Introduction
Cardiomyopathy, a severely disabling complication of
diabetes mellitus (DM), is the leading cause of mortality among
adults worldwide (1). Individuals
with cardiomyopathy are often at a risk of suffering from an
irregular heart beat and sudden cardiac mortality. Although the
cause of cardiomyopathy is poorly understood, the pathophysiology
of diabetic cardiomyopathy (DCM) is hypothesized to be
multifactorial (2). In DM, the
myocardial tissue structure changes and dysfunction that are
induced by factors other than coronary artery disease and cardiac
neuropathy are defined as DCM (3).
The first clinical manifestation of DCM is diastolic
dysfunction, which may be later accompanied by systolic
dysfunction. Myocardial cell degeneration and necrosis are the
major factors causing pathophysiological changes in DCM (4). Transforming growth factor (TGF)-β1
and connective tissue growth factor (CTGF) are highly expressed in
experimental DM hearts, which is in association with cell
proliferation, recognition, apoptosis, special differentiation and
extracellular matrix accumulation. At present, a number of studies
have shown that levels of TGF-β1 and CTGF increase in myocardial
tissues of diabetic patients, thus, these factors are expected to
become new targets for the treatment of DCM (5,6).
Urocortin was first described by Vaughan et
al as a 40-amino-acid peptide associated with the
corticotrophin-releasing factor (CRF) family, which binds to and
activates type 1 and 2 CRF receptors (CRFRs) (7). Urocortin is distributed in the
central nervous system and periphery, in sites such as the
Edinger-Westphal nucleus, adipose tissue, heart, kidney and
immunological tissue (8).
Endothelial urocortin has been shown to suppress the generation of
angiotensin II-induced reactive oxygen species production in
endothelial cells (9).
Urocortin-induced endothelium-dependent relaxation of rat arteries
has also been reported (10). In
addition, this peptide has been found in the heart and been shown
to cause marked vasodilatation of the aorta (11). Administration of urocortin for four
days was shown to have sustained beneficial hemodynamics, hormonal
and renal effects in an experimental heart failure model (12). A previous study demonstrated that
urocortin may play a protective role in ischemia-reperfusion injury
in rat hearts against oxidative stress by inhibiting the activities
of free radicals (13). Urocortin
was also found to exhibit an inhibitory effect on the activity of
serum angiotensin converting enzyme (14). Therefore, the results of these
studies strongly indicate that urocortin may have a beneficial
effect on DCM.
To the best of our knowledge, the underlying
mechanisms of urocortin in DCM remain unclear. We hypothesized that
DCM may be reversed by urocortin. Thus, in the present study, the
role of urocortin in the progression of DCM and the relevant
mechanisms involving the Akt/glycogen synthase kinase-3β signaling
pathway were investigated. The levels of glycosylated hemoglobin
(HbA1c), creatine phosphokinase isoenzyme (CK-MB), brain
natriuretic peptide (BNP), TGF-β1 and CTGF, as well as the collagen
volume fraction (CVF) and left ventricular mass index (LVWI), were
used to estimate the effect of urocortin on DCM, mediated by the
CRFR-2.
Materials and methods
Animals and supplementation
Animal care and experimental protocols were carried
out in accordance with the Guide for the Care and Use of Laboratory
Animals (NIH publication no. 85-23, revised 1996) and approved by
the Ethics Committee of Shandong University (Jinan, China). A total
of 50 male Wistar rats (weight, 250–300 g; age, 18–20 weeks) were
purchased from the Experimental Animal Center of Shanghai Animal
Institute (Shanghai, China) and used in the study.
DM was induced in 40 rats via intraperitoneal
injection of 55 mg/kg streptozotocin (STZ; Sigma-Aldrich, St.
Louis, MO, USA) dissolved in 0.1 M citrate buffer (pH 4.5). The ten
remaining animals were treated with a vehicle and were referred to
as the control group. After three days of STZ injections, the blood
glucose levels were measured using a glucometer (AccuCheck; Roche
Diagnostics, Mannheim, Germany). Rats that had blood sugar levels
of 200 mg/dl, were used for the study. The diabetic rats were
divided into four groups (10 animals per group), which included the
diabetic (DM), urocortin-treated (UCN; Sigma-Aldrich), urocortin +
astressin (UCN + AST) and urocortin + triciribine groups (UCN +
TRI). Astressin (Sigma-Aldrich) was used as a CRFR-2 antagonist,
while triciribine was used as an inhibitor of Akt. No statistically
significant differences in the non-fasting blood glucose levels
were observed among the four groups. Rats in the UCN group received
7 μg/kg urocortin intraperitoneally per day; rats in UCN +AST group
received 7 μg/kg urocortin and 35 μg/kg astressin intraperitoneally
per day; and rats in the UCN + TRI group received 7 μg/kg urocortin
and 0.5 mg/kg triciribine intraperitoneally per day for 16 weeks.
Rats in the diabetic group received the same volume of normal
saline. Age-matched male Wistar rats were used as normal controls
(10 animals). The normal control and diabetic rats were housed in a
room with a 12-h artificial light cycle with free access to an 18%
high-fat diet (standard rat diet, 8% fat) and water. The body
weight (BW) and non-fasting blood glucose levels were measured
weekly. After 16 weeks of supplementation, the animals were
anaesthetized with 1 g/kg−1 intraperitoneal urethane and
sacrificed. A blood sample was taken from the heart and the serum
was isolated for biochemical measurements. The heart was removed
under aseptic conditions and perfused with ice-cold
diethylpyrocarbonate-treated distilled water (Wuhan Boster
Biological Technology, Ltd., Wuhan, China).
Measurements of BW, LVW and LVWI
After the rats were weighed and anesthetized with
intraperitoneal urethane (1 g/kg), the thorax was rapidly opened
and the heart was excised. The heart was washed in 0.01 mol/l
phosphate-buffered saline and the LVW was detected, from which the
LVWI was calculated as LVW/BW.
Measurements of HbA1c, CK-MB and BNP
CK-MB and BNP levels in the rats were measured using
a RA-50 semi-auto analyzer, while HbA1c analysis was performed
using a Nycocard Reader (Axis-Shield, Oslo, Norway).
Myocardial pathology and CVF
observations
Heart tissues were paraffin-embedded, cut into
sections and stained with hemotoxylin and eosin. In addition,
collagen in the heart was specifically stained with Ponceau S and
the CVF was determined. In brief, images of the sections were
captured and analyzed with Image-Pro Plus 6.0 image analysis
software. Five fields were randomly selected and the CVF was
calculated as the collagen area/total area, which was followed by
averaging. However, the area of collagen surrounding the vessels
was not included in the CVF. Four arterioles in the ventricular
wall were selected and the cross-sectional area was measured.
Measurement of TGF-β1 and CTGF in the
plasma
Plasma TGF-β1 and CTGF levels were determined using
a sandwich ELISA method with a commercially available kit (BD
Opt-EIA; BD Biosciences, Franklin Lakes, NJ, USA). Appropriate
controls and standards were used, as specified by the
manufacturer’s instructions, and the data are expressed as pg/ml of
plasma.
Quantitative polymerase chain reaction
(PCR) analysis of TGF-β1 and CTGF mRNA expression
Total RNA was isolated using TRIzol reagent and
purified with an RNeasy kit (Qiagen, Valencia, CA, USA), according
to the manufacturer’s instructions. Reverse transcription of 500 ng
total RNA was performed in a total volume of 20 μl using an iScript
cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). In total, 1 μl
cDNA was amplified by PCR in 20-μl reactions containing special
primers and iQ SYBR Green Supermix (Bio-Rad). PCR was performed for
40 cycles, consisting of 95°C for 15 sec, 94°C for 5 sec, 58°C for
15 sec and 72°C for 15 sec, using the iCycle iQ Real-Time PCR
Detection System (Bio-Rad). The primers used for PCR were purchased
from Invitrogen Life Technologies (Grand Island, NY, USA) and had
the following sequences: TGF-β1 forward, 5′-GCTCGCTTTGTACAACAGCA-3′
and reverse, 5′-GAGTTCTACGTGTTGCTCCA-3′; β-actin forward,
5′-CCTCTATGCCAACACAGTGC-3′ and reverse, 5′-GTACTCCTGCTTGCTGATCC-3′;
CTGF forward, 5′-CAAGGGCCTCTTCTGTGACT-3′ and reverse,
5′-TGGAGATTTTGGGAGTACGG-3′; β-actin forward,
5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ and reverse,
5′-CTAGAAGCATTTGCGGTGGACGATGGA GGG-3′. Relative mRNA expression
levels were determined using the comparative Ct method with data
normalized against 36B4 riboprotein mRNA and calibrated to the
average ΔCt value of the untreated controls. Data are expressed as
a percentage of the control, which was set to 100%.
Western blot analysis
Proteins were extracted from the heart homogenates,
and aliquots (50 μg) were subjected to SDS-PAGE (7.5% gel) and
transferred to nitrocellulose membranes. The membranes were blocked
for 2 h at room temperature with block solution that was provided
in the enhanced chemiluminescence (ECL) kit. The membranes were
then incubated with primary antibodies overnight at 4°C. Equal
protein samples were used for western blot analysis with the
following rat antibodies targeted against: TGF-β1 (EM010-48), CTGF
(BC087839), phospho-Akt (Ser 473; GM-AT7126), Akt, phospho-GSK-3β
(Ser 9; 07835), GSK-3β (bc000251) and β-actin (all purchased from
Biogot Technology Co., Ltd., Shanghai, China). The membranes were
then washed for 30 min in wash solution (ECL kit) and incubated
with rat secondary IgG antibodies conjugated with horseradish
peroxidase in block solution (Biogot Technology Co., Ltd.). The
membranes were washed for 30 min in wash solution and the
immunoreactive bands were detected with an ECL kit.
Statistical analysis
Statistical analysis was performed using SPSS
version 14.0 statistical software (SPSS, Inc., Chicago, IL, USA)
and data are expressed as the mean ± standard deviation.
Comparisons of the mean values between multiple groups were
performed with one-way analysis of variance, where P<0.05 was
considered to indicate a statistically significant difference.
Results
Urocortin decreases the LVWI in diabetic
rats
Compared with the control group, a significant
increase in the LVWI was observed in the DM (P<0.01), UCN
(P<0.05), UCN + AST (P<0.01) and UCN + TRI (P<0.01)
groups. Notably, urocortin decreased the LVWI in diabetic rats
(P<0.01), and the LVWI was partially decreased by urocortin and
triciribine treatment as compared with urocortin treatment alone
(P<0.05). However, administering astressin with urocortin
appeared to reduce the beneficial effect of urocortin on the LVWI
(Fig. 1A).
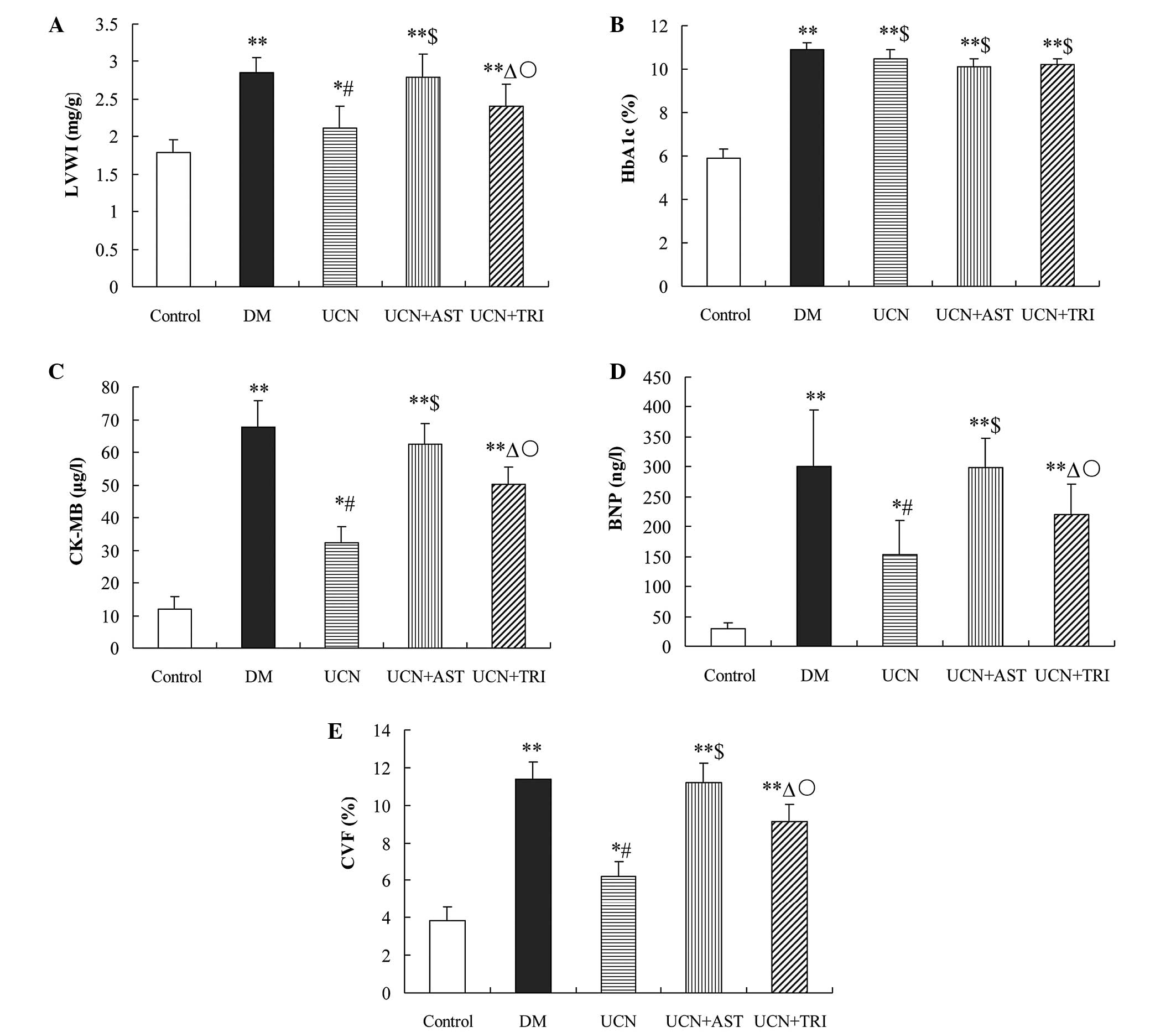 | Figure 1Effect of urocortin on (A) LVWI, (B)
HbA1c, (C) CK-MB, (D) BNP and (E) CVF and in the diabetic rats.
Levels of BNP and CK-MB, as well as the LVWI and CVF, were
decreased by urocortin and urocortin and triciribine treatment,
while astressin blunted the effect of urocortin. HbA1c levels were
not affected. *P<0.05 and **P<0.01, vs.
control; #P<0.01 and ΔP<0.05, vs. DM
group and UCN + AST; $P>0.05, vs. DM group;
○P<0.05, vs. UCN group. LVWI, left ventricular mass
index; HbAlc, glycosylated hemoglobin; CK-MB, creatine kinase
isoenzyme; BNP, brain natriuretic peptide; CVF, collagen fraction
volume; DM, diabetes mellutis; UCN, urocortin; AST, astressin. |
Urocortin decreases the levels of CK-MB
and BNP
HbA1c, CK-MB and BNP levels in the DM (P<0.01),
UCN (P<0.05), UCN + AST (P<0.01) and UCN + TRI (P<0.01)
groups were significantly increased compared with the control. No
statistically significant differences were observed in the HbA1c
levels (P>0.05) among the DM, UCN, UCN + AST and UCN + TRI
groups. Compared with the control, the CK-MB and BNP levels in the
diabetic rats markedly increased (P<0.01), however, this
increase was diminished (P<0.01) by treatment with urocortin and
was partially reduced (P<0.05) by urocortin and triciribine
treatment when compared with urocortin administration alone. This
effect of urocortin on CK-MB and BNP was significantly reversed by
astressin (Fig. 1B–D).
Myocardial CVF inhibition by
urocortin
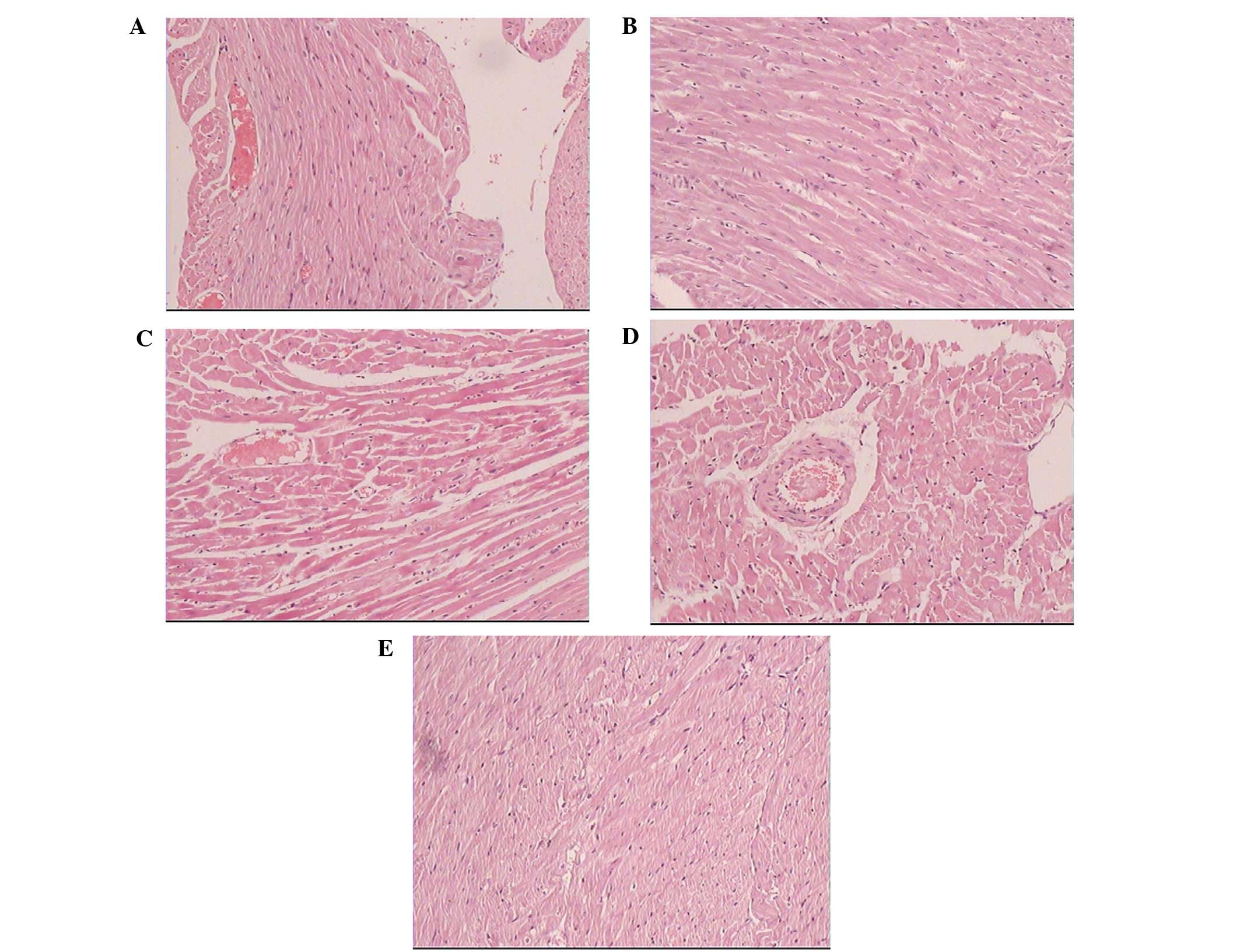
Cardiomyocytes in the normal rats presented with a
regular arrangement, clear stripes without myofilament fracture and
uniform intercellular space. By contrast, cardiomyocytes in the
diabetic group exhibited myofibrillar disarray and myocardial
fibrosis, scattered muscle fiber-degeneration, coarse granules in
the cytoplasm, nucleus swelling and deformation, loss of cardiac
muscle fiber stripes, interstitial edema and hyperplasia and
lymphocytic infiltration. The myocardial cell arrangement in the
diabetic rats treated with urocortin was more regular than that in
the diabetic group (Fig. 2).
Compared with the control group, the CVF in the DM, UCN, UCN + AST
and UCN + TRI groups significantly increased (P<0.05). However,
urocortin treatment suppressed the CVF increase (P<0.01) in the
diabetic rats, while urocortin and triciribine treatment exhibited
a partial effect on the CVF (P<0.05). The effect of urocortin
was reversed by astressin (Fig.
1E).
Urocortin decreases the levels of TGF-β1
and CTGF in the plasma of diabetic rats
Hyperglycemia is known to activate several cytokines
via oxidative stress, which may contribute to the development of
DCM. Compared with the control, TGF-β1 and CTGF levels increased
significantly (P<0.01) in the plasma of the diabetic rats.
Supplementation with urocortin markedly reduced the levels of
TGF-β1 and CTGF as compared with DM treatment (P<0.01), while
supplementation with urocortin and triciribine partially reduced
the TGF-β1 and CTGF levels when compared with urocortin treatment
alone (P<0.05). Astressin with urocortin treatment in the
diabetic rats appeared to reverse the effect of urocortin on the
levels of TGF-β1 and CTGF (Fig.
3).
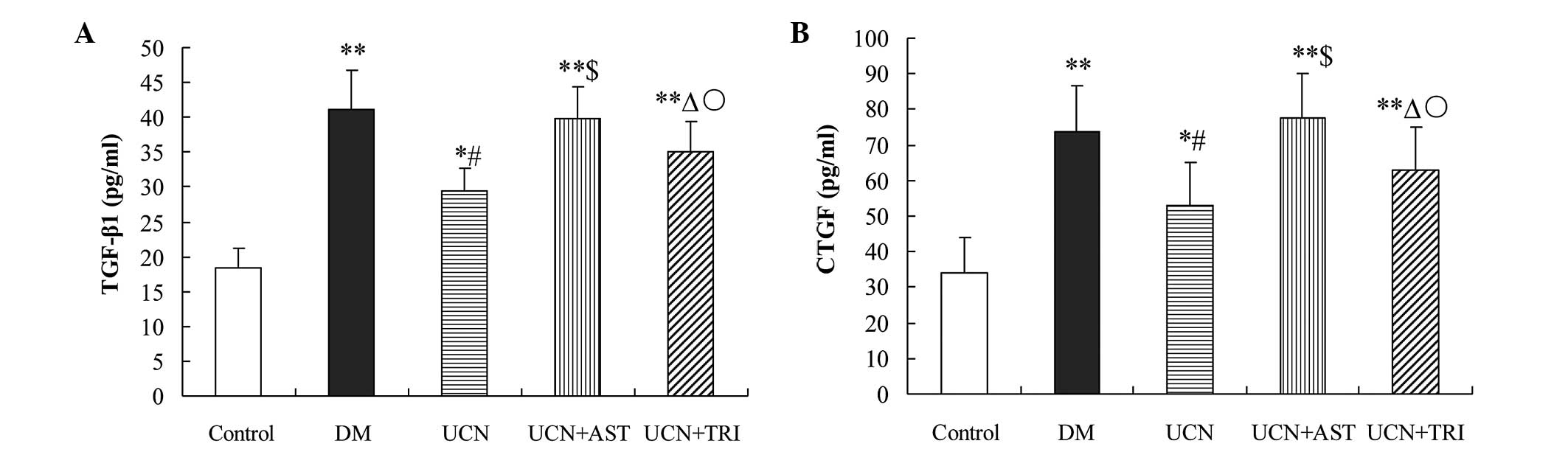 | Figure 3Urocortin and urocortin with
triciribine reduced the oversecretion of (A) TGF-β1 and (B) CTGF in
the plasma of diabetic rats. However, astressin reversed the effect
of urocortin. *P<0.05 and **P<0.01, vs.
control; #P<0.01 and ΔP<0.05, vs. DM
and UCN + AST groups; $P>0.05, vs. DM group;
○P<0.05, vs. UCN group. TGF, transforming growth
factor; CTGF, connective tissue growth factor; DM, diabetes
mellitus; UCN, urocortin; AST, astressin. |
Urocortin decreases the expression of
TGF-β1 and CTGF in diabetic rats
To investigate the mechanism by which urocortin
exhibits beneficial effects on myocardial fibrosis in diabetic
rats, the expression levels of TGF-β1 and CTGF were analyzed, since
they had been shown to be involved in myocardial fibrosis. The
effect of urocortin on TGF-β1 and CTGF expression was investigated
in cardiac muscular tissues obtained from each group. Quantitative
PCR analysis revealed that TGF-β1 and CTGF mRNA expression levels
in the DM (P<0.01), UCN (P<0.05), UCN + AST (P<0.01) and
UCN + TRI (P<0.01) groups were higher compared with the control
animals. However, urocortin decreased the TGF-β1 and CTGF mRNA
expression levels in diabetic rats (P<0.01), and supplementation
with urocortin and triciribine partially decreased the TGF-β1 and
CTGF mRNA expression levels when compared with urocortin treatment
alone (P<0.05). Furthermore, western blot analysis demonstrated
that urocortin decreased the protein expression levels of TGF-β1
and CTGF in diabetic rats (P<0.01). However, astressin with
urocortin treatment completely eliminated the inhibitory effect of
urocortin on TGF-β1 and CTGF overexpression (Fig. 4).
 | Figure 4Urocortin and urocortin with
triciribine treatment inhibited TGF-β1 and CTGF expression in the
cardiac muscular tissues of diabetic rats, while astressin
administration eliminated the effects of urocortin. mRNA expression
levels of (A) TGF-β1 and (B) CTGF in the various groups as detected
by quantitative PCR. (C) Protein expression levels of TGF-β1 and
CTGF in the various groups, as detected by western blot analysis.
*P<0.05 and **P<0.01, vs. control;
#P<0.01 and ΔP<0.05, vs. DM and UCN +
AST groups; $P>0.05, vs. DM group;
○P<0.05, vs. UCN group. TGF, transforming growth
factor; CTGF, connective tissue growth factor; PCR, polymerase
chain reaction; DM, diabetes mellitus; UCN, urocortin; AST,
astressin. |
Urocortin activates the Akt/GSK-3β
signaling pathway
Using western blot analysis, Akt phosphorylation was
shown to be significantly inhibited in the diabetic hearts. In
addition, marked activation of GSK-3β was observed in the diabetic
hearts. By contrast, urocortin induced a significant increase
(P<0.01) in the phosphorylation of Akt and GSK-3β in the
myocardium, while astressin inhibited the effect of urocortin on
Akt and GSK-3β phosphorylation (Fig.
5).
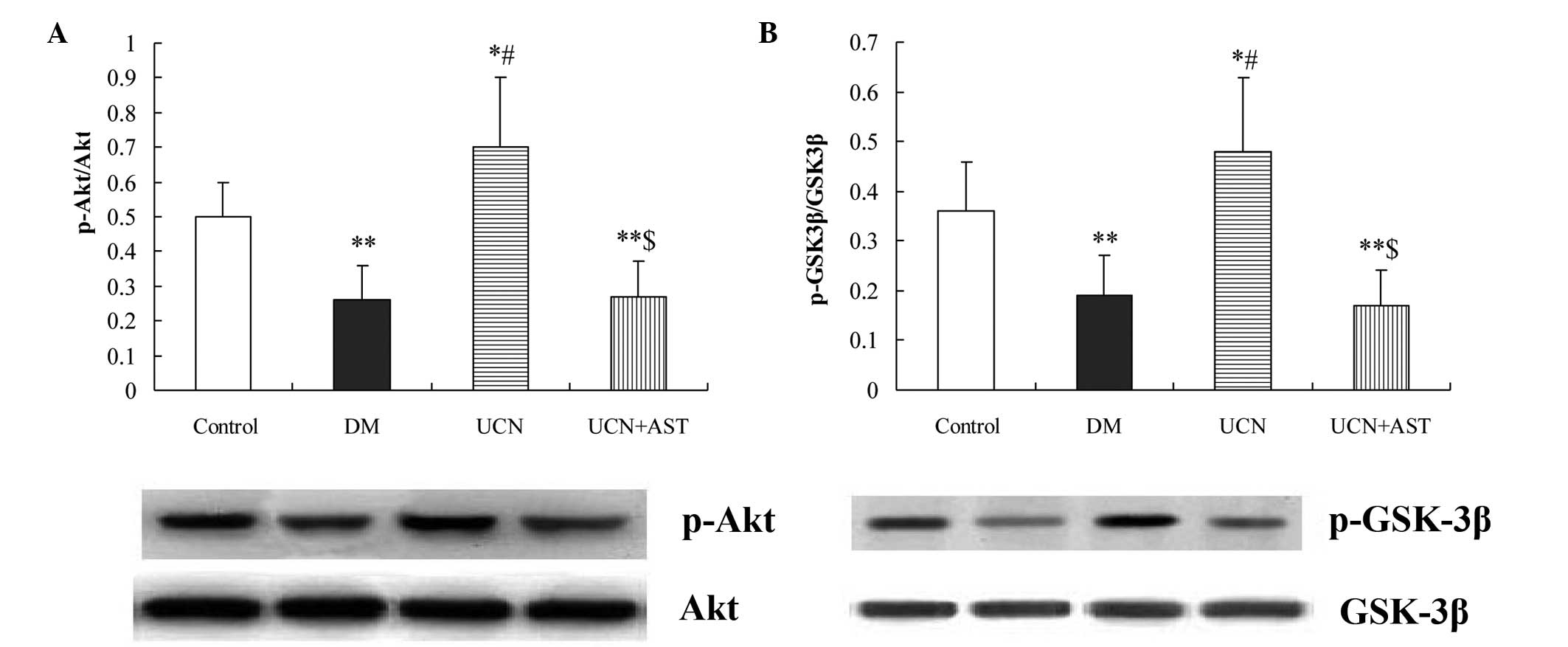 | Figure 5Urocortin activated Akt and
inactivated GSK-3β in the cardiac muscular tissues of diabetic
rats, while astressin eliminated the effects of urocortin. Western
blot analysis of (A) p-Akt and Akt and (B) p-GSK-3β and GSK-3β.
*P<0.05 and **P<0.01, vs. control;
#P<0.01, vs. DM and UCN + AST groups;
$P>0.05, vs. DM group. GSK, glycogen synthase kinase;
p-Akt, phospho-Akt; p-GSK, phospho-glycogen synthase kinase; DM,
diabetes mellitus; UCN, urocortin; AST, astressin. |
Discussion
DCM is an important cardiovascular complication that
causes cardiac dysfunction in DM patients. The main pathological
changes include myocardial cell focal hypertrophy, degeneration,
necrosis, apoptosis and myocardial remodeling. A series of
pathophysiological changes caused by myocardial interstitial
remodeling play an important role in the pathogenesis of DCM
(15).
The major novel observation of the present study was
that urocortin may be beneficial in reversing the effects of DCM.
Consistent with pervious studies, the 16-week untreated diabetic
rats in the study were characterized by excess collagen
accumulation and cardiac hypertrophy. In the untreated diabetic
hearts, increased BNP and CK-MB accumulation, coupled with elevated
LVWIs and CVFs, were observed. In addition, enhanced myocardial
expression of TGF-β1 and CTGF, coupled with inactivated Akt/GSK-3β
signaling, were observed, which eventually culminated in cardiac
fibrosis. By contrast, urocortin was found to prevent the
development of these characteristic alterations of DCM, and these
beneficial effects involved CRFR-2. The underlying mechanisms also
involved the inhibitory effects of urocortin on the overexpression
and secretion of TGF-β1 and CTGF via the activation of the
Akt/GSK-3β signaling pathway in the diabetic hearts.
In response to high levels of glucose, the
expression of the potent profibrotic factor, TGF-β1, significantly
increases, which leads to fibrotic consequences. TGF-β1 is an
important fibrogenic factor that promotes the synthesis and
secretion of collagen I and III which induces myocardial fibrosis
(16). In the development of
extracellular matrix accumulation, CTGF may function as a downsteam
mediator of TGF-β1 (17). In the
present study, LVWI, BNP, CK-MB and CVF levels, as well as TGF-β1
and CTGF mRNA and protein expression levels, were significantly
higher in the diabetic group when compared with the control group,
indicating that the increase in TGF-β1 and CTGF expression levels
in the DM rats was closely associated with myocardial
remodeling.
Akt promotes cell survival by inhibiting several
targets that are involved in apoptotic signaling cascades. GSK-3β,
a major substrate of Akt, not only has central functions in
glycogen metabolism and insulin function, but also plays a crucial
role in transmitting apoptotic signals, DM-induced inflammation and
fibrosis (18). A previous study
demonstrated that the activation of GSK-3β played a pivotal role in
DM-induced energy metabolic derangement and, consequently,
pathological remodeling in the heart (19). In DM, Akt phosphorylation can be
reduced by the elevated circulation of free fatty acids and
inflammatory cytokines, which leads to the activation of GSK-3β
(20). In addition, Akt has a
clearly defined role in the regulation of cardiovascular functions,
including cardiac growth, contractile function and coronary
angiogenesis (21). The results of
the present study demonstrated that triciribine, an inhibitor of
Akt, partially weakened the effects of urocortin on DCM and the
Akt/GSK-β pathways involved in mediating the effects of urocortin
on DCM.
Urocortins are endogenous vasoactive peptides that
have been shown to exert powerful beneficial neurohormonal,
hemodynamic and renal effects in an experimental heart failure
model (22). Urocortins function
predominantly through two receptor subtypes, CRFR-1 and CRFR-2. The
receptors possess seven transmembrane domains and are G-protein
coupled. CRF-2(a) receptors constitute the dominant peripheral
CRFR-2 form, particularly in the heart and vasculature. Receptor
concentrations are high in the left ventricle and intramyocardial
vessels (23). However, a recent
study revealed that the CRF-2 receptor exhibits much more potent
effects on the cardiovascular system compared with CRF (24). The CRF-2 receptor enhances cardiac
contractility, coronary blood flow, heart rates and cardiac output.
A number of in vitro and ex vivo investigations have
shown that in cases of ischemia/reperfusion, atherosclerosis and
hypertension, urocortin can protect cardiac cells from severe
injury (25). As a novel small
molecular active peptide, urocortin exerts protective effects via
autocrine and/or paracrine signaling pathways (26,27).
The majority of studies have reported that urocortin binds to
CRFR-2 and promotes an increase in cAMP levels, thus, activating
protein kinase A (28,29). Brar et al found that the
mitogen-activated protein kinase and phosphatidylinositol 3-kinase
pathways were involved in the protective mechanisms of urocortin
(30,31). In a recent study, urocortin was
shown to directly activate AMP-activated protein kinase in ex
vivo-perfused mouse hearts and decrease injury and contractile
dysfunction during ischemia/reperfusion. In addition, the study
revealed that stimulation of CRFR-2 by CRF and urocortin induced
the release of BNP, however, this had an inotropic effect on the
heart, which is consistent with the observations of the present
study (32).
Considering the role of myocardial interstitial
remodeling in diabetic development, the present study aimed to
investigate the beneficial effects of urocortin on DCM remodeling.
Firstly, it was found that 16 weeks of intervention with urocortin
in diabetic rats significantly reduced the levels of BNP and CK-MB,
the LVWI and myocardial CVF, as well as the mRNA and protein
expression levels of TGF-β1 and CTGF. Secondly, urocortin was shown
to significantly promote the phosphorylation of Akt and GSK-3β in
the myocardium. However, in the UCN + AST group, the effects of
urocortin were prevented, which indicated that the effects were
closely associated with the CRFRs. Thirdly, triciribine partially
eliminated the effects of urocortin on DCM, indicating that the
Akt/GSK-β signaling pathways were partially involved in mediating
the effects of urocortin.
In conclusion, the results of the present study
support the hypothesis that urocortin markedly inhibits the
development of cardiac dysfunction, myocardial fibrosis and
inflammation in diabetic rats. Urocortin was shown to protect
cardiac functions, reverse ventricular remodeling, inhibit
hypertrophy and decrease the collagen content. The predominant
underlying mechanism may be that urocortin binds to CRFR-2, which
then inhibits the expression of TGF-β1 and CTGF. The Akt/GSK-β
signaling pathways are partially involved in mediating the effects
of urocortin. These observations provide new guidance for the
clinical treatment of DCM. However, it is not known whether these
observations have a bearing on the potential clinical application
of urocortin as a novel agent, with a potent protective function in
the treatment of human DM. In addition, whether alterations in
these control pathways contribute to the development of
cardiovascular disease remains unknown.
The present study has demonstrated that urocortin
significantly improves cardiac function. Urocortin inhibits
myocardial fibrosis and inflammation in diabetic rats, which is
associated with the inhibition of TGF-β1 and CTGF expression and
the Akt/GSK-β signaling pathways. These results provide new
guidance for the clinical treatment of DCM.
Acknowledgements
The study was supported by grants from the Natural
Science Foundation of China (no. 81201037) and the Natural Science
Foundation of Liaoning Province (nos. 2012921020 and
2010225034).
References
|
1
|
Jaffe AS, Spadaro JJ, Schechtman K,
Roberts R, Geltman EM and Sobel BE: Increased conjestive heart
failure after myocardial infarction of modest extent in patients
with diatetes mellitus. Am Heart J. 108:31–37. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Falcão-Pires I and Leite-Moreira AF:
Diabetic cardiomyopathy: understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar
|
|
3
|
Huynh K, McMullen JR, Julius TL, Tan JW,
Love JE, et al: Cardiac specific IGF-1 receptor transgenic
expression protects against cardiac fibrosis and diastolic
dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes.
59:1512–1520. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang ZY, Prins JB and Marwick TH: Diabetic
cardiomyopathy: evidence, mechanisms, and therapeutic implications.
Endocr Rev. 25:543–567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar :
|
|
6
|
Dai QM, Lu J and Liu NF: Fluvastatin
attenuates myocardial interstitial fibrosis and cardiac dysfunction
in diabetic rats by inhibiting over-expression of connective tissue
growth factor. Chin Med J (Engl). 124:89–94. 2011.
|
|
7
|
Vaughan J, Donaldson C, Bittencourt J,
Perrin MH, Lewis K, Sutton S, et al: Urocortin, a mammalian
neuropeptide related to fish urotensin I and to
corticotropin-releasing factor. Nature. 378:287–292. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fekete EM and Zorrilla EP: Physiology,
phamacology, and therapeutic relevance of urocortins in mammals:
ancient CRF paralogs. Front Neuroendocrinol. 28:1–27. 2007.
View Article : Google Scholar
|
|
9
|
Honjo T, Inoue N, Shiraki R, Kobayashi S,
Otsui K, Takahashi M, et al: Endothelial urocortin has potent
antioxidative properties and is upregulated by inflammatory
cytokines and pitavastatin. J Vasc Res. 43:131–138. 2006.
View Article : Google Scholar
|
|
10
|
Huang Y, Chan FL, Lau CW, Tsang SY, He GW,
Chen ZY and Yao X: Urocortin-induced endothelium-dependent
relaxation of rat coronary artery: role of nitric oxide and
K+ channels. Br J Pharmacol. 135:1467–1476. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takahashi K: Distribution of urocortins
and corticotropin-releasing factor receptors in the cardiovascular
system. Int J Endocrinol. 2012:3952842012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rademaker MT, Charles CJ, Espiner EA,
Frampton CM, Lainchbury JG and Richards AM: Four-day urocortin-I
administration has sustained beneficial haemodynamic, hormonal and
renal effects in experimental heart failure. Eur Heart J.
26:2055–2062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu CN, Yang C, Liu XY and Li S: In vivo
protective effects of urocortin on ischemia-reperfusion injury in
rat heart via free radical mechanisms. Can J Physiol Pharmacol.
83:459–465. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang C, Xu Y, Mendez T, Wang F, Yang Q and
Li S: Effects of intravenous urocortin on angiotensin-coverting
enzyme in rats. Vascul Pharmacol. 44:238–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tarquini R, Lazzeri C, Pala L, Rotella CM
and Gensini GF: The diabetic cardiomyopathy. Acta Diabetol.
48:173–181. 2011. View Article : Google Scholar
|
|
16
|
Ma YX, Li WH and Xie Q: Rosuvastatin
inhibits TGF-beta1 expression and alleviates myocardial fibrosis in
diabetic rats. Pharmazie. 68:355–358. 2013.PubMed/NCBI
|
|
17
|
Umezono T, Toyoda M, Kato M, Miyauchi M,
et al: Glomerular espression of CTGF, TGF-beta 1 and type IV
collagen in diabetic nephropathy. J Nephrol. 19:751–757.
2006.PubMed/NCBI
|
|
18
|
Liu Y, Tanabe K, Baronnier D, Patel S,
Woodgett J, et al: Conditional ablation of GSK-3β in islet beta
cells results in expanded mass and resistance to fat
feeding-induced diabetes in mice. Diabetologia. 53:2600–2610. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Feng W, Xue W, et al: Inactivation
of GSK-3β by metallothionein prevents diabetes-related changes in
cardiac energy metabolism, inflammation, nitrosative damage, and
remodeling. Diabetes. 58:1391–1402. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu W, Wu J, Cai F, Xiang J, Zha W, et al:
Curcumin alleviates diabetic cardiomyopathy in experimental
diabetic rats. PloS One. 7:e520132012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Katare RG, Caporali A, Oikawa A, Meloni M,
et al: Vitamin B1 analog benfotiamine prevents diabetes-induced
diastolic dysfunction and heart failure through Akt/Pim-1-mediated
survival pathway. Circ Heart Fail. 3:294–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bale TL, Hoshijima M, Gu Y, et al: The
cardiovascular physiologic actions of urocortin II: acute effects
in murine heart failure. Proc Natl Acad Sci USA. 101:3697–3702.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Emeto TI, Moxon JV, Rush C, Woodward L and
Golledge J: Relevance of urocortins to cardiovascular disease. J
Mol Cell Cardiol. 51:299–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liew HK, Hsu CW, Wang MJ, et al:
Therapeutic benefit of urocortin in rats with intracerebral
hemorrhage. J Neurosurg. 116:193–200. 2012. View Article : Google Scholar
|
|
25
|
Rademaker MT, Charles CJ, Nicholls G and
Richards M: Urocortin 2 sustains haemodynamic and renal function
during introduction of beta-blockade in experimental heart failure.
J Hypertens. 29:1787–1795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gruson D, Ginion A, Lause P, Ketelslegers
JM, Thissen JP and Bertrand L: Urotensin II and urocortin trigger
the expression of myostatin, a negative regulator of cardiac
growth, in cardiomyocytes. Peptides. 33:351–353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rademaker MT, Charles CJ, Nicholls MG and
Richards AM: Interactions of enhanced urocortin 2 and
mineralocorticoid receptor antagonism in experimental heart
failure. Circ Heart Fail. 6:825–832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parkes DG, Weisinger RS and May CN:
Cardiovascular actions of CRH and urocortin: an update. Peptides.
22:821–827. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miki I, Seya K, Motomura S and Furukawa K:
Role of corticotropin-releasing factor receptor type 2 in
urocortin-induced vasodilation of rat aortas. J Pharmacol Sci.
96:170–176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brar BK, Jonassen AK, Stephanou A, et al:
Urocortin protects against ischemic and reperfusion injury via a
MAPK-dependent pathway. J Biol Chem. 275:8508–8514. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rademaker MT, Charles CJ, Ellmers LJ,
Lewis LK, Nicholls MG and Richards AM: Prolonged urocortin 2
administration in experimental heart failure: sustained
hemodynamic, endocrine, and renal effects. Hypertension.
57:1136–1144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ikeda K, Fujioka K, Manome Y and Tojo K:
Clinical perspectives of urocortin and related agents for the
treatment of cardiovascular disease. Int J Endocrinol.
2012:1986282012. View Article : Google Scholar : PubMed/NCBI
|