Introduction
Sepsis, which results in multiple organ failure,
remains a leading cause of mortality and morbidity in intensive
care units (1,2). Uncontrolled hyperinflammatory and
inappropriate cytokine responses during early sepsis are
hypothesised to be the cause of multiple organ dysfunction syndrome
(MODS) during sepsis. Thus, controlling inflammation during early
sepsis may reduce organ injury and prevent mortality following
septic insult. Sepsis is the leading cause of mortality in
critically ill patients, and the incidence of sepsis is increasing
(3,4). The mortality rate of severe sepsis is
very high (up to 70%) and the calculated costs exceed $15 billion
per year in the United States (3).
The incidence rate of severe sepsis during hospitalisation almost
doubled within the last decade and is considerably greater than
previously predicted (5).
Lipopolysaccharide (LPS), which triggers a systemic inflammatory
response (SIR) (1,2), can stimulate RAW264.7 and other
inflammatory cells to produce a variety of inflammatory mediators,
including tumour necrosis factor-α (TNF-α) and interleukin (IL)-1,
6 and 8. These inflammatory mediators enhance the activity of
nuclear factor-κB (NF-κB), resulting in a large number of secondary
inflammatory mediators, which aggravates the waterfall effect of
SIR, further promoting the development of systemic inflammatory
response syndrome (SIRS) into MODS (3).
TNF-α-induced protein 8-like 2 (TIPE2) is necessary
for maintaining immune homeostasis (6) and is a member belonging to
TNF-α-induced protein-8 (TNFAIP8) family, which is highly expressed
in inflammatory tissues, thus, mainly exhibits a negative
regulatory effect on the innate immune response (7). Several studies have shown that TIPE2
can inhibit NF-κB expression by virtue of multiple channels,
thereby reducing the generation of TNF-α, IL-6 and other
proinflammatory mediators and alleviating the sepsis inflammatory
response (8).
Statin drugs, including hydroxyl methyl glutaric
phthalein coenzyme A (HMG-CoA) reductase inhibitors, are currently
the most widely applied and important lipid-regulating drugs. A
recent study demonstrated that statins not only exhibit
lipid-lowering effects for heart protection, but also inflammatory
effects (9). A previous study
revealed that statins can inhibit the production of TNF-α, IL-6 and
other inflammatory mediators in rats with sepsis (10).
The aim of the present study was to investigate the
immune regulatory mechanism of atorvastatin. Since TIPE2 plays a
crucial role during the regulatory process of inflammatory immune
disorders in severely infected patients, the present study aimed to
determine whether atorvastatin inhibited NF-κB expression via the
enhancement of TIPE2 expression in LPS-induced RAW264.7 cells, and
further decreased the production of proinflammatory factors,
including TNF-α and IL-6.
Materials and methods
Materials
A murine RAW264.7 macrophage cell line was obtained
from the American Type Culture Collection (Rockville, MD, USA).
Dulbecco’s modified Eagle’s medium (DMEM), trypsin, foetal bovine
serum (FBS) and LPS were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Enhanced chemiluminescence (ECL) reagents and NF-κB and
TIPE2 polyclonal antibodies were purchased from BD Pharmingen. (San
Jose, CA, USA). Anti-rabbit IgG horseradish peroxidase-conjugated
secondary antibodies were obtained from R&D Systems Inc.
(Minneapolis, MN, USA). HECAMEG was obtained from Vegatec
(Villejuif, France), and TRIzol and electrophoresis reagents were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Atorvastatin was purchased from Pfizer (New York City, NY,
USA).
TIPE2 knockdown
TIPE2 siRNA (5′-GAAGTGAAA CTCAGGTCCG-3′) or
scrambled control siRNA (5′-AGT GAAACAGTGCAGCTGC-3′) was cloned
into the lentiviral vector, pLL3.7, which carries green fluorescent
protein (GFP) cDNA. Lentiviruses were produced by cotransfecting
293T cells with pLL3.7 plasmids and packaging vectors (11). To knockdown TIPE2 expression,
RAW264.7 were infected with recombinant lentiviruses that carried
the TIPE2 siRNA. Cells infected with the same number of recombinant
lentiviruses carrying the control siRNA served as wildtype
controls. GFP was used to track the virus-infected cells. For the
RAW264.7 cell line, infected cells constituted >90% of the total
number of cells and the transfected cells were used for the
following experiments.
Cell culture
TIPE2 siRNA and control RAW264.7 macrophages were
grown in DMEM supplemented with 5% FBS, 100 μg/ml penicillin, 100
μg/ml streptomycin and 50 μg/l amphotericin B. Cells were
subcultured in six-well plates and maintained until subconfluence.
The medium was then replaced with serum-free culture medium for 24
h prior to the addition of LPS and/or other reagents. The cells
were incubated with various concentrations of LPS for 9 h, or 10
ng/ml LPS with various concentrations of atorvastatin for different
time periods. For the atorvastatin experiments, the cells were
incubated in serum-free medium containing 10 ng/ml LPS in the
presence or absence of atorvastatin for 9 h.
RNA isolation and reverse
transcription
Total RNA was isolated from cultured RAW264.7
macrophages using the single-step acid guanidinium
thiocyanate/phenol/chloroform extraction method. Total RNA (1 μg)
was incubated with 200 units Moloney-Murine Leukemia Virus reverse
transcriptase in buffer containing 50 mmol/l Tris-HCl (pH 8.3), 75
mmol/l KCl, 3 mmol/l MgCl2, 20 units RNase inhibitor, 1
μmol/l poly(dT) oligomer and 0.5 mmol/l each dNTP in a final volume
of 20 μl. The reaction mixture was incubated at 42°C for 1 h and
then at 94°C for 5 min to inactivate the enzyme. A total of 80 μl
diethyl pyrocarbonate-treated water was added to the reaction
mixture prior to storage at −70°C.
Quantitative polymerase chain reaction
(qPCR)
RAW264.7 macrophages were homogenised in TRIzol
reagent using a Mixer 301. Total RNA was extracted according to the
manufacturer’s instructions. RNA samples were electrophoresed in
agarose gels and visualised with ethidium bromide for quality
control. In total, 3 μg RNA was reverse transcribed with reverse
transcriptase for 1 h at 37°C for cDNA synthesis. Quantitative
changes in the mRNA expression levels were assessed with a CFX
Connect Real-Time PCR Detection System (Bio-Rad, Hercules, USA)
using SYBR-Green detection (Aria-tous, Iran). The PCR master mix
consisted of 0.5 units Taq polymerase, 2 μl each primer and
3 μl each cDNA sample in a final volume of 20 μl. All the
amplifications were repeated three times. The oligonucleotide
primer sequences are shown in Table
I. β-actin was used as an endogenous control and each sample
was normalised on the basis of the β-actin content. The relative
quantification of the mRNA expression levels of the target genes
was calculated using the 2−ΔΔCt method.
 | Table IPrimer sequences of the genes used to
validate the microarray analysis by qPCR. |
Table I
Primer sequences of the genes used to
validate the microarray analysis by qPCR.
| Gene | Primer | Product (bp) |
|---|
| TIPE2 | F:
5′-GGGAACATCCAAGGCAAG-3′
R: 5′-AGCTCATCTAGCACCTCACT-3′ | 195 |
| MIF | F:
5′-ATGCCTATGTTCATCGTGAAC-3′
R: 5′-GGCTACGACGAAGGTGGAAC-3′ | 341 |
| NF-κB | F:
5′-GCACGGATGACAGAGGCGTGTATAAGG-3′
R: 5′-GGCGGATGATCTCCTTCTCTCTGTCTG-3′ | 420 |
| IκB | F:
5′-TGCTGAGGCACTTCTGAG-3′
R: 5′-CTGTATCCGGGTGCTTGG-3′ | 421 |
| β-actin | F:
5′-GATTACTGCTCTGGCTCCTGC-3′
R: 5′-GACTCATCGTACTCCTGCTTGC-3′ | 190 |
Western blot analysis
Protein expression levels of TIPE2, nitric oxide
synthase (NOS), haem oxygenase (HO-1) and cyclooxygenase-2 (COX-2)
from cultured RAW264.7 cells were quantified using western blot
analysis. Briefly, proteins from a 50-μg sample were separated
using 10% sodium dodecyl sulphate polyacrylamide gel
electrophoresis and transferred onto polyvinylidene fluoride
membranes. The membranes were incubated overnight with mouse
monoclonal anti-TIPE2 or anti-HO-1 antibodies (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) or rabbit monoclonal
anti-NOS and anti-COX-2 antibodies (Abcam, Cambridge, MA, USA) at
4°C overnight. Following incubation with a secondary antibody,
immunoreactive bands were visualised using an ECL detection system.
Data were quantified using densitometry following scanning with
TINA software (Raytest Isotopenmessgeräete GmbH, Straubenhardt,
Germany).
Immunohistochemistry
Immunostaining was performed on RAW264.7 cells
following antigen retrieval using Retrievagen A (Zymed
Laboratories, Inc., South San Francisco, CA, USA) at 100°C for 20
min, and endogenous peroxidases were quenched with 3%
H2O2. RAW264.7 cells were blocked with 2%
bovine serum albumin in phosphate-buffered saline, which was
followed by staining with primary anti-NF-κB p65 (BD Pharmingen,
San Jose, CA, USA) antibodies at room temperature for 1 h. RAW264.7
cells were washed and incubated with secondary antibodies (R&D
Systems, Minneapolis, MN, USA), and developed using VECTASTAIN ABC
(Vector Laboratories, Inc., Burlingame, CA, USA) and
3,3′-diaminobenzidine (Vector Laboratories, Inc.). Image-Pro Plus
analysis software (Media Cybernetics, Inc., Rockville, MD, USA) was
used to calculate the NF-κB p65-positive expression in RAW264.7
cells, which was expressed as positive units.
TNF-α, IL-10, IL-6 and prostaglandin E2
(PGE2) assays
Following incubation, the supernatant was collected
for the measurement of TNF-α, IL-6 and PGE2. TNF-α, IL-6 and PGE2
levels were assayed in RAW264.7 cells using enzyme-linked
immunosorbent assay kits (Assay designs, Inc., Ann Arbor, MI, USA),
according to the manufacturer’s instructions.
Determination of nitric oxide (NO)
production
Levels of the NO derivative, nitrite, were
determined using the test. A nitrite detection kit was used
according to the manufacturer’s instructions. The samples were
assayed in triplicate and a standard curve using NaNO2
was generated for each experiment for quantification. Briefly,
100-ml samples of medium or standard NaNO2 was mixed
with 100 ml Griess reagent in a 96-well plate. After 15 min, the
optical density was measured with a microplate reader at 540
nm.
Detection of reactive oxygen species
(ROS) production by chemiluminescence
Chemiluminescence of RAW264.7 macrophages was
evaluated using the microplate method in Hank’s balanced salt
solution (pH 7.4) at room temperature. RAW264.7 cells were
incubated with various concentrations of atorvastatin for 9 h in
the presence of 10 ng/ml LPS. Next, luminol (Sigma-Aldrich)
solution was added to the wells containing 2×105 cells,
resulting in a final concentration of 110 μM. After 5 min, phorbol
myristate acetate (PMA) solution was added to obtain a
concentration of 0.8 μM, in order to stimulate the macrophages. The
final volume of each sample was 200 μl. Chemiluminescence was
determined for 5 min with luminol alone and following stimulation
with PMA for 30 min. The measurement system was equipped with a LB
960 CentroXS3 microplate luminometer (Berthold Technologies GmbH
& Co., Wildbad, Germany).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical significance was determined using one-way analysis of
variance and post-hoc (Bonferroni) test deviations. P<0.05 was
considered to indicate a statistically significant difference.
Results
TIPE2 siRNA interference increases NF-κB
p65 expression and the secretion of TNF-α and IL-6 in LPS-induced
RAW264.7 macrophages
To investigate the effect of TIPE2 on NF-κB p65
expression and the secretion of TNF-α and IL-6, RAW264.7 cells or
TIPE2-siRNA RAW264.7 cells were incubated with LPS (10 ng/ml) for
various time periods. The expression of NF-κB p65 and the secretion
of IL-6 and TNF-α in the two groups increased with LPS at each time
point; however, when compared with the RAW264.7 macrophages, the
NF-κB p65 expression and secretion of IL-6 and TNF-α were
significantly elevated in the TIPE2-siRNA RAW264.7 macrophages at
6, 9 and 12 h (Figs. 1B and
2C and D). Thus, TIPE2
downregulated the expression of NF-κB p65 and decreased the
production of IL-6 and TNF-α in a time-dependent manner (Figs. 1B and 2C and D). To determine the
concentration-dependent effects of LPS, RAW264.7 cells or
TIPE2-siRNA RAW264.7 cells were incubated at various concentrations
of LPS for 9 h. LPS increased the expression of NF-κB p65 and the
production of IL-6 and TNF-α in a concentration-dependent manner
(Figs. 1A and 2A and B). However, when compared with the
RAW264.7 macrophages, the NF-κB p65 expression and secretion of
IL-6 and TNF-α were significantly elevated in the TIPE2-siRNA
RAW264.7 macrophages treated with 5, 10 and 15 ng/ml LPS.
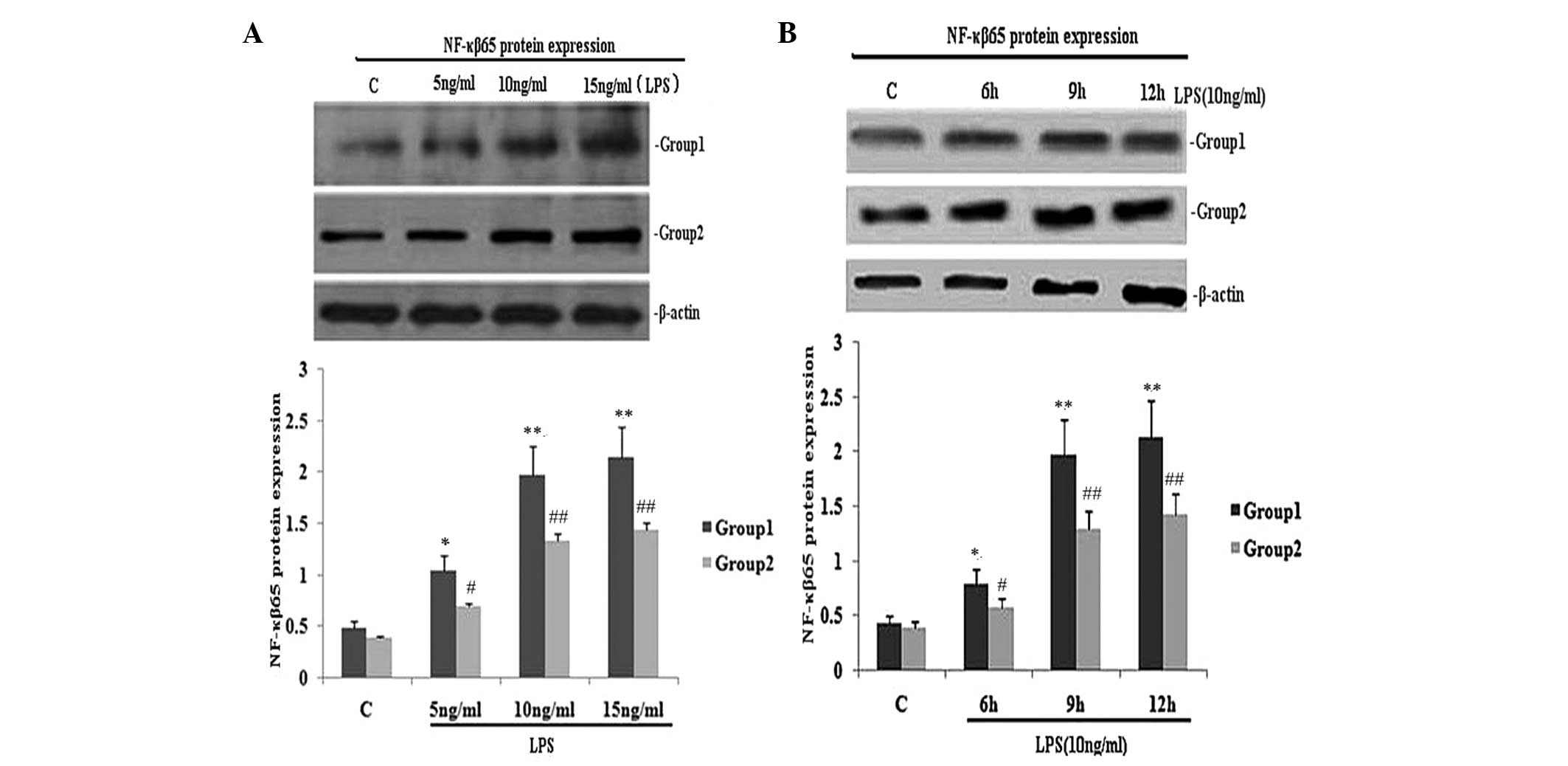 | Figure 1Effect of TIPE2 on NF-κB p65 protein
expression in LPS induced RAW264.7 cells. TIPE2 siRNA and control
RAW264.7 cells were incubated with various concentrations of LPS
for 9 h, or 10 ng/ml LPS for various time periods. NF-κB p65
protein expression was measured using western blot analysis. Values
are expressed as the mean ± standard deviation of three independent
experiments. Groups: 1, TIPE2 siRNA RAW264.7 cells; 2, RAW264.7
cells; C, control. (A) Dose and (B) time dependent effects of LPS
on NF-κB p65 protein expression in RAW264.7 cells and TIPE2 siRNA
RAW264.7 cells. (A) P<0.05, group 1 (5, 10 and 15 mg/ml) vs.
group 2 (5, 10 and 15 mg/ml); group 1: *P<0.05,
**P<0.01, vs. control; group 2:
#P<0.05, ##P<0.01, vs. control. (B)
P<0.05, group 1 (6, 9 and 12 h) vs. group 2 (6, 9 and 12 h);
group 1: *P<0.05, **P<0.01, vs.
control; group 2: #P<0.05, ##P<0.01,
vs. control. TIPE2, tumour necrosis factor-α induced protein 8 like
2; NF-κB, nuclear factor κB; LPS, lipopolysaccharide. |
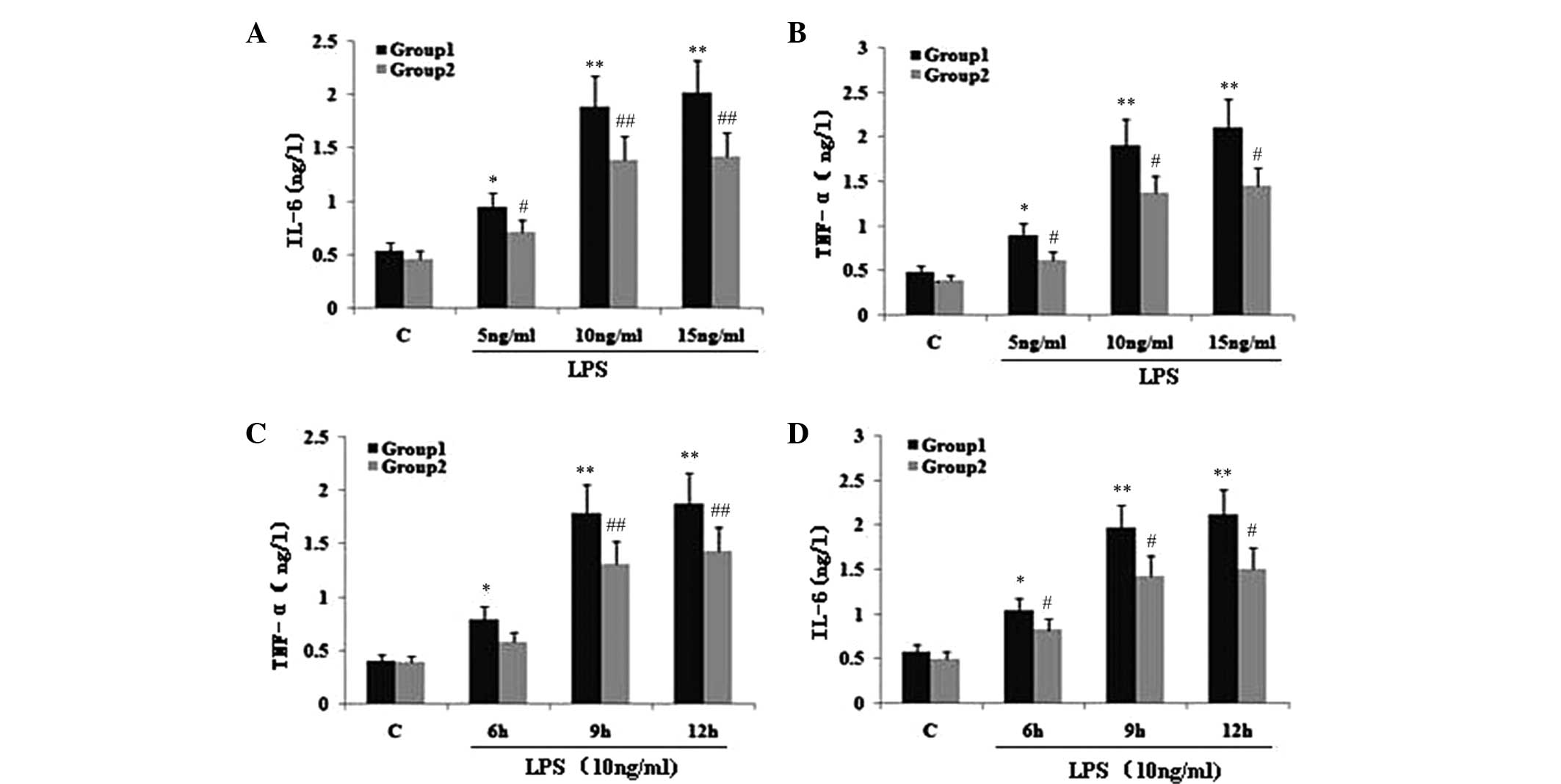 | Figure 2Effect of TIPE2 on proinflammatory
factors in LPS induced RAW264.7 cells. TIPE2 siRNA and control
RAW264.7 cells were incubated with various concentrations of LPS
for 9 h, or 10 ng/ml LPS for various time points. TNF-α and IL-6
levels in the supernatant were assayed using ELISA. Values are
expressed as the mean ± standard deviation of three independent
experiments. Groups: 1, TIPE2 siRNA RAW264.7 cells; 2, RAW264.7
cells; C, control. (A,B) Dose and (C,D) time dependent effects of
LPS on IL-6 and TNF-α levels in RAW264.7 cells and TIPE2 siRNA
RAW264.7 cells. (A,B) P<0.05, group 1 (5, 10 and 15 mg/ml) vs.
group 2 (5, 10 and 15 mg/ml); group 1: *P<0.05,
**P<0.01, vs. control; group 2:
#P<0.05, ##P<0.01, vs. control. (C,D)
P<0.05, group 1 (6, 9 and 12 h) vs. group 2 (6, 9 and 12 h);
group 1: *P<0.05, **P<0.01, vs.
control; group 2: #P<0.05, ##P<0.01,
vs. control. TIPE2, TNF-α induced protein 8 like 2; LPS,
lipopolysaccharide; TNF-α, tumour necrosis factor α; IL,
interleukin; ELISA, enzyme linked immunosorbent assay. |
LPS upregulates TIPE2 expression in a
dose- and time-dependent manner in RAW264.7 macrophages
To determine the dose-dependent effect on TIPE2
expression in LPS-induced RAW264.7 macrophages, RAW264.7 cells were
incubated with various concentrations of LPS for 9 h. LPS increased
the expression of TIPE2 mRNA in a concentration-dependent manner
(Fig. 3B). Concentrations as low
as 5 ng/ml LPS were effective in inducing the mRNA expression of
TIPE2. To determine the time-dependent effect, RAW264.7 cells were
incubated with 10 ng/ml LPS for various time periods. The
expression of TIPE2 was increased by LPS as early as 6 h, which was
the earliest time point investigated. Maximum induction was reached
after 9 h incubation, which remained stable for at least 12 h,
which was the longest time point investigated (Fig. 3A).
Atorvastatin upregulates LPS-induced
TIPE2 and IκB mRNA expression and inhibits LPS-induced NF-κB and
MIF mRNA expression in RAW264.7 macrophages
To determine whether atorvastatin affected the
LPS-induced gene expression of TIPE2, IκB, MIF and NF-κB, RAW264.7
cells were incubated with various concentrations of atorvastatin
for 9 h in the presence of 10 ng/ml LPS. TIPE2, IκB, MIF and NF-κB
expression levels were measured using qPCR analysis. PCR analysis
indicated that the LPS-induced mRNA expression of TIPE2 and IκB
increased significantly with atorvastatin (Figs. 4 and 5). Consistent with this observation, the
LPS-induced mRNA expression of NF-κB and MIF was also suppressed by
atorvastatin (Figs. 4 and 5). Therefore, atorvastatin upregulated
the LPS-induced expression of TIPE2, but decreased NF-κB and MIF
expression in a dose-dependent manner.
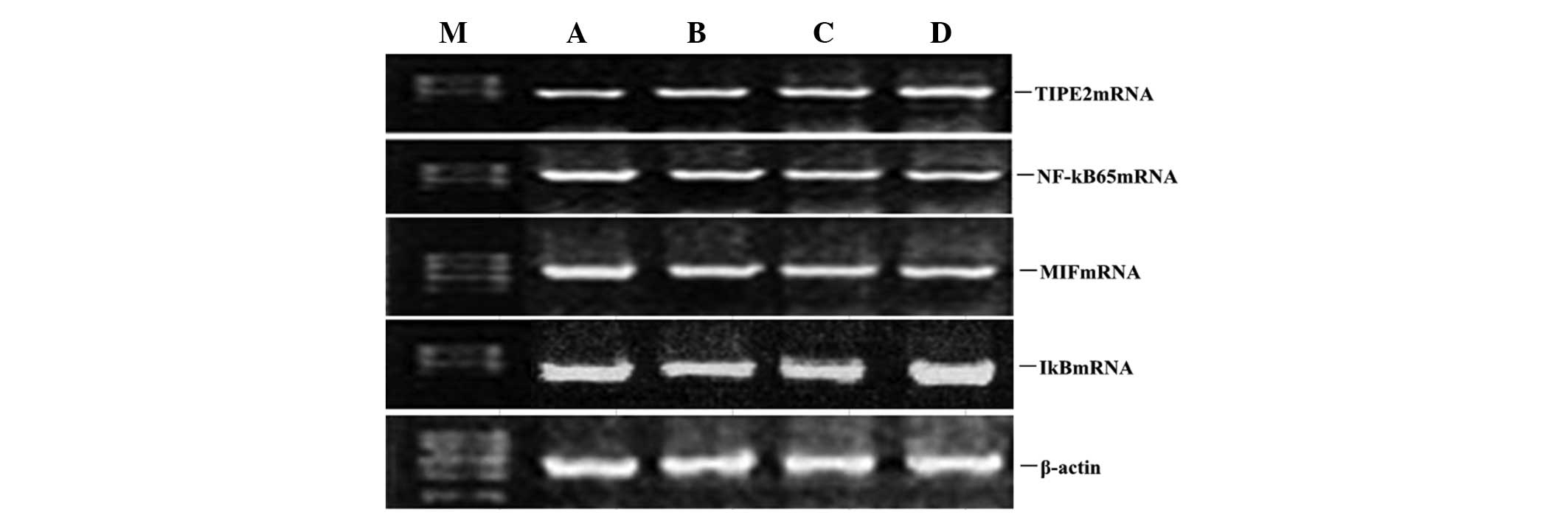 | Figure 4Dose-dependent effect of atorvastatin
on LPS-induced TIPE2, MIF, NF-κB and IκB expression in RAW264.7
cells. RAW264.7 cells were incubated with various concentrations of
atorvastatin for 9 h in the presence of 10 ng/ml LPS. TIPE2, MIF,
NF-κB and IκB mRNA expression levels were measured using qPCR.
Representative qPCR analysis shows the mRNA expression levels of
TIPE2, MIF, NF-κB and IκB in RAW264.7 cells. M, mark; A, LPS (10
ng/ml); B, LPS (10 ng/ml) + atorvastatin (10 μM); C, LPS (10 ng/ml)
+ atorvastatin (15 μM); D, LPS (10 ng/ml) + atorvastatin (20 μM);
TIPE2, tumour necrosis factor-α-induced protein 8-like 2; LPS,
lipopolysaccharide; NF-κB, nuclear factor-κB; MIF, macrophage
migration inhibitory factor; qPCR, quantitative polymerase chain
reaction. |
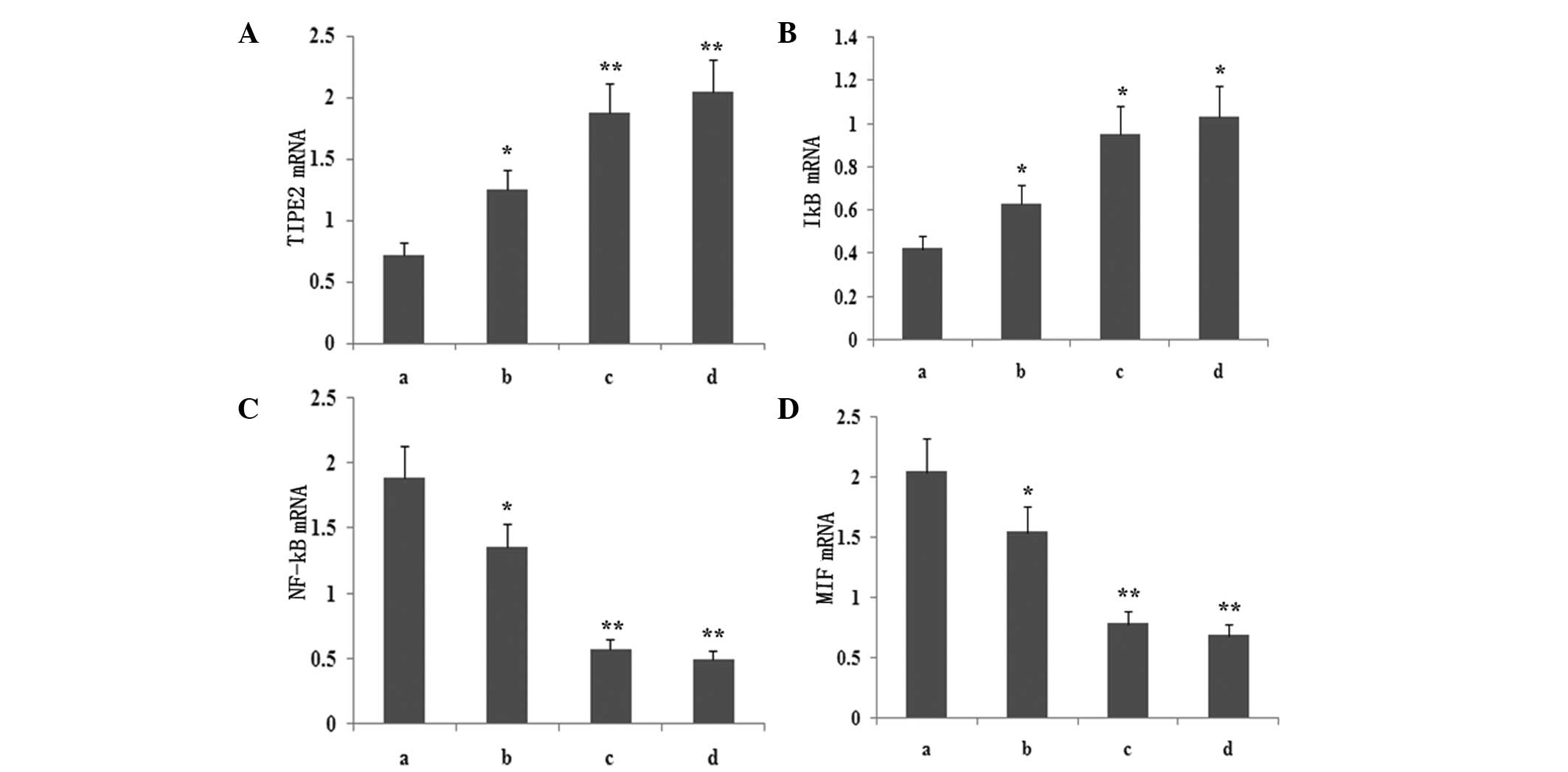 | Figure 5Dose-dependent effect of atorvastatin
on LPS-induced (A) TIPE2, (B) IκB, (C) NF-κB and (D) MIF expression
in RAW264.7 cells. RAW264.7 cells were incubated with various
concentrations of atorvastatin for 9 h in the presence of 10 ng/ml
LPS. TIPE2, MIF, NF-κB and IκB mRNA expression levels were measured
using qPCR. Data are expressed as the mean ± standard error of the
mean of three independent experiments.*P<0.05 and
**P<0.01, vs. ATV (10, 15 and 20 μM). a, LPS (10
ng/ml); b, LPS (10 ng/ml) + atorvastatin (10 μM); c, LPS (10 ng/ml)
+ atorvastatin (15 μM); d, LPS (10 ng/ml) + atorvastatin (20 μM);
TIPE2, tumour necrosis factor-α-induced protein 8-like 2; LPS,
lipopolysaccharide; NF-κB, nuclear factor-κB; MIF, macrophage
migration inhibitory factor; qPCR, quantitative polymerase chain
reaction. |
Atorvastatin increases TIPE2 protein
expression, and inhibits COX-2 and NOS protein expression in
RAW264.7 macrophages
To observe whether atorvastatin affected LPS-induced
TIPE2, COX-2 and NOS protein expression, RAW264.7 cells were
incubated with various concentrations of atorvastatin for 9 h in
the presence of 10 μg/ml LPS. Protein expression levels of TIPE2,
COX-2 and NOS were determined using western blot analysis. The
results indicated that the LPS-induced protein expression of COX-2
and NOS was significantly inhibited, and TIPE2 was further
increased by atorvastatin in a dose-dependent manner (Figs. 6 and 7).
 | Figure 6Dose-dependent effect of atorvastatin
on LPS-induced TIPE2, COX-2 and NOS expression in RAW264.7 cells.
RAW264.7 cells were incubated with various concentrations of
atorvastatin for 9 h in the presence of 10 ng/ml LPS. TIPE2, COX-2
and NOS expression levels were measured using western blot
analysis. Representative western blot showing the protein
expression levels of TIPE2, COX-2 and NOS in RAW264.7 cells. A, LPS
(10 ng/ml); B, LPS (10 ng/ml) + atorvastatin (10 μM); C, LPS (10
ng/ml) + atorvastatin (15 μM); D, LPS (10 ng/ml) + atorvastatin (20
μM); TIPE2, tumour necrosis factor-α-induced protein 8-like 2; LPS,
lipopolysaccharide; COX-2, cyclooxygenase-2; NOS, nitric oxide
synthase. |
 | Figure 7Dose-dependent effect of atorvastatin
on LPS-induced (A) TIPE2, (B) COX-2 and (C) NOS expression in
RAW264.7 cells. RAW264.7 cells were incubated with various
concentrations of atorvastatin for 9 h in the presence of 10 ng/ml
LPS. TIPE2, COX-2 and NOS expression levels were measured using
western blot analysis. Data are expressed as the mean ± standard
error of the mean of three independent
experiments.*P<0.05 and **P<0.01, vs.
atorvastatin (10, 15 and 20 μM). a, LPS (10 ng/ml); b, LPS (10
ng/ml) + atorvastatin (10 μM); c, LPS (10 ng/ml) + atorvastatin (15
μM); d, LPS (10 ng/ml) + atorvastatin (20 μM); TIPE2, tumour
necrosis factor-α-induced protein 8-like 2; LPS,
lipopolysaccharide; COX-2, cyclooxygenase-2; NOS, nitric oxide
synthase. |
Atorvastatin inhibits the production of
inflammatory mediators in RAW264.7 macrophages
To observe whether atorvastatin affected LPS-induced
IL-6, TNF-α, PGE2 and NO expression, RAW264.7 cells were incubated
with various concentrations of atorvastatin for 9 h in the presence
of 10 ng/ml LPS. The levels of IL-6, TNF-α, PGE2 and NO in the
supernatant were assayed. As shown in Fig. 8, atorvastatin reduced IL-6, TNF-α,
PGE2 and NO production in a dose-dependent manner.
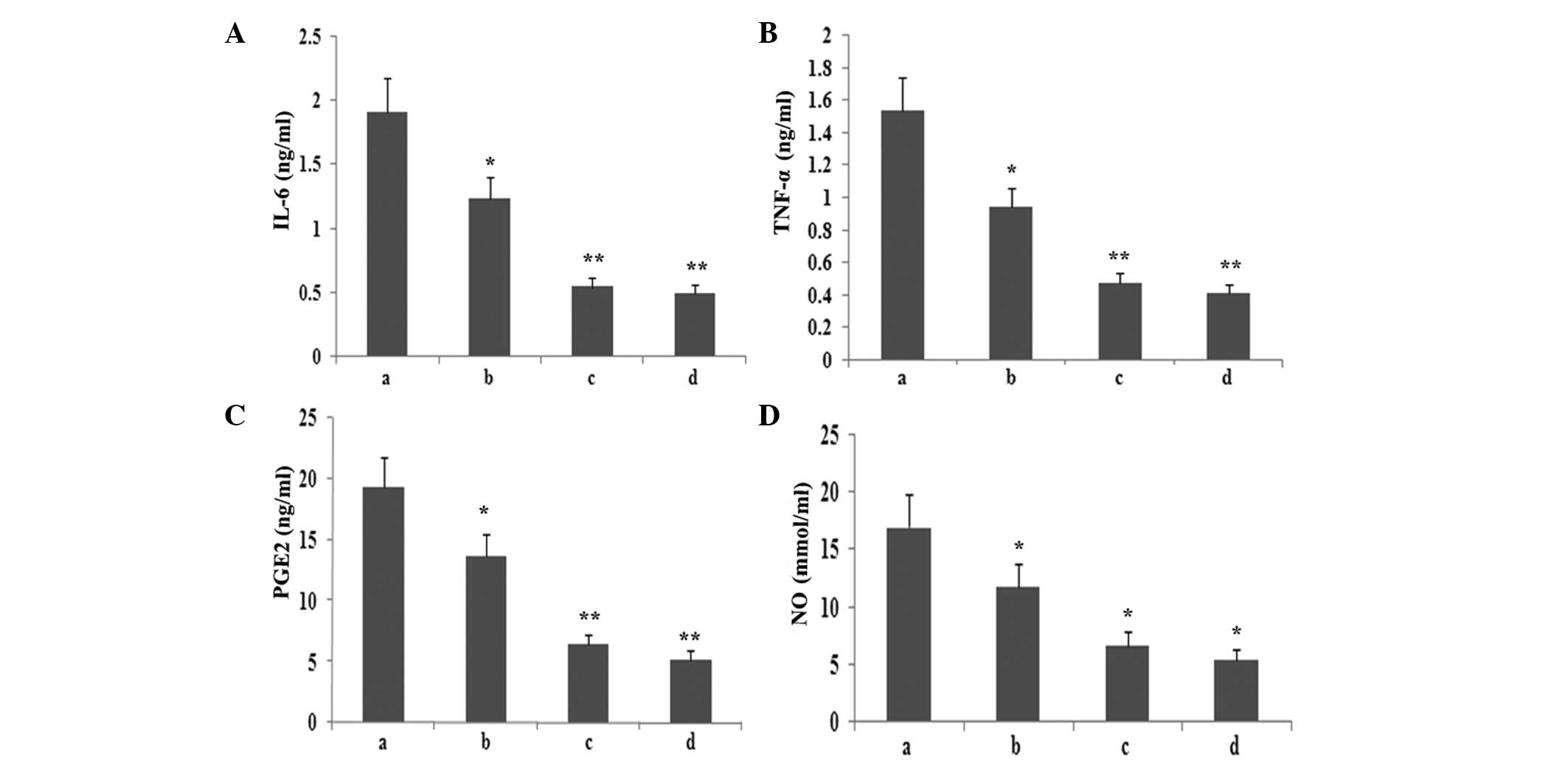 | Figure 8Effect of atorvastatin on the
LPS-induced production of IL-6, TNF-α, NO and PGE2 in RAW264.7
cells. RAW264.7 cells were incubated with various concentrations of
atorvastatin for 9 h in the presence of 10 ng/ml LPS. Following
incubation, the supernatant was collected, and IL-6, TNF-α, NO and
PGE2 levels were assayed. Data are expressed as the mean ± standard
error of the mean of three independent experiments.
*P<0.05 and **P<0.01, vs. LPS control.
a, LPS (10 ng/ml); b, LPS (10 ng/ml) + atorvastatin (10 μM); c, LPS
(10 ng/ml) + atorvastatin (15 μM); d, LPS (10 ng/ml) + atorvastatin
(20 μM); TNF-α, tumour necrosis factor-α; IL, interleukin; LPS,
lipopolysaccharide; PGE2, prostaglandin E2, NO, nitric oxide. |
Atorvastatin increases HO-1 expression
and reduces ROS production
To investigate whether atorvastatin affected
LPS-induced HO-1 and ROS expression, RAW264.7 cells were incubated
with various concentrations of atorvastatin for 9 h in the presence
of 10 ng/ml LPS. As shown in Fig.
9, atorvastatin upregulated HO-1 expression, but reduced the
production of ROS in a dose-dependent manner.
Discussion
A largely unopposed early proinflammatory response
occurs in severe sepsis, which results in cell death (12). Serum levels of proinflammatory
cytokines, including TNF-α, IL-1β and IL-6, peak at early time
points during sepsis (13).
Monocytes-macrophages are important cells of the innate immune
system and are widely present in blood, spleen, liver, lung and
other tissues. Upon severe infections, macrophages produce a large
number of inflammatory mediators, such as IL-1, TNF-α, NO, IL-8 and
IL-6 (14). Excessive inflammatory
factors produced by activated macrophages are associated with the
pathophysiology of various acute inflammatory diseases, including
acute respiratory distress syndrome (ARDS), SIRS and MODS (15–17).
The present study revealed that LPS can stimulate the production of
various inflammatory factors in a time- and dose-dependent manner,
while inhibiting NF-κB expression decreases the generation of
TNF-α, IL-6 and other inflammatory factors, which is conducive to
inhibiting the further development of sepsis.
NF-κB is considered to play a key role in the
production of inflammatory mediators, including proinflammatory
cytokines (TNF-α and IL-1), cell adhesion molecules (intercellular
cell adhesion molecule-1 and vascular cell adhesion molecule-1),
immunoreceptors and acute phase protein production (18,19).
In resting cells, NF-κB is localised to the cytoplasm as a
heterodimer consisting of two polypeptides of 50 (p50) and 65 kDa
(p65), which are noncovalently associated with cytoplasmic
inhibitory proteins, such as IκB. In response to LPS exposure,
viral infection, the expression of specific viral products or other
physiological stimuli, IκB undergoes a series of biological events,
namely rapid phosphorylation in the N-terminal domain by a large
multikinase complex, polyubiquitination and degradation by the 26 S
proteasome, which enables translocation of the NF-κB heterodimer to
the nucleus (20). Once NF-κB
reaches the nucleus, it activates the transcription of several
inflammatory enzymes, including COX-2 and inducible NOS (iNOS), by
interacting with κB sites in their promoter regions. Furthermore,
NO derived from iNOS and PGE2, which is synthesised by COX-2, plays
a pivotal role in the pathogenesis of acute and chronic
inflammation. Activated NF-κB facilitates the secretion of
proinflammatory mediators, such as TNF-α and IL-6. The present
study also found that LPS promoted NF-κB p65 activation, inhibited
IκB activation, increased proinflammatory mediator production,
upregulated COX-2 and NOS expression and enhanced PGE2 and NO
expression. However, the increase in TIPE2 expression caused by
atorvastatin treatment reduced the production of TNF-α, IL-6, PGE2
and NO via blocking NF-κB p65 activation and upregulating IκB
activation.
TIPE2, as a negative regulatory protein,
predominantly functions in the regulation of the inflammatory
mediator response process (21).
Several studies have demonstrated that in TIPE2-deficient rats,
weight loss, splenomegaly, leukocytosis and multiple-organ
spontaneous inflammatory mediator reactions in multiple organs
occur. Following LPS attacks, the excessive response increases,
thus, sepsis occurs, which results in premature mortality.
According to our previous study, TNF-α and IL-1, -6 and -12 levels
are high, and are accompanied with higher levels of IL-10. In
addition, the number of T, B and dendritic cells significantly
increase (22,23). As indicated by previous studies,
TIPE2 may be involved in the regulation of multiple signalling
transduction pathways (24,25).
TIPE2 can inhibit the activation of c-jun N-terminal kinase (JNK)
and p38 mitogen-activated protein kinase (MAPK), and decrease the
activation of transcription factor activator protein-1, such that
it negatively regulates the JNK and p38 MAPK signalling
transduction pathways. In addition, TIPE2 plays a role in the
extracellular signal-regulated kinase signalling pathway. It is
well established that in a resting state, NF-κB exists as a
trimeric complex in cells, connecting the two subunits, p65 and
p50, with IκB. Upon TIPE2 deletion or mutation, the inhibitor IκB
is degraded from the trimeric complex via protein kinase C
phosphorylation, thus, NF-κB can be freely translocated from the
cytoplasm to the nucleus. By virtue of the binding site, NF-κB
initiates the transcription and translation of a variety of
cytokine genes, including TNF-α, IL-1 and IL-6, thereby inducing
the activation of inflammatory cells (26). The present study demonstrated that
increased NF-κB expression resulted in enhanced expression levels
of IL-6, TNF-α and TIPE2 in RAW264.7 cells via a compensatory
effect. However, further enhancement of TIPE2 expression
significantly lowered NF-κB expression and the production of
proinflammatory mediators.
MIF is a proinflammatory mediator that plays a
crucial role in sepsis. In human plasma, MIF normally circulates at
levels ranging between 2 and 10 ng/ml, but fluctuates in a diurnal
rhythm, which reflects neuroendocrine control (27). Plasma MIF concentrations are
elevated to extremely high levels in various inflammatory
disorders, which is the first indication that MIF may be involved
in systemic infection and sepsis. Healy et al reported that
macrophages are peripheral sources of MIF that respond to
inflammatory stimuli in vivo (28). Subsequently, Donnelly and Rogers
(29) demonstrated that increased
concentrations of MIF are present in the lungs of individuals with
ARDS, and that alveolar macrophages are a source of protein. These
studies also demonstrated that MIF enhances, and anti-MIF
antibodies attenuate, the secretion of the proinflammatory
cytokines, TNF-α and IL-8, from alveolar macrophages. In the
present study, LPS was shown to increase MIF expression; however,
the increase in MIF expression was inhibited by regulating TIPE2
via atorvastatin treatment.
HO-1 is an inducible enzyme that catalyses the
rate-limiting step in the conversion of free haem to carbon
monoxide, free iron and biliverdin, which is subsequently
catabolised into bilirubin via biliverdin reductase (30). In addition to the primary role in
haem degradation, HO-1 has also been recognised to play other
important roles in inflammation via HO-1 knockout mice and a human
case of genetic HO-1 deficiency (31). According to references, the gaseous
molecule CO is responsible for most of the anti-inflammatory
actions of HO-1. CO inhibited the production of pro-inflammatory
cytokines, such as TNF-α, IL-1β by activating macrophages.
Therefore, HO-1 and its enzymatic metabolites are critical
regulators of inflammation, with activate macrophages function as
critical targets. An imbalance between ROS and the antioxidant
defence system results in oxidative stress, which is closely
associated with the pathogenesis of sepsis (32). Increased levels of ROS cause direct
tissue injury and promote inflammatory responses via the regulation
of diverse proinflammatory mediators. In the present study, LPS
inhibited HO-1 expression, increased ROS activity and promoted an
inflammatory response. However, atorvastatin upregulated HO-1
expression via increasing TIPE2 expression, which decreased ROS
activity and prevented an inflammatory response.
Statins are drugs that lower lipid levels typically
via the inhibition of HMG-CoA reductase. Research on statins is no
longer limited to investigating the lipid lowering effects, with an
increasing number of studies investigating the anti-inflammatory
effects. Due to the anti-inflammatory effects, statins can inhibit
the release of inflammatory factors, thus, statins may exert
effects on the inflammatory response against sepsis (33,34).
Chello et al (35) used
cecal ligation and puncture (CLP) to generate a rat model of
sepsis. The study indicated that the average survival rate of mice
following statin pretreatment is four times greater compared with
controls. Furthermore, Chello et al (36) indicated that simvastatin, in
addition to its direct role in the neutrophil proinflammatory
effect, can also decrease the levels of C reactive protein (CRP),
TNF-α, IL-6 and other proinflammatory factors, thus, being
conducive to anti inflammatory effects. Chaudhry et al
(37) found that cerivastatin
significantly reduced the release of TNF-α and IL-6, improving the
survival rate of rats with sepsis. In addition, cerivastatin
accelerated bacterial removal in rats 24 h after infection with
Salmonella typhi and 48 h with Staphylococcus aureus. The
anti inflammatory mechanism of statins is associated with NF-κB.
Statins function to reduce the isoprenylation of Rho proteins,
preventing Rho proteins from binding to the cell membrane, which
decreases the release of NF-κB into the nucleus, thereby decreasing
inflammatory cytokine secretion (24) and achieving an anti inflammatory
response. Previous studies have found that statins can reduce the
binding of bacterial LPS induced C Rel/p65 heterodimers with tissue
factor κB sites, thereby regulating inflammatory factors at the
gene level, promoting anti inflammatory cytokine (IL-10) synthesis
and thereby playing a significant anti inflammatory role (38,39).
In a previous study, CLP based sepsis models were generated 18 days
after intragastric administration of statins on a 10 mg/kg/day
basis. After ~24 h, the serum TNF-α, IL-1 and IL-6 levels were
significantly lower compared with the control group and the number
of white blood cells and neutrophils had significantly decreased
(40), confirming that statins can
reduce the level of inflammatory factors in sepsis rats, resulting
in anti inflammatory effects (41). Gonzalez et al (42) performed studies on patients with
sepsisand demonstrated that at day 14 following oral administration
of 80 mg/day simvastatin, the level of CRP gradually decreased and
reached a normal level. However, in the control group, the CRP
level increased on a daily basis, indicating that statins can
reduce the systemic inflammatory response. As indicated by the
present study, atorvastatin can promote TIPE2 expression in
RAW264.7 cells, inhibit NF-κB, COX-2, MIF and NOS expression,
increase IκB and HO-1 expression, significantly reduce the
generation of TNF-α and IL-6 and suppress oxidation reactions in a
dose- and time-dependent manner.
In conclusion, the production of high levels of
proinflammatory mediators is a key factor during the occurrence and
development of sepsis. Atorvastatin inhibits NF-κB, MIF, COX-2 and
NOS expression, increases HO-1 and IκB expression and significantly
reduces the expression of TNF-α and NO proinflammatory factors.
Furthermore, prevented PGE2 and IL-6, oxidative stress via an
increase in TIPE2 expression, which may contribute to the
suppression of the occurrence and development of sepsis upon severe
infection.
Acknowledgements
The authors thank Professor Qinqin Huang and
Professor Shaoxi Cai for their technical assistance.
Abbreviations:
|
TIPE2
|
TNF-α-induced protein 8-like 2
|
|
NF-κB
|
nuclear factor-κB
|
|
IL
|
interleukin
|
|
TNF-α
|
tumour necrosis factor-α
|
|
LPS
|
lipopolysaccharide
|
|
PBS
|
phosphate-buffered saline
|
|
HMG-CoA
|
3-hydroxy-3-methylglutaryl
coenzyme
|
|
MIF
|
macrophage migration inhibitory
factor
|
|
SIR
|
systemic inflammatory response
|
|
SIRS
|
systemic inflammatory response
syndrome
|
|
MODS
|
multiple organ dysfunction
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
NO
|
nitric oxide
|
|
NOS
|
nitric oxide synthase
|
|
COX-2
|
cyclooxygenase-2
|
|
HO-1
|
haem oxygenase
|
|
PGE2
|
prostaglandin E2
|
|
ROS
|
reactive oxygen species
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
CLP
|
cecal ligation and puncture
|
References
|
1
|
Wynn J, Cornell TT, Wong HR, Shanley TP
and Wheeler DS: The host response to sepsis and developmental
impact. Pediatrics. 125:1031–1041. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Unuma K, Aki T, Funakoshi T, Yoshida K and
Uemura K: Cobalt protoporphyrin accelerates TFEB activation and
lysosome reformation during LPS-induced septic insults in the rat
heart. PLoS One. 8:e565262013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Juskewitch JE, Prasad S, Salas CF and
Huskins WC: Reliability of the identification of the systemic
inflammatory response syndrome in critically ill infants and
children. Pediatr Crit Care Med. 13:e55–e57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walley KR, Lukacs NW, Standiford TJ,
Strieter RM and Kunkel SL: Balance of inflammatory cytokines
related to severity and mortality of murine sepsis. Infect Immun.
64:4733–4738. 1996.PubMed/NCBI
|
|
5
|
Cinel I and Opal SM: Molecular biology of
inflammation and sepsis: a primer. Crit Care Med. 37:291–304. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Woodward MJ, de Boer J, Heidorn S, Hubank
M, Kioussis D, Williams O and Brady HJ: TNFAIP8 is an essential
gene for the regulation of glucocorticoid-mediated apoptosis of
thymocytes. Cell Death Differ. 17:316–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Z, Fayngerts S, Wang P, et al: TIPE2
protein serves as a negative regulator of phagocytosis and
oxidative burst during infection. Proc Natl Acad Sci USA.
109:15413–15418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Freundt EC, Bidere N and Lenardo MJ: A
different TIPE of immune homeostasis. Cell. 133:401–402. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Antonopoulos AS, Margaritis M, Lee R,
Channon K and Antoniades C: Statins as anti-inflammatory agents in
atherogenesis: molecular mechanisms and lessons from the recent
clinical trials. Curr Pharm Des. 18:1519–1530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Truwit JD: Statins: a role in infected
critically ill patients? Crit Care. 15:1452011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rubinson DA, Dillon CP, Kwiatkowski AV, et
al: A lentivirus-based system to functionally silence genes in
primary mammalian cells, stem cells and transgenic mice by RNA
interference. Nat Genet. 33:401–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu L, Gokden N and Mayeux PR: Evidence for
the role of reactive nitrogen species in polymicrobial
sepsis-induced renal peritubular capillary dysfunction and tubular
injury. J Am Soc Nephrol. 18:1807–1815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dombrovskiy VY, Martin AA, Sunderram J and
Paz HL: Rapid increase in hospitalization and mortality rates for
severe sepsis in the United States: a trend analysis from 1993 to
2003. Crit Care Med. 35:1244–1250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vigil KJ, Adachi JA and Chemaly RF: Viral
pneumonias in immunocompromised adult hosts. J Intensive Care Med.
25:307–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hlava N, Niemann CU, Gropper MA and
Melcher ML: Postoperative infectious complications of abdominal
solid organ transplantation. J Intensive Care Med. 24:3–17. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao J, Yang X, Auh SL, Kim KD, Tang H and
Fu YX: Do adaptive immune cells suppress or activate innate
immunity? Trends Immunol. 30:8–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim KD, Zhao J, Auh S, Yang X, Du P, Tang
H and Fu YX: Adaptive immune cells temper initial innate responses.
Nat Med. 13:1248–1252. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
López-Rincón G, Pereira-Suárez AL, Del
Toro-Arreola S, et al: Lipopolysaccharide induces the expression of
an autocrine prolactin loop enhancing inflammatory response in
monocytes. J Inflamm (Lond). 10:242013.PubMed/NCBI
|
|
19
|
Jeong IK, Oh da H, Park SJ, et al:
Inhibition of NF-κB prevents high glucose-induced proliferation and
plasminogen activator inhibitor-1 expression in vascular smooth
muscle cells. Exp Mol Med. 43:684–692. 2011.
|
|
20
|
Lin CC, Shih CH, Yang YL, et al: Thrombin
induces inducible nitric oxide synthase expression via the MAPK,
MSK1, and NF-κB signaling pathways in alveolar macrophages. Eur J
Pharmacol. 672:180–187. 2011.PubMed/NCBI
|
|
21
|
Zhang LJ, Liu X, Gafken PR, Kioussi C and
Leid M: A chicken ovalbumin upstream promoter transcription factor
I (COUP-TFI) complex represses expression of the gene encoding
tumor necrosis factor alpha-induced protein 8 (TNFAIP8). J Biol
Chem. 284:6156–6168. 2009. View Article : Google Scholar
|
|
22
|
Sun H, Gong S, Carmody RJ, et al: TIPE2, a
novel negative regulator of innate and adaptive immunity that
maintains immune homeostasis. Cell. 133:415–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Laliberté B, Wilson AM, Nafisi H, et al:
TNFAIP8: a new effector for Galpha(i) coupling to reduce cell death
and induce cell transformation. J Cell Physiol. 225:865–874.
2010.PubMed/NCBI
|
|
24
|
Antosz H and Choroszyńska D: Negative
regulation of Toll-like receptor signalling. Postepy Hig Med Dosw
(Online). 67:339–350. 2013.(In Polish).
|
|
25
|
Zhang X, Wang J, Fan C, et al: Crystal
structure of TIPE2 provides insights into immune homeostasis. Nat
Struct Mol Biol. 16:89–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li D, Song L, Fan Y, et al:
Down-regulation of TIPE2 mRNA expression in peripheral blood
mononuclear cells from patients with systemic lupus erythematosus.
Clin Immunol. 133:422–427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Zeng X, Chen S, et al:
Characterization, epitope identification and mechanisms of the
anti-septic capacity of monoclonal antibodies against macrophage
migration inhibitory factor. Int Immunopharmacol. 11:1333–1340.
2011. View Article : Google Scholar
|
|
28
|
Healy ZR, Liu H, Holtzclaw WD and Talalay
P: Inactivation of tautomerase activity of macrophage migration
inhibitory factor by sulforaphane: a potential biomarker for
anti-inflammatory intervention. Cancer Epidemiol Biomarkers Prev.
20:1516–1523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Donnelly LE and Rogers DF: Novel targets
and drugs in inflammatory lung disease. Curr Opin Pharmacol.
8:219–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tu YP, Chuang SJ, Chen SC, et al:
Simvastatin induces the expression of hemeoxygenase-1 against
ischemia-reperfusion injury on the testes in rats. Toxicol Lett.
207:242–250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park JS, Jung JS, Jeong YH, et al:
Antioxidant mechanism of isoflavone metabolites in hydrogen
peroxide-stimulated rat primary astrocytes: critical role of heme
oxygenase-1 and NQO1 expression. J Neurochem. 119:909–919. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nassar NN, Li G, Strat AL and Abdel-Rahman
AA: Enhanced hemeoxygenase activity in the rostral ventrolateral
medulla mediates exaggerated hemin-evoked hypotension in the
spontaneously hypertensive rat. J Pharmacol Exp Ther. 339:267–274.
2011. View Article : Google Scholar
|
|
33
|
Kagami S, Kanari H, Suto A, et al: HMG-CoA
reductase inhibitor simvastatin inhibits proinflammatory cytokine
production from murine mast cells. Int Arch Allergy Immunol.
146(Suppl 1): 61–66. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trezzi M, Blackstone EH, Sun ZY, et al:
Statin therapy is associated with fewer infections after cardiac
operations. Ann Thorac Surg. 95:892–900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chello M, Anselmi A, Spadaccio C, et al:
Simvastatin increases neutrophil apoptosis and reduces inflammatory
reaction after coronary surgery. Ann Thorac Surg. 83:1374–1380.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chello M, Mastroroberto P, Patti G,
D’Ambrosio A, Morichetti MC, Di Sciascio G and Covino E:
Simvastatin attenuates leucocyte-endothelial interactions after
coronary revascularisation with cardiopulmonary bypass. Heart.
89:538–543. 2003. View Article : Google Scholar
|
|
37
|
Chaudhry MZ, Wang JH, Blankson S and
Redmond HP: Statin (cerivastatin) protects mice against sepsis
related death via reduced proinflammatory cytokines and enhanced
bacterial clearance. Surg Infect (Larchmt). 9:183–194. 2008.
View Article : Google Scholar
|
|
38
|
Fraunberger P, Gröne E, Gröne HJ and Walli
AK: Simvastatin reduces endotoxin induced nuclear factor kappaB
activation and mortality in guinea pigs despite lowering
circulating low density lipoprotein cholesterol. Shock. 32:159–163.
2009. View Article : Google Scholar
|
|
39
|
Lin H, Xiao Y, Chen G, et al: HMG CoA
reductase inhibitor simvastatin suppresses Toll like receptor 2
ligand induced activation of nuclear factor kappa B by preventing
RhoA activation in monocytes from rheumatoid arthritis patients.
Rheumatol Int. 31:1451–1458. 2011. View Article : Google Scholar
|
|
40
|
Meijvis SC, van de Garde EM, Rijkers GT
and Bos WJ: Treatment with anti inflammatory drugs in community
acquired pneumonia. J Intern Med. 272:25–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fogerty MD, Efron D, Morandi A, Guy JS,
Abumrad NN and Barbul A: Effect of preinjury statin use on
mortality and septic shock in elderly burn patients. J Trauma.
69:99–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
González CM, Luna AH, Morales MP, Sanchez
JA, Guzman CO and Granillo GF: Statin anti-inflammatory therapy in
septic patients. Critical Care. 12(Suppl5): P26–P35. 2008.
|
















