with an increase in
caspase-1 activity (16).
Statins, while recognized for their
cholesterol-lowering benefits in coronary heart disease prevention,
exhibit pleiotropic effects extending beyond lipid modulation.
These include improved endothelial function, atherosclerotic plaque
stabilization, reduced oxidative stress and inflammation and
inhibition of thrombogenic response. Furthermore, statins
demonstrate benefits beyond the cardiovascular system, impacting
the immune, central nervous and skeletal systems. These pleiotropic
effects are partially attributed to the inhibition of isoprenoids,
particularly affecting small GTP-binding proteins such as Rho, Ras,
and Rac, essential for intracellular signalling (17). Beyond their established
cardiovascular benefits, statins have shown promise in tackling
critical conditions including cancer and inflammatory bowel
disease. Their potential extends to improving vascular tone,
suggesting a broader therapeutic scope than previously recognized
(18,19). This protective effect extends beyond
cholesterol reduction, highlighting the pleiotropic benefits of
statins. Rajangam et al (20) demonstrated that acute rosuvastatin
administration significantly reduced doxorubicin-induced
cardiotoxicity in rats, evidenced by decreased cardiac marker
enzymes and improved antioxidant activity. Statins also exhibit
pleiotropic properties with potential applications in various
neurological conditions, including epilepsy. Interestingly, recent
research suggests that statins may interact synergistically with
certain antiepileptic drugs. For example, Rajangam et al
(21) demonstrated that
atorvastatin potentiated the anticonvulsant effects of lacosamide
in both electroshock and chemo-convulsant models of epilepsy in
mice. This synergistic effect was linked to increased plasma
concentrations of atorvastatin and a potential modulation of
neuronal sodium channels (21).
Further demonstrating the pleiotropic benefits of statins,
rosuvastatin mitigated scopolamine-induced amnesia in mice,
improving cognitive function in elevated plus maze and Morris water
maze tests (22).
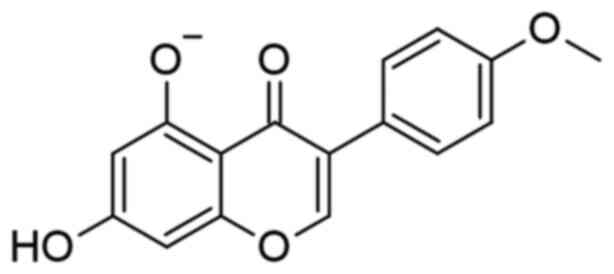
Biochanin-A (BA) is one of the essential isoflavones
that can potentially prevent and treat numerous diseases by acting
on various human systems. BA is a plant-derived chemical found in
soy, red clover, peas and alfalfa. It has shown excellent research
outcomes in treating cancers, neurological disorders, diabetes,
airway disorders, hyperlipidemia, and numerous other
disorders/diseases (23). The
chemical structure of BA is shown in Fig. 1. By increasing SIRT-1 expression in
pancreatic tissue, BA considerably lowered blood glucose levels,
glucose tolerance, insulin resistance, and enhanced insulin
sensitivity, demonstrating a significant impact on type 2 diabetes
mellitus in a study on high-fat diet diabetic rats (24). It has a significant effect on type 2
diabetes mellitus, which might be linked with SIRT-1. Another study
revealed the ability of BA to prevent diabetes by diminishing the
genes transforming growth factor-β1 and protease-activated
receptors 2, which are responsible for diabetic nephropathy and
reducing blood glucose levels of diabetic animals (25). While BA has demonstrated
anti-diabetic effects in various studies (24,26-31)
its potential in mitigating statin-induced diabetes remains
unexplored. Moreover, the molecular mechanisms underlying such an
effect remain to be elucidated.
Sirtuins are members of Sir2 genes which is a
nicotinamide adenine dinucleotide (NAD)+-dependent
deacetylase family of proteins. Among seven different sirtuins,
sirtuin-1 (SIRT-1) has direct and indirect involvement in insulin
signaling, and insulin resistance causing Type 2 diabetes mellitus
(32). By increasing SIRT-1
expression, the insulin sensitivity of skeletal muscle can be
improved. Previous studies have highlighted the role of SIRT-1 in
metabolism and shown that its regulation can influence insulin
resistance (33), and metformin
could be acting through SIRT-1 for insulin resistance by its
upregulation (34). It has been
revealed that diabetic patients have SIRT-1 downregulated in muscle
tissue (35). SIRT-1 is a protein
that is dysregulated when a person is on a high-calorie diet or
when there is chronic exposure to high free fatty acids (FFA)
(36), which is the case in
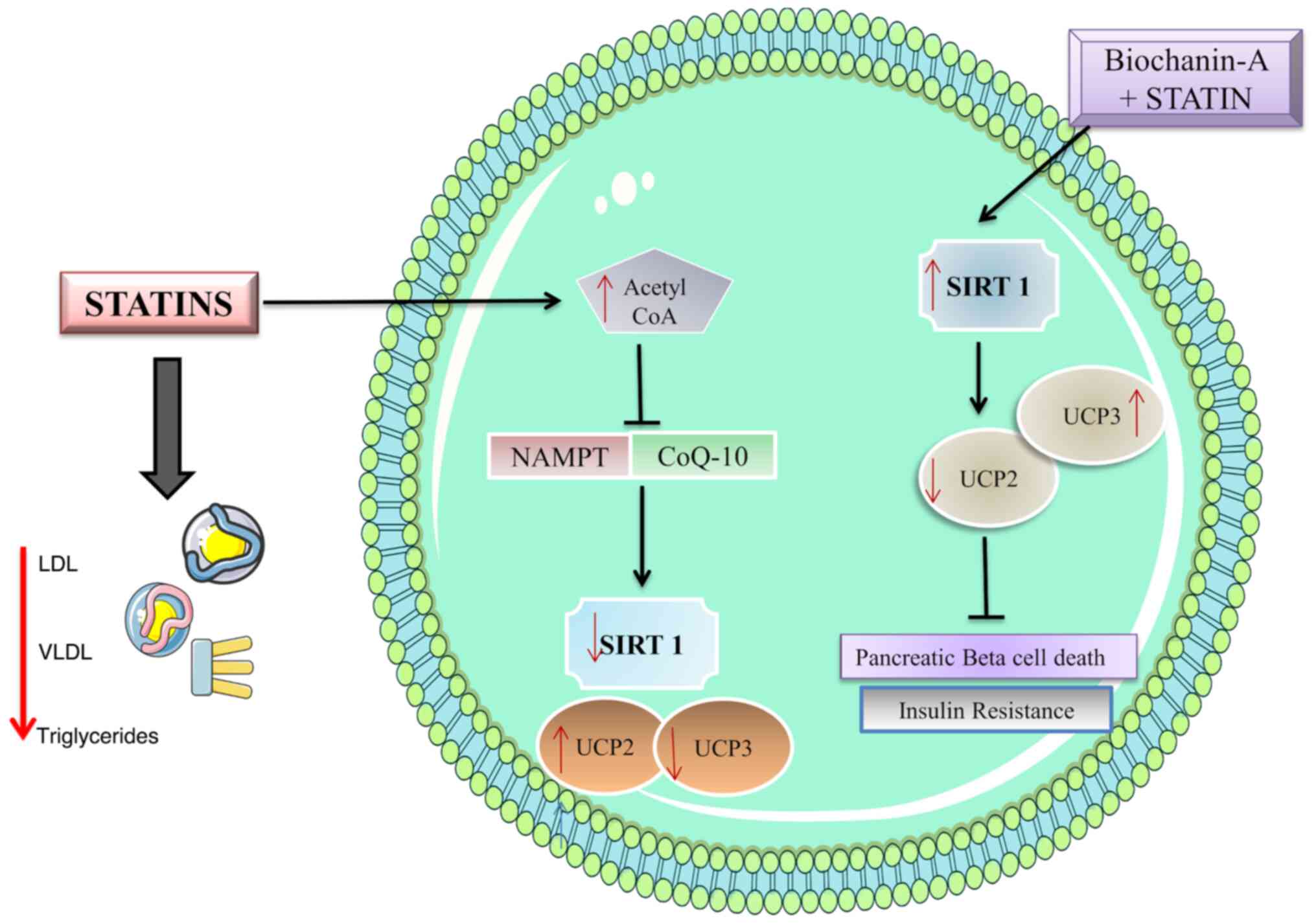
patients who are taking statin. The increase in acetyl Co-A will
lead to the accumulation of FFA and result in the downregulation of
SIRT-1 (33,37,38).
SIRT-1 inhibition can cause upregulation of uncoupling proteins
(UCP) UCP2 and UCP3 which will result in insulin resistance and
pancreatic beta cell death.
BA has not been investigated specifically for its
use in statin-induced diabetes. To date, only hydroxychloroquine
has been tested successfully for the same (39), but its potential mechanism has not
been researched; only its action has been confirmed. Mechanism for
statin-induced diabetes has not been well established. The present
study addresses these critical gaps by investigating the mechanism
of statin-induced diabetes and, for the first time, establishing
the role of SIRT-1 and UCPs in this context.
Statins cause insulin resistance due to the
accumulation of acetyl CoA which is diverted to the synthesis of
FFA (15). This excess accumulation
of FFA is further exacerbated by the downregulation of SIRT-1,
preventing the metabolism of the fatty acids (33,36-38).
These accumulated fatty acids prevent the phosphorylation of
Insulin receptors resulting in insulin resistance. The possible
molecular pathway involving SIRT-1 is depicted in Fig. 2.
BA works to upregulate SIRT-1 in statin-induced
diabetes which in turn halts the apoptosis of pancreatic beta
cells. It also functions to prevent insulin resistance in muscle
cells by the same mechanism. The upregulation of SIRT-1 causes the
downregulation of UCP2(40) which
plays a role in insulin resistance in pancreatic beta cells and
muscle cells. Statin treatment is known to lower the UCP3 level
(15,41,42).
Insulin resistance was assessed using L6 skeletal muscle cells and
pancreatic beta cell apoptosis in MIN-6 cells. MIN-6 cells, derived
from mouse pancreatic beta-cells, are a well-established in
vitro model for studying beta-cell function and dysfunction.
Their responsiveness to glucose, as evidenced by glucose-stimulated
insulin secretion, makes them particularly relevant for
investigating statin-induced diabetes, which is often characterized
by impaired beta-cell function. Furthermore, previous studies,
including the authors' own preliminary findings, have identified
that MIN-6 cells exhibit sensitivity to statin exposure, mimicking
aspects of statin-induced beta-cell dysfunction observed in
vivo. This sensitivity makes them a suitable model for
investigating the potential protective effects of BA in
statin-induced diabetes.
L6 skeletal muscle cells were chosen for our study
because skeletal muscle is a major site of glucose disposal in the
body, accounting for a significant portion of insulin-stimulated
glucose uptake. Dysregulation of glucose metabolism in skeletal
muscle is a key contributor to insulin resistance and type 2
diabetes. Importantly, statins have been shown to influence
skeletal muscle function and insulin sensitivity, highlighting the
relevance of L6 cells in the context of statin-induced diabetes. By
including L6 cells, it was aimed to capture potential systemic
effects of both statins and BA on glucose metabolism beyond
pancreatic beta-cells.
Prior to our study, there was limited understanding
of the mechanism behind statin-induced diabetes. While BA has shown
promise as an anti-diabetic agent in preclinical studies, its
specific potential to mitigate statin-induced diabetes was largely
unexplored. The present study directly addresses this gap by
investigating the effects of BA on cellular models of
statin-induced diabetes. Although some studies have explored the
influence of BA on metabolic pathways, its precise mechanisms of
action in the context of statin-induced diabetes were not
well-defined. The current research delves into these molecular
mechanisms, focusing particularly on SIRT-1, UCP2 and lipogenic
pathways, to elucidate how BA might counteract the diabetogenic
effects of statins at a cellular level.
The present study contributes significantly to the
field by providing the first evidence, to the best of our
knowledge, of BA's potential to prevent statin-induced diabetes in
an in vitro setting. Novel insights into the molecular
mechanisms through which BA exerts its protective effects are
provided, highlighting potential therapeutic targets for further
investigation. These findings lay the groundwork for future in
vivo and clinical studies to further validate the therapeutic
potential of BA and ultimately translate these findings into
clinical practice.
Statins continue to be essential in treating
cardiovascular conditions since their substantial benefits surpass
potential side effects. Therefore, statins are likely to maintain
their critical role in cardiac treatment strategies. However,
diabetes is a serious chronic disorder with severe complications,
and preventing added comorbidities is essential. For this purpose,
if an additional compound can prevent diabetes, it would be of
great benefit to millions of patients. The study would also help
establish the mechanism of statin-induced diabetes and explore the
role of SIRT-1 and UCPs in the disease.
Materials and methods
Cell culture and chemicals
The mouse pancreatic beta cell line (MIN-6) and rat
myoblast (L6) cell lines were procured from the American Type
Culture Collection (ATCC). The cells were cultured in Dulbecco's
Modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.). The MIN-6 culture medium was supplemented with 15%
heat-inactivated fetal bovine serum (FBS), 1%
antibiotic-antimycotic solution and 1X β-mercaptoethanol (all from
Gibco; Thermo Fisher Scientific, Inc.) in an atmosphere of 5%
CO2 and 95% humidity at 37˚C. Media was changed every 48
h. The cell was dissociated with cell dissociating solution (0.2%
trypsin, 0.02% EDTA, glucose-free PBS).
L6 were maintained using DMEM with 10% FBS,
penicillin (100 IU/ml) and streptomycin (100 µg/ml) (both from
Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
of 5% CO2 at 37˚C until confluent. The cell was
dissociated with cell dissociating solution (0.2% trypsin, 0.02%
EDTA, glucose-free PBS). When L6 myoblasts reached confluence, the
medium was switched to the differentiation medium containing DMEM
and 2% horse serum (Gibco; Thermo Fisher Scientific, Inc.). After 4
additional days, the differentiated L6 cells had fused into
myotubes. L6 myotubes were used for treatment. All the chemicals
were obtained from Invitrogen; Thermo Fisher Scientific, Inc.
unless otherwise stated.
Cell viability assay
The effect of BA and atorvastatin on the viability
of L6 and MIN-6 cells was determined by sulforhodamine B (SRB)
assay (43). Briefly, a confluent
monolayer of cells was trypsinized, counted, and seeded in a flat
bottom 96-well plate at a density of 5x103 cells/well
with a culture medium. After 24 h, the residual medium was
replaced, and the cells were incubated in a new culture medium
containing different concentrations of atorvastatin and BA. The
pre-treatment with 0.0001 to 10 µM of BA for 24 h and 100 nM of
atorvastatin for 48 h in the L6 cells and pre-treatment with 6.25
to 200 µM of BA for 24 h and 50 µM of atorvastatin for 48 h in
MIN-6 cells were carried out for the study. After desired
incubation, cells were fixed with 50% (w/v) cold TCA, and then
plates were kept at 4˚C for 1 h. Following 1 h incubation, plates
were washed with distilled water four times and allowed to dry at
room temperature (RT). Dried plates were stained with 0.4% (w/v)
SRB in 1% (v/v) acetic acid for 30 min in dark. Excess and unbound
SRB dye was removed by repeated washing with 1% (v/v) acetic acid,
and plates were allowed to air dry. Absorbance was measured at 540
nm using a microplate reader by solubilizing the bound dye with 10
mM (w/v) Tris HCl.
Glucose uptake assay
A total of 5x103 L6 cells were seeded per
well in a 96-well plate using 100 µl of DMEM supplemented with 10%
FBS. The cell culture was maintained by replacing the medium every
2-3 days. On the 5th day, the cells were differentiated into
myotubes by substituting the medium with DMEM containing 2% horse
serum. Subsequent daily medium replacements with low serum
facilitated myotube maturation over 3 days. Prior to the assay, the
medium was switched to serum-free DMEM. On the assay day, cells
were treated with various conditions, including insulin or samples
lacking serum or glucose, and incubation was performed for
specified durations. Glucose uptake was assessed using a
2-deoxyglucose (2DG) uptake assay (Glucose uptake Glo-Kit; cat no.
J1341; Promega Corporation) wherein cells were exposed to 0.1 mM
2DG in PBS for 30 min at 25˚C. The reaction was halted by adding
stop buffer followed by neutralization buffer, and then
2-Deoxy-D-glucose 6-phosphate (2DG6P) detection reagent was added.
After 1 h of incubation at 25˚C, luminescence was measured using a
luminometer with integration ranging from 0.3 to 1 sec. The Glucose
Uptake Rate was calculated according to the following formula: Rate
of glucose uptake=[(2DG6P x (volume of sample) ÷ (number of cells x
time of uptake)].
FFA estimation assay
DMEM, FBS, penicillin-streptomycin, trypsin and the
FFA Quantification Kit (cat no. MAK044; MilliporeSigma) were
utilized. L6 cells (1x106) were harvested and extracted
using 200 µl of chloroform containing 1% Triton-100, employing a
micro-pestle. The resulting samples underwent centrifugation at
13,000 x g for 10 min at 4˚C to eliminate insoluble material. The
organic phase was carefully transferred to a new tube and air-dried
at 50˚C to eliminate chloroform, discarding the resultant pellet.
Subsequently, samples were subjected to drying using a nitrogen
evaporator to remove any residual organic solvent.
The dried lipids were reconstituted by dissolving
them in 200 µl of the FFA assay buffer, followed by sonication or
vortex until a homogeneous mixture was achieved. FFA quantification
was performed according to the manufacturer's protocol (cat. no.
MAK044). In brief, 50 µl of the sample was mixed with 2 µl of ACS
and incubated for 30 min at 37˚C. This was followed by adding a
master mix of 50 µl to each reaction. The samples were thoroughly
mixed and further incubated for 30 min at 37˚C in the dark.
Absorbance measurements were received at 570 nm.
Cholesterol estimation assay
Cells (1x106) were extracted using 200 µl
of a mixture containing chloroform, isopropanol and IGEPAL
(7:11:0.1) in a micro-homogenizer. After extraction, samples were
centrifuged at 13,000 x g for 10 min at 4˚C to eliminate insoluble
material. The resulting organic phase was carefully transferred to
a new tube and air-dried at 50˚C to remove chloroform, with the
pellet subsequently discarded. Samples were dried using a nitrogen
evaporator to eliminate residual organic solvent.
The dried lipids were reconstituted by dissolving
them in 200 µl of Cholesterol Assay Buffer (1X PBS with 50 mM
cholic acid), followed by sonication or vortex until a homogeneous
mixture was obtained. Cholesterol estimation was performed using a
modified protocol employing the Agape kit (cholesterol
quantification kit; cat. no. MAK043; MilliporeSigma). Specifically,
10 µl of the sample was mixed with 200 µl of cholesterol assay
reagent and incubated for 10 min at 37˚C. Absorbance was then
measured at 510 nm.
Insulin release assay
The MIN-6 cell line, sourced from ATCC, was cultured
in DMEM supplemented with 10% inactivated FBS, glutamine, BME,
penicillin (100 IU/ml) and streptomycin (100 µg/ml) until
confluent. After dissociation with cell dissociating solution
(0.25% trypsin, 0.02% EDTA, in PBS), cells were seeded at
0.8x106 cells/well in a 6-well plate and incubated for
24 h at 37˚C in a 5% CO2 incubator. The cells were then
treated with BA samples for 24 h, followed by statin treatment at
50 µM for 48 h. Subsequently, the media was replaced with Krebs's
buffer containing 5.8 mM glucose and incubated for 4 h. Cell
culture supernatants were collected and subjected to ELISA for
insulin quantification according to the manufacturer's protocol
(Mouse INS1 ELISA kit; cat. no. RAB0817; MilliporeSigma). In the
ELISA procedure, standards and samples were added to appropriate
wells and incubated for 2.5 h at RT or overnight at 4˚C. After
washing, detection antibody and Streptavidin solution were added
successively, followed by incubation and washing steps. TMB
one-step substrate reagent was added, and after 30 min of
incubation in the dark, the reaction was stopped with a stop
solution. Absorbance was measured at 450 nm.
Mitochondrial membrane potential (MMP)
assay
The MIN-6 cells were treated with 50 and 100 µM BA
for 24 h, followed by 50 µM atorvastatin treatment for 48 h
post-incubation. After treatment, the media was removed, and cells
were gently washed with 1X PBS. Subsequently, the cells were
stained with Rhodamine 123 (cat. no: R8004; MilliporeSigma) and
analyzed using a BD FACSCanto™ II flow cytometer (BD
Biosciences). The data were analyzed using FlowJo™
software (version 10.8; BD Biosciences).
Western blotting
The procedure involved initial sample preparation,
where cells or tissues were harvested and homogenized in a RIPA
buffer (cat. no. 89901; Thermo Fisher Scientific, Inc.) containing
a protease inhibitor cocktail (cat. no. 11873580001; Roche
Diagnostics). Following centrifugation to remove debris, protein
concentration was determined using Bradford or BCA assay.
Subsequently, proteins were separated via SDS-PAGE electrophoresis,
loaded onto the gel alongside molecular weight markers, and
electrophoresed until dye migration was complete. Transfer of
proteins to a PVDF or nitrocellulose membrane was carried out using
a wet or semi-dry transfer apparatus with an appropriate transfer
buffer. Post-transfer, the membrane underwent blocking with 5%
non-fat milk or BSA (cat. no. A7906; MilliporeSigma) in TBST
containing 0.1% Tween-20 to prevent non-specific binding before
being incubated with primary antibodies overnight at 4˚C. After
washing, the membrane was probed with HRP-conjugated secondary
antibodies and rewashed to remove unbound antibodies. Protein bands
were visualized using ECL Western Blotting Detection Reagent (cat.
no. RPN2106; GE Healthcare Life Sciences), and images were captured
with a ChemiDoc™ Imaging System (Bio-Rad Laboratories,
Inc.). Band intensity analysis was performed using ImageJ software
(version 1.53; National Institutes of Health). Statistical analysis
was conducted using GraphPad Prism software (version 8.0; GraphPad
Software, Inc.; Dotmatics) to ascertain significant differences
between experimental groups. Positive and negative controls were
included to validate assay specificity, while housekeeping proteins
served as loading controls for normalization. Experiments were
conducted with sufficient biological and technical replicates to
ensure robustness and reproducibility of results.
Protein estimation for insulin receptor pathway and
UCPs in L6 cells was conducted by BCA protein assay kit
(Invitrogen; Thermo Fisher Scientific, Inc.). The cells were
conventionally sub-cultured and counted using a Hemocytometer. The
cell count was adjusted to 1x105 cells/2 ml. A total of
2 ml of cell suspension was added to each dish in a 6-well plate
and incubated until the cells reached confluence. Cells were
further cultured in DMEM supplemented with 2% horse serum for 7
days with alternate-day media change. The cells, post harvesting,
were washed twice using 1X PBS. Cells were lysed to extract total
protein using 300 µl RIPA buffer containing 1X protease inhibitor.
Cell contents were incubated for 30 min by gentle mixing every 5
min at 4˚C. Post incubation, the cells were centrifuged at 14,123 x
g for 15 min for 4˚C to obtain the protein lysates. The supernatant
containing the protein lysate was carefully transferred to a fresh
tube, and the protein concentration was determined using the BCA
Protein Assay Kit according to the manufacturer's instructions. A
total of 100 µg protein samples from each cell lysate were mixed
with 5X loading dye and heated for 2 min at 95°C.
Protein samples were loaded and separated on 8-, 10- and 15%
SDS-PAGE gel using Mini protean Tetra cell (Bio-Rad Laboratories,
Inc.). Nitrocellulose membrane (0.2 µM) was equilibrated in
transfer buffer for 10 min at RT. Protein transfer was conducted
for 15 min in Turbo Transblot (Bio-Rad Laboratories, Inc.)
apparatus at 2.5 A and 25 V. Blots were blocked in 3% BSA in TBST
for 1 h at RT followed by incubation with respective primary
antibodies (antibody details provided in Table I, Table
II and Table III) at
appropriate dilutions overnight at 4˚C. The blots were washed three
times with TBST for 5 min at RT. The blots were then incubated for
1 h at RT with HRP-conjugated secondary antibodies (anti-Rabbit
IgG; HRP-conjugated; 1:10,000; cat. no. E-AB-1003; Elabscience
Biotechnology, Inc.). After three washes with TBST for 5 min at
room temperature, the blots were rinsed with ECL reagent for 1 min
in the dark, and the images were captured between 0.5-15 sec
exposure in a Chemidoc XRS + imaging system. The SDS-PAGE gel
profile with the percentage of separating and stacking gel, and
primary antibody details for different protein markers in the L6
cell line are provided in Fig. 3
and Table I.
 | Table ISDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
different protein markers in the L6 cell line. |
Table I
SDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
different protein markers in the L6 cell line.
| Protein
markers | Separating gel
percentage (%) | Stacking gel
percentage (%) | Dilution | Cat. no. | Supplier | Exposure time
(sec) |
|---|
| GAPDH | 10 | 5 | 1:1,000 | E-AB-20072 | Elabscience
Biotechnology, Inc. | 15 |
| IRS-1 | 8 | 5 | 1:500 | E-AB-31831 | | 1 |
| Pan Akt | 10 | 5 | 1:500 | E-AB-12213 | | 15 |
| GLUT4 | 10 | 5 | 1:500 | E-AB-31558 | | 10 |
| PPAR-Gamma | 10 | 5 | 1:1,000 | E-AB-60059 | | 15 |
| UCP2 | 15 | 5 | 1:1,000 | E-AB-70257 | | 30 |
| UCP3 | 10 | 5 | 1:1,000 | E-AB-17514 | | 5 |
| Table IISDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
different protein markers in the MIN-6 cell line. |
Table II
SDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
different protein markers in the MIN-6 cell line.
| Protein
markers | Separating gel
percentage (%) | Stacking gel
percentage (%) | Dilution | Cat. no. | Supplier | Exposure time
(sec) |
|---|
| GAPDH | 10 | 5 | 1:1,000 | E-AB-20072 | Elabscience
Biotechnology, Inc. | 1 |
| p53 | 10 | 5 | 1:1,000 | E-AB-32466 | | 10 |
| Nicotinamide
phospho ribosyl-transferase | 10 | 5 | 1:600 | E-AB-14435 | | 10 |
| Sirtuin-1 | 10 | 5 | 1:2,000 | E-AB-32901 | | 20 |
| Table IIISDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
caspase-3 protein markers in the MIN6 cell line. |
Table III
SDS-PAGE gel profile with the
percentage of separating and stacking gel, and antibody details for
caspase-3 protein markers in the MIN6 cell line.
| Protein
markers | Separating gel
percentage (%) | Stacking gel
percentage (%) | Dilution | Cat. no. | Supplier | Exposure time
(sec) |
|---|
| GAPDH | 10 | 5 | 1:1,000 | E-AB-20072 | Elabscience
Biotechnology, Inc. | 0.5-10 |
| Caspase-3 | 10 | 5 | 1:1,000 | E-AB-66940 | | 0.5-60 |
MIN-6 cells were cultured using a similar procedure
as the L6 cells with the following modifications: The cell count
was adjusted to 10x106 cells/2 ml before plating in P35
dishes, and 20 µg of protein samples from each cell lysate were
loaded per lane for subsequent analysis. Apoptosis markers, SIRT-1
and nicotinamide phospho-ribosyl-transferase (NAMPT) protein
expression were analyzed in the MIN-6 cell line.
The SDS-PAGE gel profile and details of SDS PAGE
loading for different protein markers in MIN-6 cell line are
presented in Fig. 4 and Table II. The SDS-PAGE gel profile with
the percentage of separating and stacking gel, and primary antibody
details for caspase-3 protein markers in the MIN-6 cell line is
given in Fig. 5 and Table III.
Statistical analysis
Experimental results are expressed as the mean ±
SEM. One-way ANOVA using GraphPad Prism software (version 8 and
8.4.2; Dotmatics) was used for statistical analysis, followed by
Dunnett's multiple comparison test to assess the significance
between groups or unless stated otherwise. P<0.05 was considered
to indicate a statistically significant difference. All assays were
performed with three independent experiments (n=3), except for
western blots, where two independent experiments (n=2) were
conducted for the analysis of protein markers.
Results
Effect of BA and atorvastatin on the
viability of L6 and MIN-6 cells
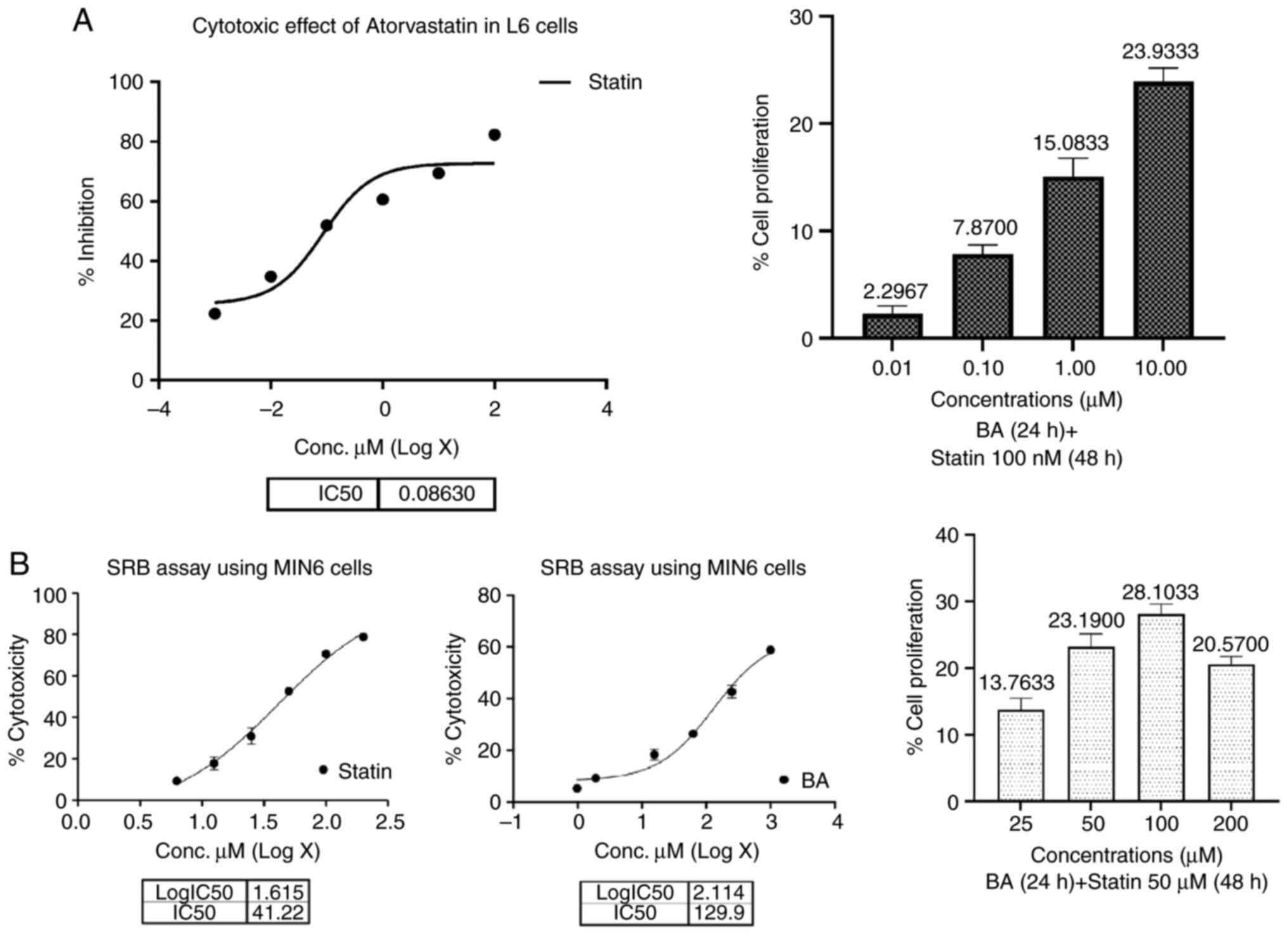
The cytotoxic effects of atorvastatin on L6 and
MIN-6 cells were assessed via the SRB assay, revealing an
IC50 value of 86.3 nM for L6 cells and 41.22 µM for
MIN-6 cells. Treatment of L6 cells with BA at varying
concentrations resulted in negligible cytotoxicity, with a maximum
of 19.78% observed at 10 µM. Subsequent co-treatment with 100 nM
atorvastatin and BA led to a significant increase in cell
proliferation, reaching 23.93% at 10 µM concentration in L6 cells
(Fig. 6A). Similarly, in MIN-6
cells, BA demonstrated a protective effect against statin-induced
cytotoxicity, with cell proliferation of up to 28.10% observed at
100 µM concentration of atorvastatin (Fig. 6B). Notably, BA exhibited a
dose-dependent response in both cell lines, with lower
concentrations failing to induce proliferation. These findings
suggest that BA may effectively counteract statin-induced
cytotoxicity and promote cell proliferation in both L6 and MIN-6
cells. For subsequent studies, concentrations of 10 and 1 µM BA in
combination with 100 nM of atorvastatin were utilized in L6 cells,
while 50 µM statin-induced cytotoxicity treated with 100 and 50 µM
BA was employed in MIN-6 cells to further elucidate its protective
mechanisms.
Effect of BA and atorvastatin on the
glucose uptake of L6 cells
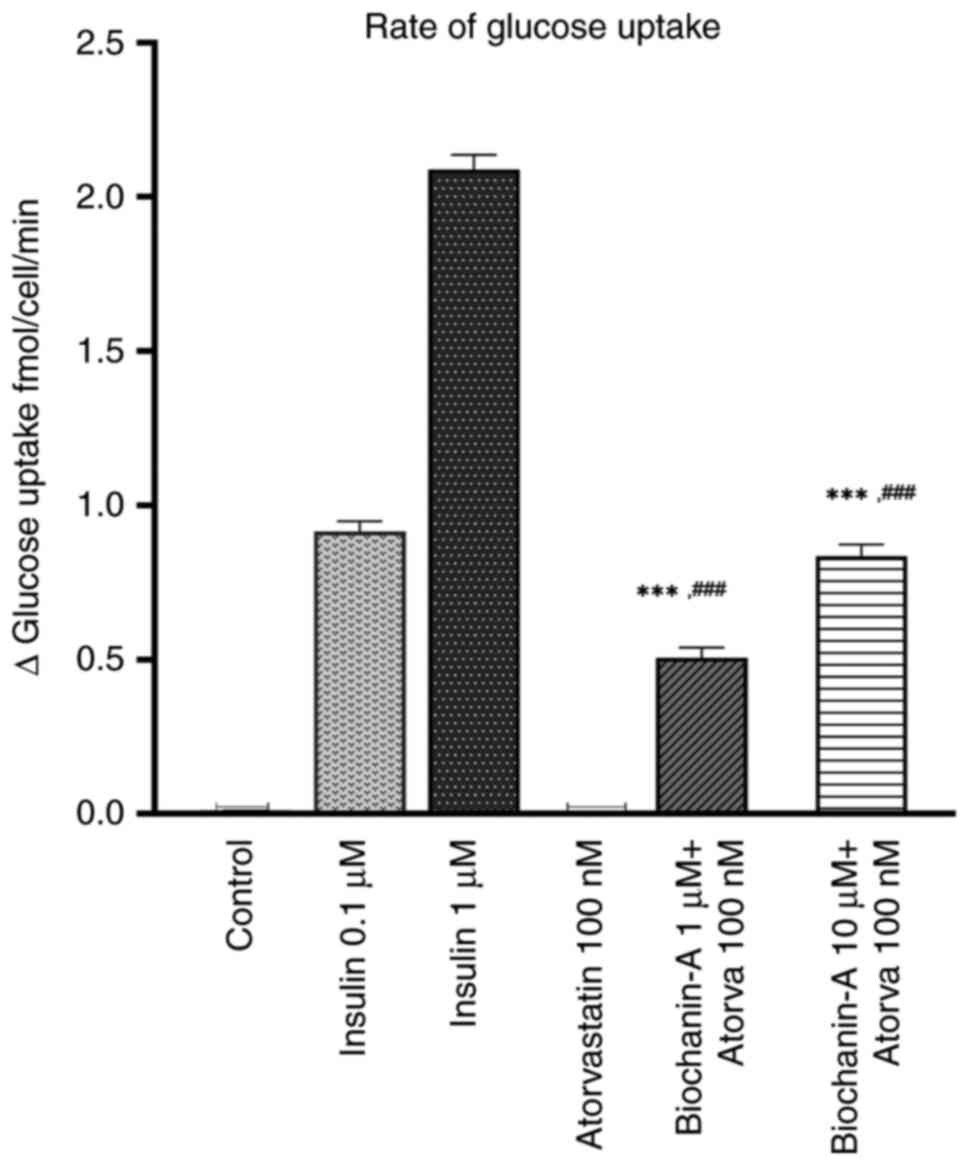
In the present study, the effect of BA and
atorvastatin on glucose uptake in L6 skeletal muscle cells was
assessed using the 2-deoxyglucose uptake assay. The results
demonstrated a significant increase in glucose uptake upon
treatment with insulin (Fig. 7).
Specifically, treatment with 1 µM insulin resulted in a 2.07-fold
increase in glucose uptake compared with the untreated control
(P<0.0001). When L6 cells were pre-treated with BA at
concentrations of 1 and 10 µM for 24 h and subsequently treated
with atorvastatin (100 nM) for 48 h, a fold increases in glucose
uptake of 0.49 and 0.82, respectively, was observed compared with
control cells. These findings indicate that while BA did increase
glucose uptake, its effectiveness was less than that of insulin
treatment alone.
Notably, atorvastatin treatment inhibited
insulin-mediated glucose uptake in L6 cells. However, pre-treatment
with BA mitigated the inhibitory effects of atorvastatin on glucose
uptake, suggesting that BA has a protective role against the
negative impact of atorvastatin on cellular glucose regulation.
These results were significant (P<0.0001 when compared with the
effect of insulin and statin alone), as determined by Tukey's
comparison test (Fig. 7).
Effect on insulin receptor pathway by
western blotting on L6 cells
To evaluate the impact of atorvastatin and BA on the
insulin receptor pathway, western blot analysis was conducted to
assess the protein expression levels of key factors, including
IRS-1, pan-Akt, GLUT-4 and peroxisome proliferator-activated
receptor (PPAR)-gamma in L6 cells (Fig.
8A). The insulin receptor substrate-1 is key in the signal
transmission cascade initiated by insulin. An increase in IRS-1 can
enhance insulin signaling and consequently boost cellular glucose
absorption, which might ameliorate insulin resistance. Western blot
results showed that L6 cells pre-treated with 10 µM BA demonstrated
a significant upregulation of IRS-1, with levels rising 2.42-fold
(Fig. 8A). This increase suggests
that BA promotes insulin signaling, potentially enhancing glucose
uptake. Dose dependency was noted, as the effect was specific to
the 10 µM concentration. Atorvastatin did not significantly affect
IRS-1 expression.
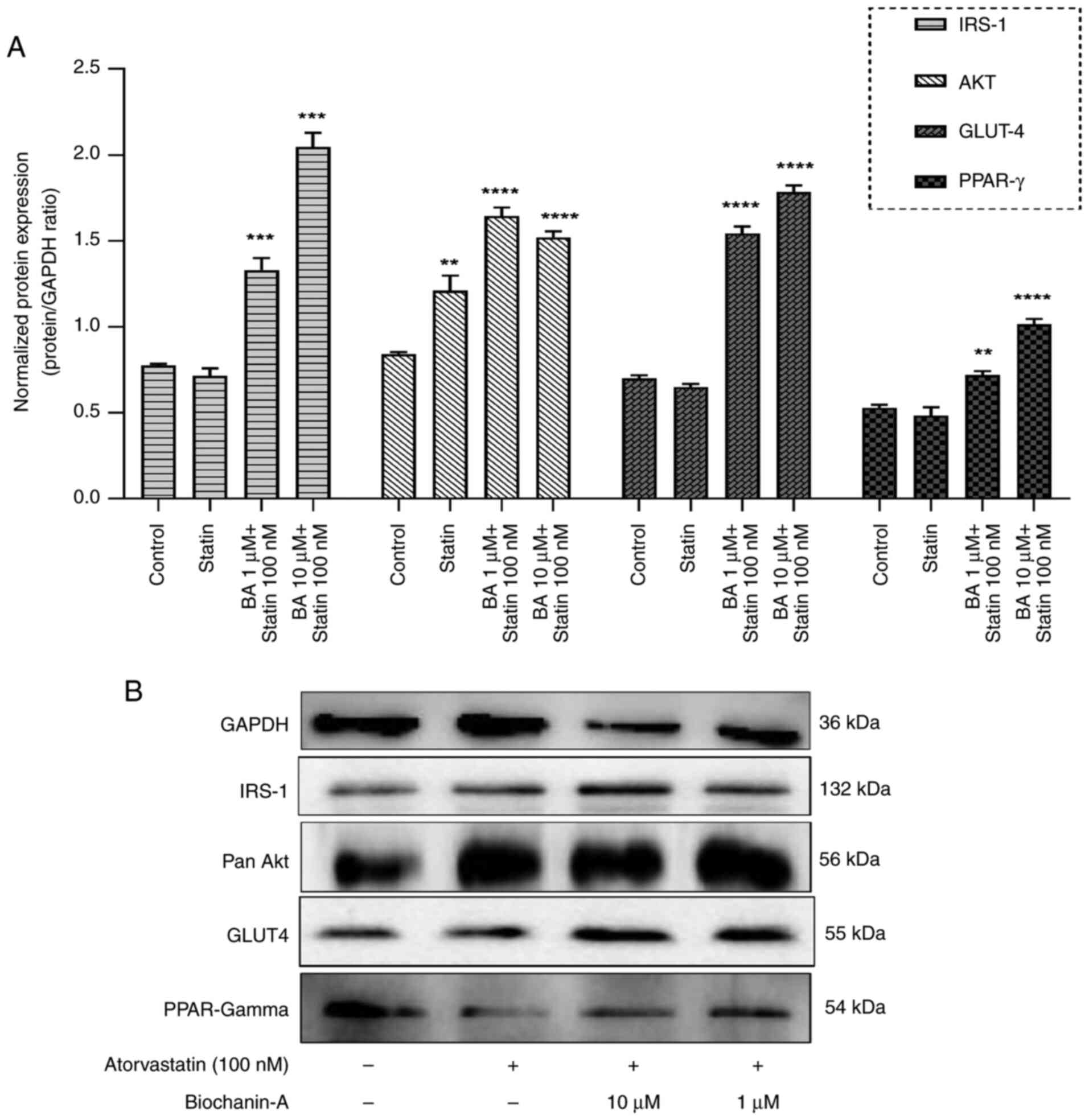 | Figure 8Western blot analysis of insulin
receptor pathway proteins. (A) L6 cells were pre-treated with BA (1
or 10 µM) for 24 h and Atorvastatin (100 nM) for 48 h. Cell lysates
were subjected to western blot analysis using antibodies against
IRS-1, AKT, GLUT4 and PPAR-gamma (loading control). Pre-treatment
with BA upregulated the levels of these proteins at 10 µM.
Densitometric analysis of protein expression normalized to GAPDH.
Data are presented as the mean ± SEM (n=2). **P<0.01,
***P<0.001 and ****P<0.0001 compared
with control group, as indicated. Statistical significance was
determined by one-way ANOVA followed by Dunnett's post hoc test.
(B) Representative western blot images of IRS-1, AKT, GLUT4 and
PPAR-gamma in L6 cells treated with atorvastatin alone and BA
pre-treatment at 1 and 10 µM, followed by atorvastatin exposure.
The bands shown in this panel originate from different western
blots conducted under identical experimental conditions. A shared
GAPDH control was used for comparison across the blots, which was
necessary due to the 10-well capacity of the mini blot module. BA,
Biochanin-A; PPAR, peroxisome proliferator-activated receptor. |
The western blot analysis focused on pan-Akt, a
crucial protein kinase involved in metabolism and cell survival,
revealed that L6 skeletal muscle cells treated solely with
atorvastatin displayed a significant change in pan-Akt levels but
lesser when compared with the combination of BA groups. By
contrast, cells pre-treated with BA followed by atorvastatin
exhibited a significant 1.79-fold increase in pan-Akt expression,
suggesting that BA may enhance Akt activation within the insulin
signaling pathway (Fig. 8A).
Western blot analyses demonstrated that
pre-treatment of L6 cells with 10 µM BA led to a substantial
increase in GLUT4 protein levels, with a 2.76-fold upregulation
observed (Fig. 8A). This effect of
BA on GLUT4 expression was dose-dependent, indicating a potential
mechanism for increased glucose uptake and metabolism through the
insulin signaling pathway.
PPAR-gamma is a nuclear receptor that plays a
critical role in regulating insulin sensitivity and glucose
homeostasis. PPAR activation in mature adipocytes induces the
expression of a number of genes involved in the insulin signaling
cascade, thereby improving insulin sensitivity. PPAR-gamma
activation results in a marked improvement in type 2 diabetic
patients of insulin and glucose parameters resulting from an
improvement of whole-body insulin sensitivity. BA at a dose of 10
µM in combination with atorvastatin 100 nm showed a dose-dependent
effect by upregulating the PPAR-gamma protein by 1.71-fold
(Fig. 8A). This indicates that BA
can activate PPAR-gamma significantly and modulate insulin
signaling despite the statin effect of downregulating this protein
by 0.71-fold.
Pre-treatment of L6 cells with 10 µM BA
significantly upregulated IRS-1, pan-Akt and GLUT4 expression, by
2.42-fold, 1.79-fold and 2.76-fold, respectively. This suggests
that BA enhances insulin signaling and glucose uptake.
Additionally, BA (10 µM) in combination with atorvastatin (100 nm)
increased PPAR-gamma protein expression by 1.71-fold, indicating
its ability to modulate insulin sensitivity despite atorvastatin's
downregulation of PPAR-gamma. These findings highlight the
potential of BA to improve insulin sensitivity and glucose
homeostasis through multiple pathways. The western blot images of
these markers are specified in Fig.
8B.
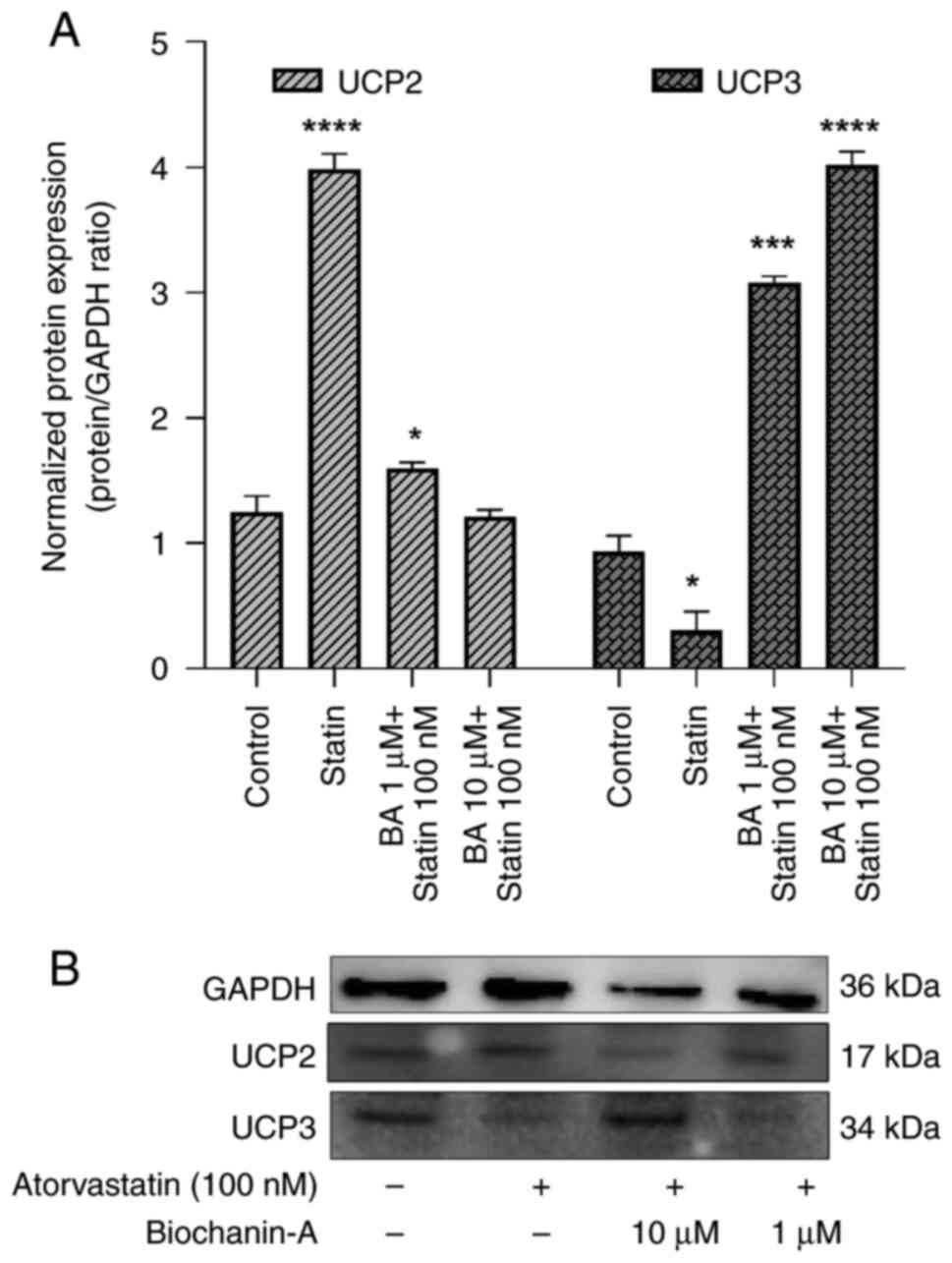
Role of UCPs in modulating sSIRT-1 by
BA and atorvastatin-treated L6 cells
In evaluating the biochemical pathway interactions
between BA and atorvastatin, a key focus was on the UCP levels
(Fig. 9). Our in vitro
experiments on L6 cells revealed a significant upregulation of UCP2
by atorvastatin. Western blotting demonstrated that atorvastatin
elevated UCP2 expression by 3.43-fold compared with the untreated
control group. This upregulation of UCP2, in the absence of BA, was
associated with a decrease in SIRT-1 protein levels, aligning with
our original hypothesis.
Subsequent treatment with BA in combination with
atorvastatin produced a reversal of this effect, with normalization
of UCP2 expression to 1-1.08-fold relative to the control. This
suggests a mitigating influence of BA on the atorvastatin-induced
overexpression of UCP2, which may be through the modulation of the
SIRT-1 pathway (Fig. 9A and
B).
Western blot analysis of BA and atorvastatin on UCP3
proteins in L6 muscle cells revealed that pre-treatment with 10 µM
of BA significantly increased UCP3 levels, with a 2.6-fold
enhancement compared with untreated controls. Conversely, cells
exposed to 100 nM of atorvastatin alone exhibited a marked decrease
in UCP3 expression, down to 0.31-fold of control levels.
The statin treatment downregulated UCP3 expression
observably and BA treatment revealed upregulation of UCP3 in a
dose-dependent manner. Overall, the results suggest that the UCP2
and UCP3 expression is inversely associated under the influence of
these compounds. The data indicate that BA may serve a protective
role, modulating the negative effects statins have on UCP3
expression.
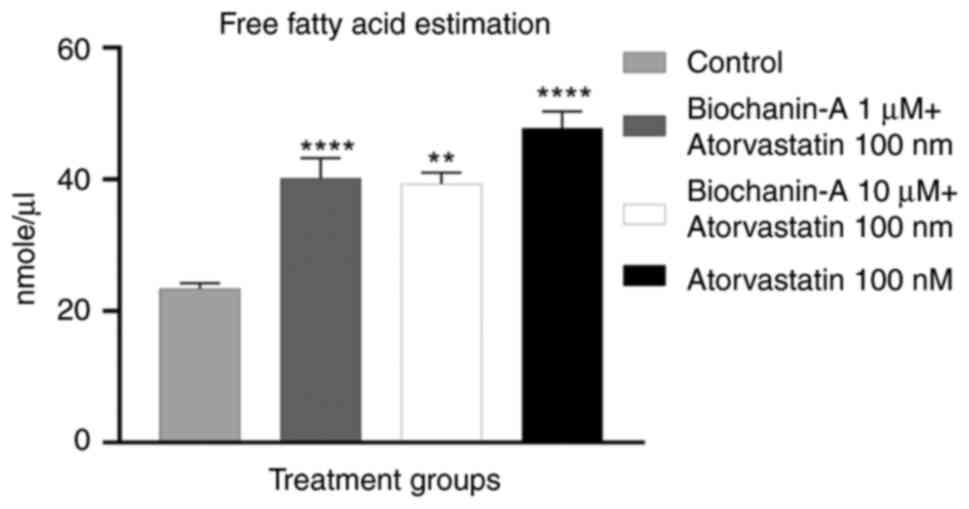
Effect of BA on statin-induced FFA and
cholesterol quantification. FFA quantification
The result of the FFA estimation assay demonstrated
that the cells treated with atorvastatin alone and its combination
with BA had an increase in FFA content compared with the control
group. The L6 cells treated with statin alone at 100 nM showed a
maximum of 47.78 nM/µl whereas BA at 1 and 10 µM respectively
showed 40.18 and 39.33 nM/µl compared with control in FFA
concentration. It was found that there was a significant change in
FFA content in the statin-alone and combination groups when
compared with the control group indicating a modulation of FFA
accumulation by BA (Fig. 10).
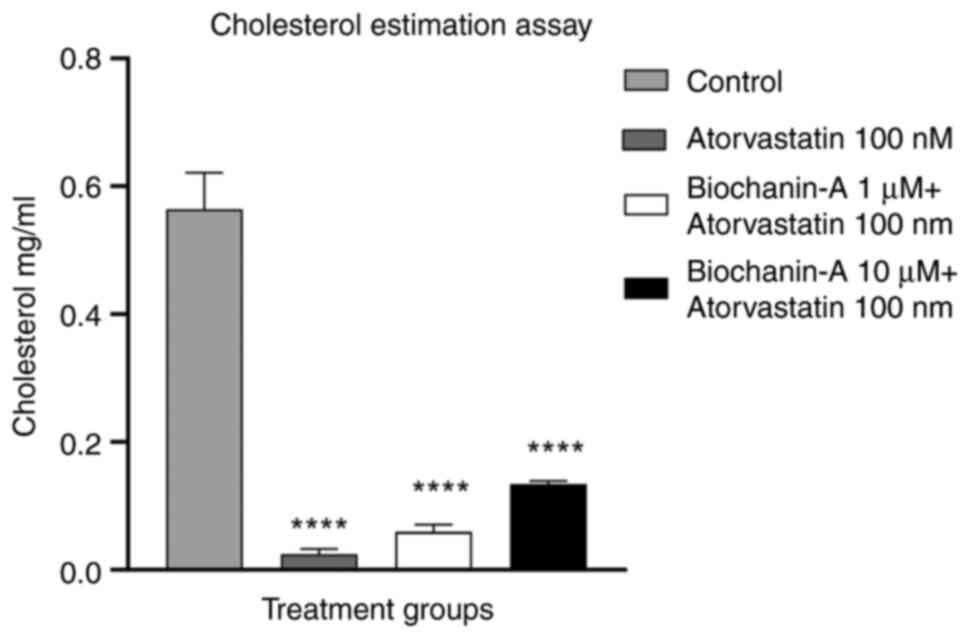
Cholesterol quantification. The result of the
cholesterol quantification assay revealed that the cells treated
with atorvastatin alone and its combination with BA had a reduction
in cholesterol content compared with the normal control group. The
L6 cells treated with statin alone at 100 nM showed a minimum of
0.02 mg/ml, and BA at 1 and 10 µM respectively showed 0.59 and
0.133 mg/ml levels of cholesterol concentration. These findings
illustrate that the combination of BA with atorvastatin does not
hamper the cholesterol-lowering efficacy of atorvastatin (Fig. 11).
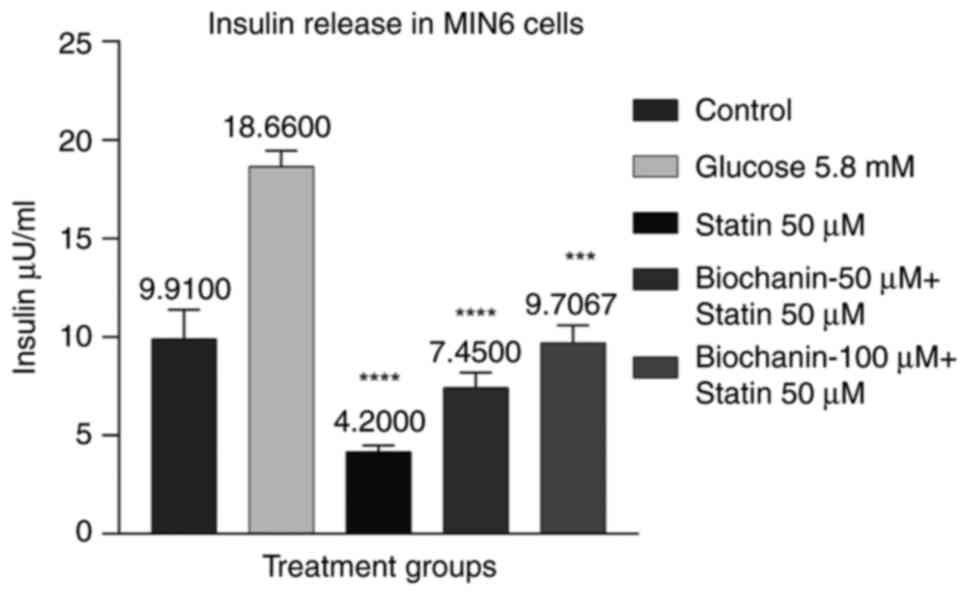
Effect of BA on statin-induced insulin
release in MIN-6 cells
Cells were pre-treated with BA at 50 and 100 µM to
evaluate the effect on insulin secretion against statin treatment
with response to glucose stimulation at 5.8 mM. The results suggest
the dose-dependent increase in insulin levels at BA 50 and 100 µM
with 7.45 and 9.7 µU/ml compared with statin treatment with 4.2
µU/ml. Insulin level in untreated control was observed to be 9.9
µU/ml. BA treatment at 100 µM shows a significant increase in
insulin levels compared with Statin (P<0.01).
The results suggest that BA can improve insulin
release against the statin effect of lowered insulin release, which
indicates that BA is an effective treatment choice in
statin-induced diabetes (Fig.
12).
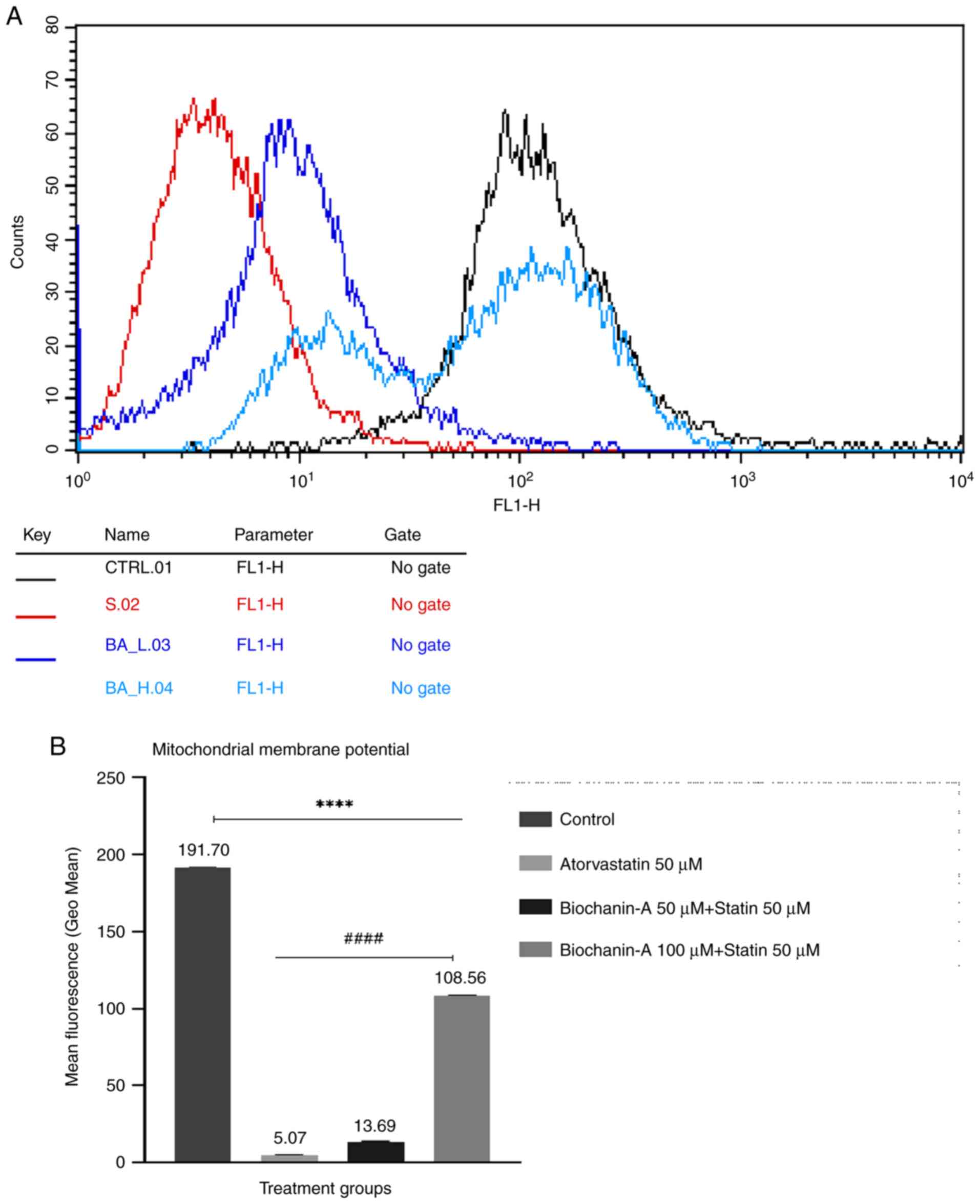
Effect on MMP by flow cytometry
The results of the MMP assay using Rhodamine dye
suggest that atorvastatin produced a decrease in cell fluorescence
which indicates loss of MMP. However, when combined with BA, there
was a significant increase in cell fluorescence compared with
atorvastatin alone, indicating a potential protective effect of BA.
The pre-treatment with BA showed a dose-dependent enhancement of
MMP in MIN-6 cells compared with the normal control. These results
suggest that BA may have a beneficial role in preserving
mitochondrial function, potentially offering therapeutic benefits
in conditions associated with mitochondrial dysfunction (Fig. 13A and B).
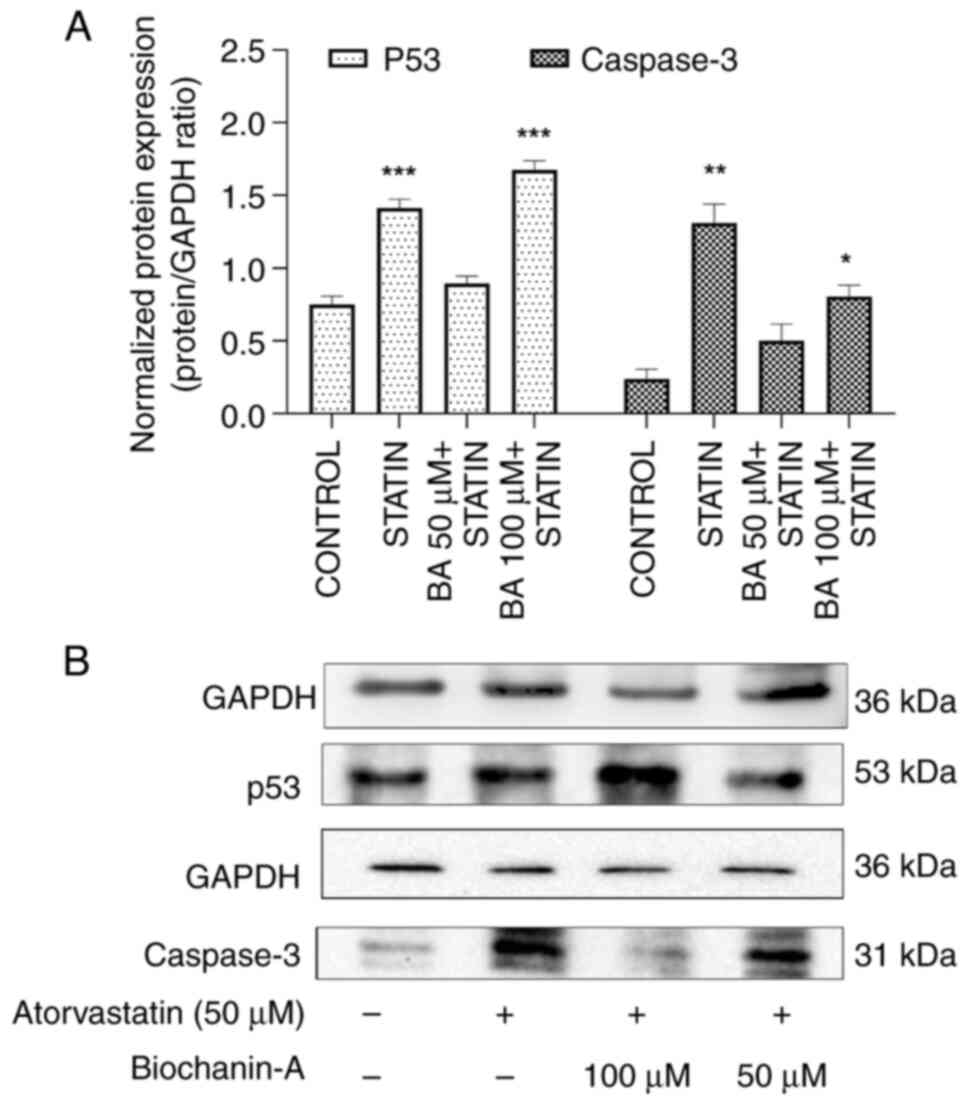
Impact of BA and statin treatment on
apoptosis marker expression in MIN-6 cells. p53 protein
expression
An upregulation of the p53 marker in the
atorvastatin-alone treatment group compared with the normal control
group was revealed using western blotting. The pre-treatment of BA
with atorvastatin showed a decrease in p53 levels at a 50 µM dose.
However, when the BA dose was doubled to 100 µM, an increase in p53
levels was evident, suggesting that high doses of BA might trigger
cellular responses to counteract DNA damage.
These findings indicate a complex interaction where
low doses of BA protect against apoptosis induced by statin
treatment and potentially protecting pancreatic beta cells from
statin-induced toxicity. This dual action of BA, with an initial
downregulation followed by upregulation of p53 at higher
concentrations, warrants further exploration into the pathway
mechanisms, particularly the analysis of p53 downstream targets
(Fig. 14A and B).
Caspase-3 protein expression. The western
blot results of MIN-6 cells pre-treated with BA at concentrations
of 50 and 100 µM suggest a significant downregulation of Caspase-3
by 1.26 and 1.91-fold, respectively, compared with the control
group. The BA treatment exhibited a dose-dependent effect on
Caspase-3 protein expression. By contrast, atorvastatin at a
concentration of 50 µM showed an upregulation of Caspase-3 by
4.03-fold compared with the control group. These results suggest
that atorvastatin induces apoptosis in the MIN-6 cells at 50 µM
concentration (Fig. 14A and
B).
Interestingly, BA in combination with atorvastatin
prevented cell apoptosis. This observation indicates a protective
effect of BA against atorvastatin-induced apoptosis. Furthermore,
this analysis confirms the reason for the increased levels of p53
observed in the present study. The elevated levels of p53 indicate
the activation of a feedback mechanism at higher combination doses
of BA.
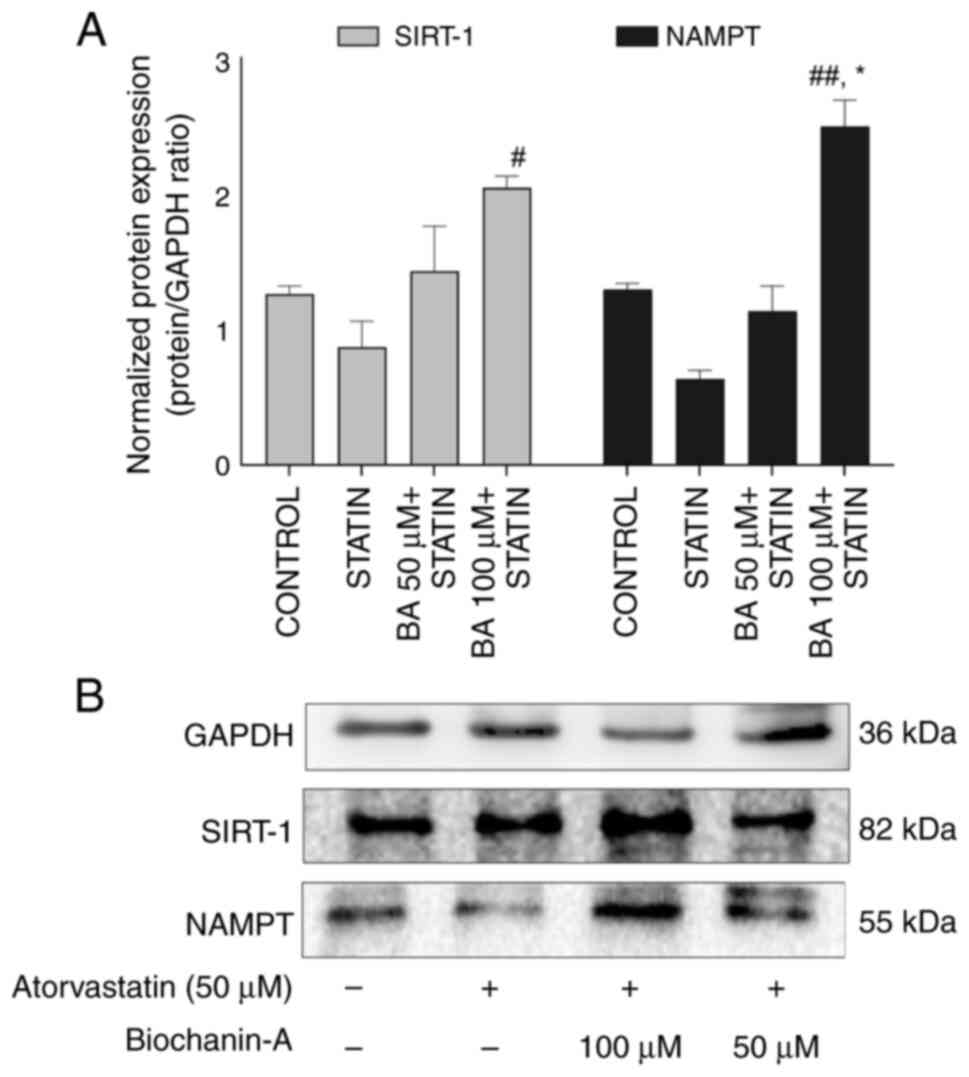
Effect of BA and statin in modulating
SIRT-1 by western blotting. SIRT-1 protein expression
The western blot findings for MIN-6 cells subjected
to pre-treatment with BA in combination with statin at
concentrations of 100 and 50 µM indicate a respective upregulation
of SIRT-1 protein by 1.62 and 1.13-fold compared with the control.
Additionally, the upregulation observed in the BA combination
groups with atorvastatin was 2.35-fold and 1.64-fold higher at 100
and 50 µM respectively, compared with atorvastatin alone at 50 µM.
Notably, the combination group with 100 µM BA demonstrated a
significant increase in upregulation, indicating a dose-dependent
effect when compared with the statin-alone-treated group.
Atorvastatin treatment resulted in a reduction in the expression of
SIRT-1 compared with the control and BA combination groups. The BA
treatment improved the SIRT-1 expression in the combination group
of atorvastatin indicating that BA can be considered as SIRT-1
modulator/activator (Fig. 15A and
B).
NAMPT protein expression
The western blot results of MIN-6 cells pre-treated
with BA at 100 µM concentrations suggest a significant upregulation
of the NAMPT protein by 1.92-fold compared with the control and
3.92-fold when compared with atorvastatin alone group. Atorvastatin
at a concentration of 50 µM resulted in a 0.49-fold regulation of
NAMPT expression compared with the normal control group. BA
combination groups upregulated NAMPT expression, whereas
atorvastatin alone downregulated its expression, highlighting the
contrasting effects of these treatments (Fig. 15A and B).
Discussion
Atorvastatin is a widely prescribed medication that
effectively lowers cholesterol levels and reduces the risk of
cardiovascular disease by inhibiting HMG-coA reductase enzyme
involved in cholesterol production (44). However, previous research has
suggested a potential link between statin use and an increased risk
of developing diabetes mellitus (4-8).
This has raised concerns about the long-term impact of statins on
glucose metabolism and has prompted the exploration of alternative
approaches to mitigate this risk. In light of the risk-benefit
analysis, statins are certainly here to stay. Thus, it is essential
to discover an agent that could prevent these undesirable effects
of statins without compromising their efficacy when used in
combination. Several studies have demonstrated a significant
association between statin use and the development of new-onset
diabetes. The exact mechanisms underlying this relationship are not
fully understood, but proposed hypotheses include impaired insulin
secretion, increased insulin resistance, and disruptions in
intracellular glucose metabolism (11). Understanding the precise pathways
through which statins may impact glucose homeostasis is crucial for
improved risk stratification and management of patients on statin
therapy.
The present study evaluated the effect of BA in
preventing statin-induced type-2 diabetes mellitus by analyzing
insulin resistance in L6 skeletal muscle cells and pancreatic beta
cell death in MIN-6 cells. The emphasis on skeletal muscle is due
to its central role in glucose control and energy balance, as it is
the main site for postprandial glucose uptake. Disruptions in this
system can lead to type 2 diabetes (45). The use of L6 and MIN-6 cell lines
offers a dual approach, providing insights into both insulin action
within muscle tissue and beta cell health, two critical
determinants in disease progression.
By employing the SRB assay (43), the optimal concentrations of
atorvastatin and BA to induce functional inhibition in MIN-6 and L6
cell lines were determined, paving the way for further
investigations. Subsequently, experiments were conducted following
the established workflow in our laboratory. Similar to a prior
study demonstrating the stimulatory impact of BA on 2-NBDG glucose
uptake assay (46), the current
findings from the 2-DG assay revealed an improved glucose uptake in
L6 cells among the group pre-treated with BA. Multiple studies have
shown that atorvastatin inhibits the glucose uptake of the cells
(47-49).
The present findings agree with the literature, as BA was able to
reduce the effect of statins decreasing glucose uptake, while
atorvastatin restricted the insulin-mediated glucose uptake of L6
cells.
It was noted that statin inhibits the
insulin-mediated glucose uptake of the cell through impairment of
the intracellular insulin receptor signaling (50) or by excessive accumulation of FFA in
skeletal muscle as a result of HMG-CoA reductase enzyme and pathway
inhibition (51). Statin decreases
the GLUT-4 translocation (49,52,53)
and impaired AKT activation (53,54) in
muscle cells leading to impairment of glucose transport in myotubes
and producing disrupted insulin receptor pathway signaling. Our
protein expression analysis revealed that there was dose-dependent
upregulation of IRS-1, GLUT-4 and PPAR-γ proteins in pre-treatment
with BA in combination with atorvastatin at 10 µM concentration.
AKT protein expression was elevated with pre-treatment of BA but
not in a dose-dependent manner. However, atorvastatin when treated
alone inhibited IRS-1 and PPAR-γ protein expression levels.
Atorvastatin produced no changes in the level of GLUT-4 but showed
an increase in the AKT expression when compared with control. The
combination of BA with atorvastatin enhanced IRS-1 expression in
skeletal muscle, improving the efficiency of the insulin signaling
pathway, and resulting in increased glucose uptake by muscle cells
in response to insulin. The number of glucose transporters on the
cell membrane is increased, leading to enhanced glucose uptake in
response to insulin, as GLUT-4 protein levels are upregulated when
BA is administered. BA in combination with a statin medication
upregulates Akt protein expression in skeletal muscle, thereby
enhancing the efficiency of the insulin signaling pathway and
facilitating glucose uptake. When PPAR gamma protein expression is
upregulated in skeletal muscle, it enhances insulin sensitivity and
glucose uptake by promoting the transcription of genes involved in
glucose metabolism and insulin signaling, thus improving overall
insulin signaling pathway efficiency.
SIRT-1 protein has involvement in insulin signaling,
and insulin resistance causing type-2 diabetes mellitus (32). The metabolic defect of skeletal
muscle is responsible for insulin resistance (55). Decreased level of SIRT-1 protein in
muscle tissue leads to insulin resistance (56). By increasing SIRT-1 expression in
skeletal muscle, insulin sensitivity can be improved (33). It is proven that diabetic patients
have downregulated SIRT-1 in muscle tissue (36).
UCPs are mitochondrial inner membrane proteins,
responsible for cellular respiration. UCP2 is present in the
pancreatic β cell, liver and heart. UCP3 is selectively expressed
in skeletal muscle cells (57).
SIRT-1 is known to repress the UCP2 gene, binding directly to UCP2
promoter (40). After glucose
stimulation, β cells fail to increase the level of ATP due to
upregulation of UCP2(58). UCP2 and
UCP3 are involved in mitochondrial function and metabolic
regulation. UCP2 negatively regulates insulin secretion in
pancreatic β cells, while UCP3, primarily in skeletal muscle, plays
a role in fatty acid metabolism (33). It was shown that UCP3 levels are
lowered by statin (15). The
accumulation of FFA is inhibited by UCP3. According to a study,
silencing of UCP-2 was associated with an upregulation of GLUT-4,
increased glucose uptake, and reduced intracellular lactate levels,
indicating improvement of oxidative glucose metabolism (59). When statin is administered, there
will be an accumulation of FFA and downregulation of SIRT-1
protein, which will cause an upregulation of UCP2 levels. BA is
known to upregulate SIRT-1 protein and thereby reverse the effect
(33). It was hypothesized that BA
may be able to inhibit statin-induced diabetes, possibly by
modulating SIRT-1 via downregulating UCP2 and upregulating
UCP3.
The expression study on L6 cells revealed that
atorvastatin enhances UCP2 protein levels compared with the
control. By contrast, the combination of BA with atorvastatin
reduced the UCP2 expression up to 1-1.08-fold compared with the
control. The increase in UCP2 expression observed in the
statin-alone group may be attributed to the reduced expression of
the SIRT-1 protein, as hypothesized. The levels of SIRT-1 were
assessed in our in vivo samples to confirm the findings.
The effect of BA on statin-induced FFA accumulation
on L6 cells was analyzed using the FFA Quantification Kit. The
colorimetric assay is based on an enzymatic cycling reaction that
selectively detects FFA in the presence of other lipids, measuring
the absorbance at 570 nm. Post incubation, the cells were treated
with BA for 24 h (1 and 10 µM) followed by atorvastatin treatment
(100 nm) for 48 h. Research indicates that an increase in
plasma-FFA concentrations is a significant factor contributing to
the occurrence of insulin resistance in skeletal muscle tissue
(60,61). Atorvastatin administration can cause
FFA accumulation in the cells due to the inhibition of the HMG CoA
reductase enzyme and further mevalonate formation in the
cholesterol synthesis pathway. This accumulation of FFA may be the
foremost reason for the insulin resistance in muscle cells
connecting to the path of SIRT-1 and UCP2. Using atorvastatin, a
hypothesized metabolic repercussion is the elevation of FFA in the
bloodstream. Atorvastatin's mechanism of action fundamentally
involves the inhibition of HMG-CoA reductase, an enzyme central to
the cholesterol synthesis pathway. This suppression leads to a
decrease in intracellular cholesterol, which sets off a regulatory
response that activates sterol regulatory element-binding proteins
(SREBP) (62). As SREBPs are
transcription factors that govern lipid biosynthesis, their
increased activity heightens the expression of lipid-associated
genes, including those facilitating fatty acid synthesis. The
subsequent rise in FFA levels may contribute to metabolic
complications, emphasizing the complexities of statin's lipid
homeostasis and its potential consequences for insulin sensitivity
(63,64). The FFA estimation assay revealed
that there was a significant change in FFA content in the
statin-alone and combination groups when compared with the control
group.
The present study utilized a cholesterol
quantification assay kit to evaluate the impact of BA and
atorvastatin on cellular cholesterol levels in L6 cells.
Cholesterol in the bloodstream exists in two primary forms: Free
cholesterol and cholesterol esters (65). This assay provides a streamlined and
efficient methodology to quantify both forms separately or combined
as total cholesterol using a coupled enzymatic reaction that
colorimetrically determines cholesterol concentration (66). The experimental data revealed that
treatment with atorvastatin, both alone and in combination with BA,
effectively reduced cholesterol content in cells when compared with
the untreated control. Importantly, the presence of BA did not
disrupt the cholesterol-lowering capability of the statin. This
suggests the potential compatibility of BA as an adjunct therapy
with statins, as it does not interfere with the primary
lipid-lowering effect of atorvastatin and might even offer
additional therapeutic benefits without compromising cholesterol
management.
The functional impact of BA on pancreatic beta-cell
activity was assessed by measuring insulin release from MIN-6
cells. Employing a mouse insulin solid-phase sandwich ELISA, which
utilizes a pre-coated antibody specific to insulin on a microplate
to capture the hormone released by the cells, insulin levels were
quantified after treatments. The results depicted a dose-dependent
augmentation in insulin secretion with BA treatment, demonstrating
increased levels of 7.45 and 9.7 µU/ml at 50 and 100 µM,
respectively. This contrasted with the lowered insulin secretion
witnessed with atorvastatin treatment alone, which amounted to 4.2
µU/ml, suggesting a suppressive effect of the statin on insulin
release. Compared with the untreated control, which had an insulin
level of 9.9 µU/ml, BA at 100 µM restored insulin secretion to a
level statistically comparable to that of untreated cells. This
significant enhancement over the atorvastatin-only treatment
(P<0.01) underlines the potential of BA as a modulatory agent
capable of countering statin-induced reduction in insulin release.
Consequently, these findings propose that BA could serve as a
protective adjunct in statin-associated diabetes, offering a novel
avenue to manage the glucose metabolic side effects associated with
statin therapy.
The MMP is a critical indicator of cellular health
and mitochondrial function (67).
In the present study, flow cytometric analysis with Rhodamine dye
revealed that atorvastatin produced a decrease in MMP, shown by
reduced cell fluorescence, which is consistent with studies of
statins exerting a negative impact on mitochondrial integrity
(42,68). Conversely, pre-treatment with BA
mitigated this effect, as evidenced by a significant increase in
fluorescence intensity in the cells treated with the combination of
atorvastatin and BA, indicating enhanced MMP. These protective
effects of BA were further substantiated by a dose-dependent
restoration of MMP in MIN-6 cells, suggesting that BA potentially
counteracts the mitochondrial dysfunction commonly associated with
statin use. This aligns with previous studies that report the
mitochondrial protective roles of isoflavones and BA (23,69,70).
Our observations propose that BA could serve as a novel therapeutic
agent to prevent or reverse mitochondrial damage, particularly in
conditions that are characterized by mitochondrial dysfunction.
This aligns with the increasing interest in plant-derived compounds
as modulators of mitochondrial health and could have far-reaching
implications for patients experiencing adverse effects from statin
therapy.
Effect on p53 and Caspase-3 as apoptosis markers on
MIN-6 cells was performed. Western blot analysis revealed an
upregulation of the p53 marker in cells treated with Atorvastatin
alone, which is consistent with the known cellular stress responses
elicited by this statin (71).
Intriguingly, co-treatment with BA at a 50 µM concentration reduced
p53 levels. This might reflect that BA may exert a protective
effect on the cells at this dose, perhaps by ameliorating the
atorvastatin-induced stress. By contrast, a higher BA dose (100 µM)
resulted in the upregulation of p53 expression relative to the
atorvastatin-alone group. The increase in p53 expression could
signify a threshold beyond which BA shifts from a protective role
to a more stimulatory one in DNA damage response against apoptosis.
The reduction of p53 expression at lower BA concentrations aligns
with the compound's putative role in mediating cellular protective
pathways. Specifically, these results can be interpreted as
evidence that BA can confer resistance against statin-induced
cellular stress in a dose-dependent manner. Given the central role
of p53 in coordinating the cell's response to DNA damage and the
subsequent decision between repair and apoptosis, the varying
effects of BA on p53 expression warrant further exploration.
Further exploring the downstream targets and consequences of p53
modulation will shed additional light on the precise molecular
interactions at play. In conclusion, the bidirectional regulation
of p53 by BA depending on dosage, suggests a complex interaction
that can either potentiate or mitigate the apoptotic signals in the
context of statin treatment. The observation of reduced apoptosis
marker levels at lower BA doses holds promise for the compound's
therapeutic potential in preventing pancreatic beta-cell apoptosis
due to statin therapy. Future studies should aim to decipher the
downstream effectors of p53 that contribute to these protective
effects, thereby enhancing our comprehension of BA's cellular
impact and informing its potential utility in clinical
settings.
Caspase-3 is an essential enzyme within the caspase
family and is known for its critical function in apoptosis, the
controlled mechanism by which cells systematically disassemble and
die (72). Activated through both
the internal (about the mitochondria) and external (connected to
death receptors) apoptotic signaling routes, caspase-3 acts as the
principal effector, responsible for apoptosis within the cell. The
activation of caspase-3 thus represents a central event in the
apoptotic program, marking the point at which various signaling
pathways converge to execute cell death (73). Both p53 and caspase-3 are
instrumental in managing the cellular response to various stress or
damage that necessitate cell removal. When p53 becomes activated
due to such stressors, it prompts the upregulation of genes
involved in cell death, including those influencing caspase-3
pathways. As a result, caspase-3 is activated to dismantle vital
cellular components, effectively carrying out cell termination.
This relationship between p53 and caspase-3 is key to determining
cellular outcomes, wherein p53-induced activation of apoptotic
agents culminates in the execution of apoptosis through
caspase-3(74). The western blot
results of MIN-6 cells pre-treated with BA at 100 and 50 µM suggest
a significant downregulation of caspase-3, compared with the
control group. The BA treatment exhibited a dose-dependent effect
on caspase-3 protein expression. By contrast, atorvastatin at a
concentration of 50 µM showed an upregulation of caspase-3 compared
with the control group. These results suggest that atorvastatin
induces apoptosis in the MIN-6 cells at 50 µM concentration.
Interestingly, BA in combination with atorvastatin prevented cell
apoptosis. This observation indicates a protective effect of BA
against atorvastatin-induced apoptosis. Furthermore, this analysis
confirms the reason for the increased levels of p53 at higher doses
of BA in combination with statin observed in the present study. The
elevated levels of p53 indicate the activation of a feedback
mechanism at higher combination doses of BA.
In summary, BA exhibits a dose-dependent
downregulation of caspase-3, suggesting its anti-apoptotic effect
on MIN-6 cells. By contrast, atorvastatin induces apoptosis, while
BA, in combination with atorvastatin, prevents cell apoptosis.
These findings provide insights into the potential protective role
of BA and its interaction with atorvastatin in regulating apoptosis
pathways in pancreatic cells.
The present study has delved into the role of
sirtuins, particularly SIRT-1, which is part of the sirtuin family,
known for its NAD+-dependent deacetylase activity. The
significance of SIRT-1 in modulating insulin signaling pathways and
its relevance to the pathogenesis of insulin resistance and type 2
diabetes mellitus has been demonstrated through our research as
well as previous studies. The regulatory influence of SIRT-1 over
metabolism and insulin resistance has been firmly established
(33,75-77).
The observed downregulation of SIRT-1 in the muscle tissues of
diabetic patients elucidates a critical aspect of metabolic
dysfunction associated with diabetes (35). The current findings align with
existing literature that proposes a correlation between
high-calorie diets or persistent exposure to elevated levels of FFA
and the subsequent downregulation of SIRT-1. This is particularly
relevant for patients undergoing statin therapy, wherein the
mechanism of action for statin-induced diabetes may partially stem
from the statins' effect on the modulation of SIRT-1.
In our experimental model using MIN-6 cells, BA,
when administered alongside atorvastatin, led to a remarkable
upregulation of SIRT-1 protein levels, as quantified by western
blot analysis. An increase in the presence of 100 and 50 µM BA was
observed compared with untreated controls. This upregulation was
also observed compared with the cells subjected to atorvastatin
alone, thereby suggesting that BA plays a potential modulatory role
in enhancing SIRT-1 expression. These results suggest that the
administration of BA may counteract the suppression of SIRT-1
induced by statin treatment, offering a promising therapeutic
prospect for individuals facing challenges with diabetes related to
statin therapy.
Given the decrease in SIRT-1 expression with
atorvastatin treatment, it was concluded that upregulation of
SIRT-1 through pharmacological agents such as BA might serve as a
viable therapeutic strategy to manage or possibly prevent
statin-induced diabetes. Therefore, BA emerges as a putative SIRT-1
activator/modulator, marking an exciting avenue for further
research and clinical exploration.
NAMPT is a critical regulator of SIRT-1 activity,
acting as a key enzyme in the NAD+ biosynthetic pathway
and directly influencing SIRT-1's deacetylase function (78). Increased levels of NAMPT lead to
elevated NAD+ availability, which is essential for
SIRT-1 activation. The interaction between NAMPT and SIRT-1
significantly impacts insulin signaling pathways; upregulation of
NAMPT enhances SIRT-1 activity, promoting insulin sensitivity and
improving glucose homeostasis (79). Conversely, dysregulation of NAMPT
expression has been associated with metabolic dysfunction,
including insulin resistance (75).
By supporting SIRT-1 activation, NAMPT can help mitigate these
negative effects, underscoring its potential therapeutic role in
conditions such as type 2 diabetes. Our protein expression analysis
aligns with this notion, as a similar trend is observed in the
present results. Pre-treatment with BA, particularly at higher
doses, upregulated NAMPT expression in a manner similar to that of
SIRT-1, validating BA as a SIRT-1 modulator in statin-induced
diabetes. UCPs are critical in the regulation of mitochondrial
function and cellular energy balance. UCP2, in particular, is
involved in the regulation of insulin secretion by pancreatic
β-cells. The activity of UCP2 is modulated by SIRT-1, which
represses the UCP2 gene by binding directly to its promoter. When
SIRT-1 is downregulated, as can occur with statin treatment, this
repression is relieved, leading to an upregulation of UCP2. An
increase in UCP2 levels can impair the β-cell's ability to generate
ATP in response to glucose stimulation, as UCP2 uncouples the
mitochondrial proton gradient, reducing the efficiency of oxidative
phosphorylation and ATP synthesis. This uncoupling can result in
diminished insulin secretion due to a lack of adequate ATP to drive
the process.
On the other hand, UCP3, also located in the
mitochondrial inner membrane, is suggested to mitigate FFA
accumulation by promoting their oxidation and reducing their toxic
effects intracellularly. Statins have been found to decrease UCP3
levels, which could exacerbate the accumulation of FFAs, leading to
lipotoxicity and further contributing to insulin resistance.
The introduction of BA appears to counteract these
effects. By upregulating SIRT-1, BA could potentially reverse the
statin-induced upregulation of UCP2, thereby restoring normal
mitochondrial function and insulin secretion in β-cells.
Additionally, the effects of BA on SIRT-1 and possibly on UCP3
could help in reducing the accumulation of FFAs, which is important
in maintaining insulin sensitivity. Therefore, in the context of
statin-induced metabolic disturbances, BA may offer a protective
effect by modulating the expression of SIRT-1 and UCP proteins,
ultimately improving the regulation of cellular energy metabolism
and insulin secretion.
The observed elevated level of UCP2 expression in
the atorvastatin-treated group compared with the normal control
aligns with the known effect of decreased SIRT-1 activity induced
by statins. Since SIRT-1 typically represses UCP2, reduced SIRT-1
activity would relieve this repression, leading to an increase in
UCP2. As UCP2 acts as a negative regulator of insulin secretion by
uncoupling oxidative phosphorylation in β-cells, this could
potentially contribute to an unfavourable metabolic profile leading
to diabetes. Conversely, BA's positive modulation of SIRT-1 appears
to counteract the effect of atorvastatin, as evidenced by the
decreased UCP2 gene expression when both are administered in
combination. This suggests that BA could be beneficial in
preventing the atorvastatin-induced upregulation of UCP2, possibly
aiding in the maintenance of β-cell function and insulin
secretion.
Regarding UCP3, decreased gene expression was noted
in the atorvastatin-treated group. Since UCP3 is involved in fatty
acid oxidation and can help to minimize the toxic effects of FFA
accumulation, downregulation by statins might exacerbate
lipid-related metabolic issues. Intriguingly, the co-treatment with
BA reversed this effect, leading to upregulation of UCP3 gene
expression. This upregulation indicates a potential protective role
of BA, which might contribute to preventing FFA accumulation and
associated insulin resistance.
The present findings suggest that BA could represent
a novel preventative strategy for statin-induced diabetes. This is
significant because statin-induced diabetes is a recognized adverse
effect of statin therapy, which is widely prescribed for
cardiovascular disease prevention. Identifying effective strategies
to mitigate this risk is crucial for improving patient outcomes.
The mechanisms by which BA exerts its protective effects,
particularly its influence on SIRT-1, UCP2, and lipogenic pathways
are particularly promising. The upregulation of SIRT-1 by BA
highlights its potential in enhancing insulin sensitivity and
protecting pancreatic β-cells, both of which are crucial for
managing diabetes. The downregulation of UCP2 further suggests that
BA may improve mitochondrial efficiency and reduce oxidative
stress, thereby improving overall metabolic health. These pathways
are known to play critical roles in glucose metabolism and insulin
sensitivity, suggesting that BA may target fundamental processes
underlying statin-induced diabetes. BA might have broader
therapeutic applications, potentially benefiting patients with
various forms of diabetes and metabolic syndrome as the pathways
modulated by BA are also relevant to other metabolic disorders. The
potential for BA to be used as an adjunct therapy alongside statins
is an exciting prospect. This could allow patients to benefit from
the well-established cardiovascular benefits of statins while
minimizing the risk of developing statin-induced diabetes. However,
it is essential to acknowledge that these findings stem from an
in vitro study. Further research, particularly in
vivo studies and ultimately clinical trials, are necessary to
confirm these findings and translate them into clinical
practice.
Overall, the present study provides encouraging
preliminary evidence to support further investigation into BA as a
potential therapeutic option for preventing statin-induced
diabetes. If confirmed in future studies, BA could have a
significant impact on public health by improving the safety and
efficacy of statin therapy.
The current findings suggest that BA alone exerts
significant anti-diabetic effects, indicating its potential as a
monotherapy in statin-induced diabetic conditions. Investigating
the use of BA as an adjunct to existing therapies for diabetic and
statin induced diabetic conditions are a promising avenue for
future research. This could involve preclinical studies exploring
synergistic effects with commonly used drugs. Based on its
mechanism of action, combining BA with agents that target SIRT-1,
UCPs, insulin release pathway and lipogenic pathway could be
particularly effective. Combining BA with statins could reduce the
statin toxicity and prevent the development of diabetes. Further
research, including clinical trials, is crucial to determine the
optimal dosage, long-term safety, and most effective clinical
application of BA.
These results suggest that BA could serve as a
mitigating agent against certain metabolic complications such as
diabetes associated with statin therapy, through its combined
effects on SIRT-1 and UCP proteins. These insights suggest that
incorporating BA with atorvastatin treatment may enhance patient
outcomes by reducing the possible side effects related to insulin
sensitivity.
The conclusion from our in vitro study
necessitates additional investigation in more complex biological
systems to confirm and substantiate the findings. Further research
should focus on validating these results in vivo, using
appropriate animal models to determine if BA can effectively and
safely prevent statin-induced diabetes. Ex vivo studies
using animal or human tissues could provide valuable insights into
the effects of BA on relevant tissues and cell types involved in
statin-induced diabetes. Incorporating more complex 3D cell culture
models, such as organoids, into future in vitro studies
could provide a more physiologically relevant system to investigate
the effects of BA. Ultimately, clinical trials are essential to
determine the therapeutic potential of BA in preventing
statin-induced diabetes in humans. Further research is needed to
determine the bioavailability and pharmacokinetics of BA in humans,
which will be crucial for designing adequate clinical trials.
Advanced mechanistic studies are warranted to fully elucidate the
molecular pathways and interactions involved in BA's protective
effects against statin-induced diabetes. Investigating the
interaction of BA with other metabolic pathways and its potential
synergistic effects when combined with other antidiabetic agents is
also recommended. Conducting long-term studies to assess the
chronic effects of BA, including its impact on insulin sensitivity,
pancreatic β-cell function and overall metabolic health will
provide additional information. By developing and optimizing BA
formulations to enhance its bioavailability and stability, ensuring
effective delivery and sustained therapeutic action is necessary.
Furthermore, dose-response studies, comparative studies, influence
of genetic and environmental factors on the response to BA
treatment, testing in different patient subpopulations and safety
studies are recommended research directions that will help to build
a robust understanding of BA's therapeutic potential and its
clinical application in managing statin-induced diabetes.
A limitation of the present study is that the
western blot experiments were conducted in duplicate due to
resource constraints associated with outsourcing. While the results
are consistent with other findings in the present study, the lack
of triplicate experiments limits statistical robustness. Ongoing
in vivo studies aim to confirm these findings and address
reproducibility concerns. Future investigations will incorporate
triplicate western blot analyses to enhance reliability. It is
important to note that the current findings offer a valuable
foundation for understanding the potential of BA; it is essential
to acknowledge that these results reflect cellular responses in an
isolated environment. Input from experts in endocrinology and
histology would add significant value to the present study. Their
perspectives could provide deeper insights into the endocrine
mechanisms and histological changes associated with BA's effects.
Extrapolating these findings to predict whole-body homeostasis
requires further investigation. The research's in vivo phase
is currently planned by the authors to address this aspect. The
in vivo phase will include a comprehensive examination of
animal tissues, focusing on endocrine function and
histopathological analysis to assess the systemic effects of BA.
This next stage will allow us to evaluate the effects of BA within
the complexity of a living organism, providing a more comprehensive
understanding of its therapeutic potential. While our in
vitro study provides a strong foundation, these future research
directions will be crucial for translating the current findings
into potential therapeutic strategies for preventing statin-induced
diabetes in humans.
In conclusion, the adverse effects of statin
therapy, particularly its association with an increased risk of
new-onset diabetes mellitus, represent a significant concern in
clinical practice. The present study has underscored the complex
interplay between statin-induced modulation of key metabolic
proteins and the consequential disturbance in glucose homeostasis.
Specifically, the downregulation of SIRT-1 and the resulting
upregulation of UCP2 in response to statin treatment emerge as
influential factors contributing to insulin resistance, while the
decrease in UCP3 exacerbates the detrimental build-up of FFA,
leading to the risk of metabolic dysfunction.
The present study sheds light on the promising role
of BA in preventing these adverse effects. The findings suggest
that BA effectively upregulates SIRT-1, thereby reducing the
untoward upregulation of UCP2. This action is paired with the
upregulation of UCP3 and insulin receptor pathway proteins,
indicating an enhancement in fatty acid oxidation and glucose
uptake, the key determinants in preserving insulin sensitivity and
metabolic balance.
Moreover, this investigation contributes to an
improved understanding of the molecular mechanisms behind
statin-induced diabetes, laying a foundation for the development of
adjunct or combination therapeutic strategies. The observed
benefits of BA offer a compelling rationale for its incorporation
into treatment regimens, representing a possible intervention to
safeguard patients from the paradoxical diabetogenic effects of
statins, while still focussing on their cardioprotective
properties. Further clinical studies are necessary to establish
optimal dosing and comprehensively assess the long-term efficacy
and safety of BA in combination with statins. Nevertheless, the
current understanding of BA's pharmacodynamics, such as its
specific interactions within metabolic pathways and its modulation
of mitochondrial function, demands a comprehensive exploration in
future studies. Despite the promising results, BA's translation
from experimental models to clinical practice is not without
challenges. The bioavailability and pharmacokinetics of BA in
humans remain to be fully elucidated. Dosage optimization,
long-term safety and potential interactions with other medications
must be thoroughly assessed. Furthermore, while extensive research
has investigated the pathways involved in the onset of prediabetes,
the identification of precise mechanisms through which BA can
affect these pathways in patients at risk for diabetes necessitates
detailed inquiry. The evidence presented highlights the preventive
strategy to manage diabetes associated with statin therapy. It is
expected that such integrative methods will contribute to a
holistic approach to cardiovascular care, along with lipid
management by preserving the metabolic health of patients.
Acknowledgements
The authors would like to thank the faculty of the
Department of Pharmacology, Manipal College of Pharmaceutical
Sciences for their hands-on training with cell culture and words of
encouragement. The authors would also like to thank Dr Nitesh Kumar
(Department of Pharmacology, NIPER, Hajipur), Dr Gopalan Kutty
(Nampurath, Department of Pharmacology, MCOPS, Manipal) and Dr
Mallikarjuna Rao (Former Principal, MCOPS, Manipal) for the critic
and suggestions provided during the conduct of the research with
their subject expertise in the field. All authors thank the Manipal
Academy of Higher Education for providing the laboratory facilities
for the execution of this project and the support provided to
ensure the timely completion of the research.
Funding
Funding: The present study was supported by the Intra mural
grant from Manipal Academy of Higher Education (Manipal, India;
grant no. MAHE/DREG/PHD/IMF/2019).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
APV and KN conceptualized the study. APV conducted
the experiments, analyzed the data, interpreted the results and
prepared the draft version of the manuscript. DKP, FB and KTG
contributed to data analysis and experiments. RS conducted
statistical analysis, contributed to data interpretation, and
supervised the study. AK provided critical intellectual input
during the study and contributed to manuscript revision. KN
supervised the study, revised the final manuscript. APV and KN
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Steinberg D and Gotto AM Jr: Preventing
coronary artery disease by lowering cholesterol levels: Fifty years
from bench to bedside. JAMA. 282:2043–2050. 1999.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lippi G, Mattiuzzi C and Cervellin G:
Statins popularity: A global picture. Br J Clin Pharmacol.
85:1614–1615. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Taylor F, Huffman MD, Macedo AF, Moore TH,
Burke M, Davey Smith G, Ward K and Ebrahim S: Statins for the
primary prevention of cardiovascular disease. Cochrane Database
Syst Rev. 2013(CD004816)2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
U.S. Food and Drug Administration (FDA):
FDA Drug Safety Communication: Important safety label changes to
cholesterol-lowering statin drugs, 2016. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-important-safety-label-changes-cholesterol-lowering-statin-drugs.
Accessed November 25, 2020.
|
|
5
|
Ramkumar S, Raghunath A and Raghunath S:
Statin therapy : Review of safety and potential side effects. Acta
Cardiol Sin. 32:631–639. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cederberg H, Stančáková A, Yaluri N, Modi
S, Kuusisto J and Laakso M: Increased risk of diabetes with statin
treatment is associated with impaired insulin sensitivity and
insulin secretion: A 6 year follow-up study of the METSIM cohort.
Diabetologia. 58:1109–1117. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sattar N, Preiss D, Murray HM, Welsh P,
Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ,
Jukema JW, et al: Statins and risk of incident diabetes: A
collaborative meta-analysis of randomised statin trials. Lancet.
375:735–742. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bang CN and Okin PM: Statin treatment,
new-onset diabetes, and other adverse effects: A systematic review.
Curr Cardiol Rep. 16(461)2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Khan MAB, Hashim MJ, King JK, Govender RD,
Mustafa H and Al Kaabi J: Epidemiology of type 2 diabetes-global
burden of disease and forecasted trends. J Epidemiol Glob Health.
10:107–111. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ganda OP: Statin-induced diabetes:
Incidence, mechanisms, and implications. F1000Res.
5(1499)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Keni R, Sekhar A, Gourishetti K, Nayak PG,
Kinra M, Kumar N, Shenoy RR, Kishore A and Nandakumar K: Role of
statins in new-onset diabetes mellitus: The underlying cause,
mechanisms involved, and strategies to combat. Curr Drug Targets.
22:1121–1128. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Abbasi F, Lamendola C, Harris CS, Harris
V, Tsai MS, Tripathi P, Abbas F, Reaven GM, Reaven PD, Snyder MP,
et al: Statins are associated with increased insulin resistance and
secretion. Arterioscler Thromb Vasc Biol. 41:2786–2797.
2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ridker PM, Pradhan A, MacFadyen JG, Libby
P and Glynn RJ: Cardiovascular benefits and diabetes risks of
statin therapy in primary prevention: An analysis from the JUPITER
trial. Lancet. 380:565–571. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Crandall JP, Mather K, Rajpathak SN,
Goldberg RB, Watson K, Foo S, Ratner R, Barrett-Connor E and
Temprosa M: Statin use and risk of developing diabetes: Results
from the diabetes prevention program. BMJ Open Diabetes Res Care.
5(e000438)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kain V, Kapadia B, Misra P and Saxena U:
Simvastatin may induce insulin resistance through a novel fatty
acid mediated cholesterol independent mechanism. Sci Rep.
5(13823)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Henriksbo BD, Lau TC, Cavallari JF, Denou
E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, et
al: Fluvastatin Causes NLRP3 inflammasome-mediated adipose insulin
resistance. Diabetes. 63:3742–3747. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Choudhary A, Rawat U, Kumar P and Mittal
P: Pleotropic effects of statins: The dilemma of wider utilization
of statin. Egypt Hear J. 75(1)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Oesterle A, Laufs U and Liao JK:
Pleiotropic effects of statins on the cardiovascular system. Circ
Res. 120:229–243. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rajangam J, Krishnan N, Palei NN, Bhatt S,
Das MK, Das S and Mathusoothanan K: Ameliorative potential of
rosuvastatin on doxorubicin-induced cardiotoxicity by modulating
oxidative damage in rats. Turk J Pharm Sci. 19:28–34.
2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Rajangam J, Lakshmanan AP, Palei NN,
Elumalai K, Kotakonda M, Prakash R and Latha P: Differential
pharmacokinetic interplay of atorvastatin on lacosamide and
levetiracetam on experimental convulsions in mice. Curr Drug Metab.
24:645–655. 2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rajangam J and Lavanya O: Effect of
rosuvastatin on learning and memory in scopolamine induced amnesia
in Mice. Trends Med. 18:1–4. 2018.
|
|
23
|
Anuranjana PV, Beegum F, K P D, George KT,
Viswanatha GL, Nayak PG, Kanwal A, Kishore A and Shenoy RR:
Mechanisms behind the pharmacological application of biochanin-A: A
review. F1000Res. 12(107)2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Oza MJ and Kulkarni YA: Biochanin A
improves insulin sensitivity and controls hyperglycemia in type 2
diabetes. Biomed Pharmacother. 107:1119–1127. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Amri J, Alaee M, Babaei R, Salemi Z,
Meshkani R, Ghazavi A, Akbari A and Salehi M: Biochanin-A has
antidiabetic, antihyperlipidemic, antioxidant, and protective
effects on diabetic nephropathy via suppression of TGF-β1 and PAR-2
genes expression in kidney tissues of STZ-induced diabetic rats.
Biotechnol Appl Biochem. 69:2112–2121. 2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Azizi R, Goodarzi MT and Salemi Z: Effect
of biochanin a on serum visfatin level of streptozocin-induced
diabetic rats. Iran Red Crescent Med J. 16(e15424)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sadri H, Goodarzi MT, Salemi Z and Seifi
M: Antioxidant effects of biochanin A in streptozotocin induced
diabetic rats. Braz Arch Biol Technol. 60(e17160741)2017.
|
|
28
|
Harini R, Ezhumalai M and Pugalendi KV:
Antihyperglycemic effect of biochanin A, a soy isoflavone, on
streptozotocin-diabetic rats. Eur J Pharmacol. 676:89–94.
2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shen P, Liu MH, Ng TY, Chan YH and Yong
EL: Differential effects of isoflavones, from Astragalus
membranaceus and pueraria thomsonii, on the activation of
PPARalpha, PPARgamma, and adipocyte differentiation in vitro. J
Nutr. 136:899–905. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mehrabadi ME and Salemi Z: Effect of
biochanin A on serum nesfatin-1 level in STZ induced type 1
diabetic rat. Diabetol Stoffwechs. 11(P225)2016.
|
|
31
|
Salemi Z, Ghasemi H, Morovati A and Sadri
H: Effects of biochanin A on Resistin, adiponectin and some stress
oxidative markers in normal and STZ-induced diabetic rats. Arch Med
Lab Sci. 4:9–16. 2020.
|
|
32
|
Cao Y, Jiang X, Ma H, Wang Y, Xue P and
Liu Y: SIRT1 and insulin resistance. J Diabetes Complications.
30:178–183. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhou S, Tang X and Chen HZ: Sirtuins and
insulin resistance. Front Endocrinol (Lausanne).
9(748)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cuyàs E, Verdura S, Llorach-Parés L,
Fernández-Arroyo S, Joven J, Martin-Castillo B, Bosch-Barrera J,
Brunet J, Nonell-Canals A, Sanchez-Martinez M and Menendez JA:
Metformin is a direct SIRT1-activating compound: computational
modeling and experimental validation. Front Endocrinol (Lausanne).
9(657)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kitada M and Koya D: SIRT1 in type 2
diabetes: Mechanisms and therapeutic potential. Diabetes Metab J.
37:315–325. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lou PH, Lucchinetti E, Scott KY, Huang Y,
Gandhi M, Hersberger M, Clanachan AS, Lemieux H and Zaugg M:
Alterations in fatty acid metabolism and sirtuin signaling
characterize early type-2 diabetic hearts of fructose-fed rats.
Physiol Rep. 5(e13388)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hou X, Xu S, Maitland-Toolan KA, Sato K,
Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, et al:
SIRT1 regulates hepatocyte lipid metabolism through activating
AMP-activated protein kinase. J Biol Chem. 283:20015–20026.
2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kilic U, Gok O, Elibol-Can B, Uysal O and
Bacaksiz A: Efficacy of statins on sirtuin 1 and endothelial nitric
oxide synthase expression: the role of sirtuin 1 gene variants in
human coronary atherosclerosis. Clin Exp Pharmacol Physiol.
42:321–330. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pareek A, Yeole P, Tenpe C, Chandurkar N
and Payghan R: Effect of atorvastatin and hydroxychloroquine
combination on blood glucose in alloxan-induced diabetic rats.
Indian J Pharmacol. 41:125–128. 2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Bordone L, Motta MC, Picard F, Robinson A,
Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A,
et al: Sirt1 regulates insulin secretion by repressing UCP2 in
pancreatic beta cells. PLoS Biol. 4(e31)2006.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Brault M, Ray J, Gomez YH, Mantzoros CS
and Daskalopoulou SS: Statin treatment and new-onset diabetes: A
review of proposed mechanisms. Metabolism. 63:735–745.
2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mollazadeh H, Tavana E, Fanni G, Bo S,
Banach M, Pirro M, von Haehling S, Jamialahmadi T and Sahebkar A:
Effects of statins on mitochondrial pathways. J Cachexia Sarcopenia
Muscle. 12:237–251. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Valentovic M: Atorvastatin. In: xPharm:
The comprehensive pharmacology reference. Elsevier; Amsterdam, The
Netherlands, pp1-6, 2007.
|
|
45
|
Merz KE and Thurmond DC: Role of skeletal
muscle in insulin resistance and glucose uptake. In: Comprehensive
Physiology. Wiley, pp785-809, 2020.
|
|
46
|
Hwang JT and Kim SH: Evaluation of
Anti-diabetic Effect of Biochanin A in C2C12 Myotube. Korean Soc
Biotechnol Bioeng J. 27:57–60. 2012.
|
|
47
|
Nowis D, Malenda A, Furs K, Oleszczak B,
Sadowski R, Chlebowska J, Firczuk M, Bujnicki JM, Staruch AD,
Zagozdzon R, et al: Statins impair glucose uptake in human cells.
BMJ Open Diabetes Res Care. 2(e000017)2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jiang Z, Yu B and Li Y: Effect of three
statins on glucose uptake of cardiomyocytes and its mechanism. Med
Sci Monit. 22:2825–2830. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sun B, Zhong Z, Wang F, Xu J, Xu F, Kong
W, Ling Z, Shu N, Li Y, Wu T, et al: Atorvastatin impaired glucose
metabolism in C2C12 cells partly via inhibiting
cholesterol-dependent glucose transporter 4 translocation. Biochem
Pharmacol. 150:108–119. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Carnagarin R, Dharmarajan AM and Dass CR:
Molecular aspects of glucose homeostasis in skeletal muscle-A focus
on the molecular mechanisms of insulin resistance. Mol Cell
Endocrinol. 417:52–62. 2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Galicia-Garcia U, Jebari S, Larrea-Sebal
A, Uribe KB, Siddiqi H, Ostolaza H, Benito-Vicente A and Martín C:
Statin treatment-induced development of type 2 diabetes: From
clinical evidence to mechanistic insights. Int J Mol Sci.
21(4725)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yaluri N, Modi S and Kokkola T:
Simvastatin induces insulin resistance in L6 skeletal muscle
myotubes by suppressing insulin signaling, GLUT4 expression and
GSK-3β phosphorylation. Biochem Biophys Res Commun. 480:194–200.
2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Sanvee GM, Panajatovic MV, Bouitbir J and
Krähenbühl S: Mechanisms of insulin resistance by simvastatin in
C2C12 myotubes and in mouse skeletal muscle. Biochem Pharmacol.
164:23–33. 2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Bonifacio A, Sanvee GM, Brecht K,
Kratschmar DV, Odermatt A, Bouitbir J and Krähenbühl S: IGF-1
prevents simvastatin-induced myotoxicity in C2C12 myotubes. Arch
Toxicol. 91:2223–2234. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Abdul-Ghani MA and DeFronzo RA:
Pathogenesis of insulin resistance in skeletal muscle. J Biomed
Biotechnol. 2010(476279)2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Elibol B and Kilic U: High levels of SIRT1
expression as a protective mechanism against disease-related
conditions. Front Endocrinol (Lausanne). 9(614)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Rousset S, Alves-Guerra MC, Mozo J, Miroux
B, Cassard-Doulcier AM, Bouillaud F and Ricquier D: The biology of
mitochondrial uncoupling proteins. Diabetes. 53 (Suppl
1):S130–S135. 2004.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Azzu V and Brand MD: The on-off switches
of the mitochondrial uncoupling proteins. Trends Biochem Sci.
35:298–307. 2010.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Kutsche HS, Schreckenberg R, Weber M,
Hirschhäuser C, Rohrbach S, Li L, Niemann B, Schulz R and Schlüter
KD: Alterations in glucose metabolism during the transition to
heart failure: The contribution of UCP-2. Cells.
9(552)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Martins AR, Nachbar RT, Gorjao R, Vinolo
MA, Festuccia WT, Lambertucci RH, Cury-Boaventura MF, Silveira LR,
Curi R and Hirabara SM: Mechanisms underlying skeletal muscle
insulin resistance induced by fatty acids: Importance of the
mitochondrial function. Lipids Health Dis. 11(30)2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Randle PJ, Garland PB, Hales CN and
Newsholme EA: The glucose fatty-acid cycle. Its role in insulin
sensitivity and the metabolic disturbances of diabetes mellitus.
Lancet. 281:785–789. 1963.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Liscurn L: Chapter 15 Cholesterol
biosynthesis. Elsevier, pp409-431, 2002.
|
|
63
|
Chavez JA and Summers SA: Lipid
oversupply, selective insulin resistance, and lipotoxicity:
Molecular mechanisms. Biochim Biophys Acta. 1801:252–265.
2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hughes RI and Aitman TJ: Genetics of the
metabolic syndrome and implications for therapy. Int Congr Ser.
1262:224–229. 2004.
|
|
65
|
Kohlmeier M: Cholesterol. In: Nutrient
Metabolism. Elsevier, pp511-526, 2003.
|
|
66
|
Li LH, Dutkiewicz EP, Huang YC, Zhou HB
and Hsu CC: Analytical methods for cholesterol quantification. J
Food Drug Anal. 27:375–386. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Gorospe CM, Carvalho G, Herrera Curbelo A,
Marchhart L, Mendes IC, Niedźwiecka K and Wanrooij PH:
Mitochondrial membrane potential acts as a retrograde signal to
regulate cell cycle progression. Life Sci Alliance.
6(e202302091)2023.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Urbano F, Bugliani M, Filippello A,
Scamporrino A, Di Mauro S, Di Pino A, Scicali R, Noto D, Rabuazzo
AM, Averna M, et al: Atorvastatin but not pravastatin impairs
mitochondrial function in human pancreatic islets and rat β-cells.
Direct effect of oxidative stress. Sci Rep. 7(11863)2017.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Koklesova L, Liskova A, Samec M, Zhai K,
Al-Ishaq RK, Bugos O, Šudomová M, Biringer K, Pec M, Adamkov M, et
al: Protective effects of flavonoids against mitochondriopathies
and associated pathologies: Focus on the predictive approach and
personalized prevention. Int J Mol Sci. 22(8649)2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kicinska A and Jarmuszkiewicz W:
Flavonoids and mitochondria: Activation of cytoprotective pathways?
Molecules. 25(3060)2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Delgado-León TG, Sálas-Pacheco JM,
Vazquez-Alaniz F, Vértiz-Hernández ÁA, López-Guzmán OD,
Lozano-Guzmán E, Martínez-Romero A, Úrtiz-Estrada N and
Cervantes-Flores M: Apoptosis in pancreatic β-cells is induced by
arsenic and atorvastatin in Wistar rats with diabetes mellitus type
2. J Trace Elem Med Biol. 46:144–149. 2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5(a008656)2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Shalini S, Dorstyn L, Dawar S and Kumar S:
Old, new and emerging functions of caspases. Cell Death Differ.
22:526–539. 2015.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010(214074)2010.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Li X: SIRT1 and energy metabolism. Acta
Biochim Biophys Sin (Shanghai). 45:51–60. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Chalkiadaki A and Guarente L: High-fat
diet triggers inflammation-induced cleavage of SIRT1 in adipose
tissue to promote metabolic dysfunction. Cell Metab. 16:180–188.
2012.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X
and Zhai Q: SIRT1 improves insulin sensitivity under
insulin-resistant conditions by repressing PTP1B. Cell Metab.
6:307–319. 2007.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Imai S and Yoshino J: The importance of
NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and
ageing. Diabetes Obes Metab. 15 (Suppl 3):S26–S33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Imai S and Kiess W: Therapeutic potential
of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes.
Front Biosci (Landmark Ed). 14:2983–2995. 2009.PubMed/NCBI View Article : Google Scholar
|