Introduction
In patients with hemophilia A, a crucial problem is
the prevalence of factor VIII, treatment for which may be
expensive. The most common complication associated with hemophilia
A is bleeding into joints, predominantly the knees, ankles and
elbows, which may lead to destruction or osteoarthritis of the
joint. Various degrees of disability may follow these initial or
recurrent hemorrhages.
Hemorrhage of the central nervous system is the most
serious manifestation of the disease. A previous study described
the case of a 12-year-old boy with severe hemophilia A, in which a
short half-life recombinant factor VII (rFVII) was administered to
the bleeding patients at 2 h intervals. Subsequently, the dosage
interval may be increased to 3, 4 or 6 h depending on the bleeding.
Large hemorrhages due to the phlebotomizing of young patients are
very rare. To the best of our knowledge, the present case is the
first report regarding the occurrence of a large hemorrhage due to
venipuncture in the elbow of a patient with hemophilia A.
Case report
A 22-year-old boy was diagnosed with hemophilia A 20
years prior to the present report, despite no relevant family
history of hemophilia. Written informed consent was obtained from
the patient. One month prior to admission in February 2013, a blood
sample was taken from a vein in the right elbow of the patient. The
venipuncture was followed by pain in the right arm, which is
associated with the development of a hemorrhage. An ulcerative
hemorrhage with local cutaneous necrosis was detected 10 days
following the venipuncture, and the patient was admitted to the
Department of Internal Medicine of West China Hospital (Chengdu,
China) for further treatment. Coagulation function tests were
conducted at 37°C. Firstly, kaolin was used to activate the XII, in
order to replace the platelet factor 3 with cephalin. Then,
Ca2+ was added to the solution to initiate clotting and
observe the coagulation time. The time required for plasma
coagulation was the activated clotting time. The activated partial
thromboplastin time (APTT) of the patient was 104.6 sec (normal,
20–40 sec), and factor VIII activity was 1.0% (normal, 60–150%). An
X-Ray of the elbow exhibited bone erosion and cyst formation.
Hemorrhage of the elbow with hemophilic arthropathy and clotting
factor VIII inhibitor formation was initially diagnosed. The
hemorrhage of the hemophilic arthropathy was caused by the
formation of clotting factor VIII. Paraffin fixing and staining
with hematoxylin and eosin, the hemorrhagic, with synovium and
hemosiderin were distinguished by different color and
morphology.
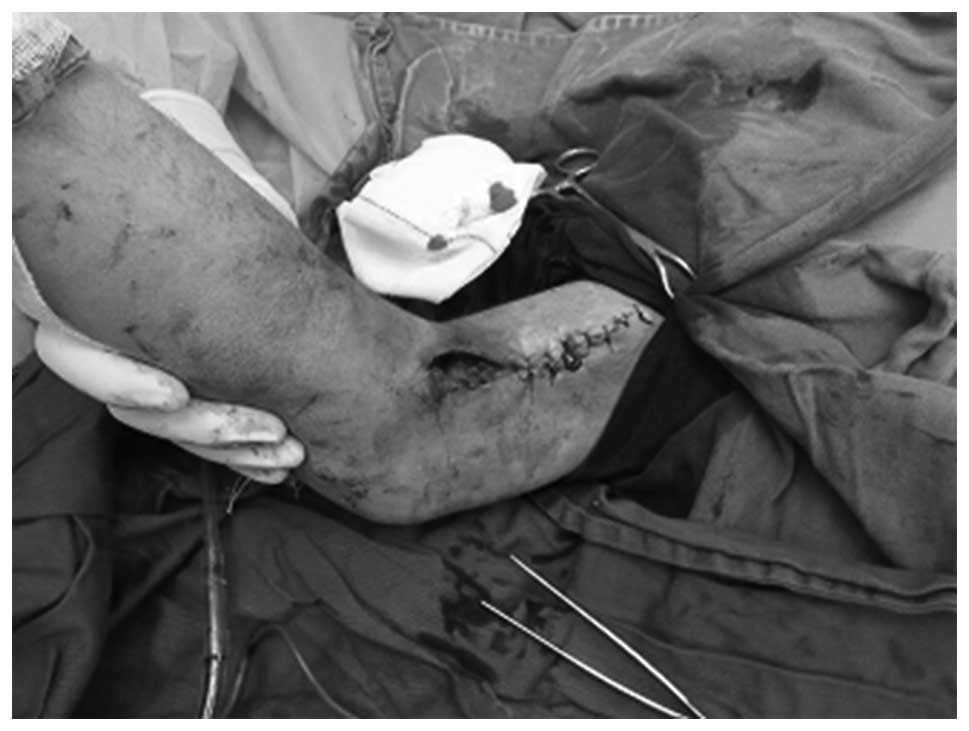
Evacuation of the hematoma in the elbow was
initially performed (Figs.
1–5). Intraoperatively, the
lesion appeared initially tough with various stages of liquid and
solid dark blue hemorrhage. The lesion, which adhered closely to
the brachial artery, was completely removed. In addition, part of
the musculus biceps brachii and biceps tendon were removed.
Considering the large size of the skin wound (~3×6 cm), the surgeon
could not suture the incision; instead, the incision was covered
with petrolatum gauze. Coagulation function tests demonstrated
that, prior to the surgery, the APTT was 102.3 sec and remained
unchanged following surgery. A total of 2,800 units of clotting
factor VIII were prescribed every 12 h for the following 4 days,
and the APTT was measured to be 102.3 sec following the continuous
infusion of factor VIII.
A second operation was performed 10 days after the
initial operation. The lesion appeared to be covered with
granulation tissue, with necrotic tissue detected in the basal area
of the lesion. In the second operation, the skin wound (~3×6 cm)
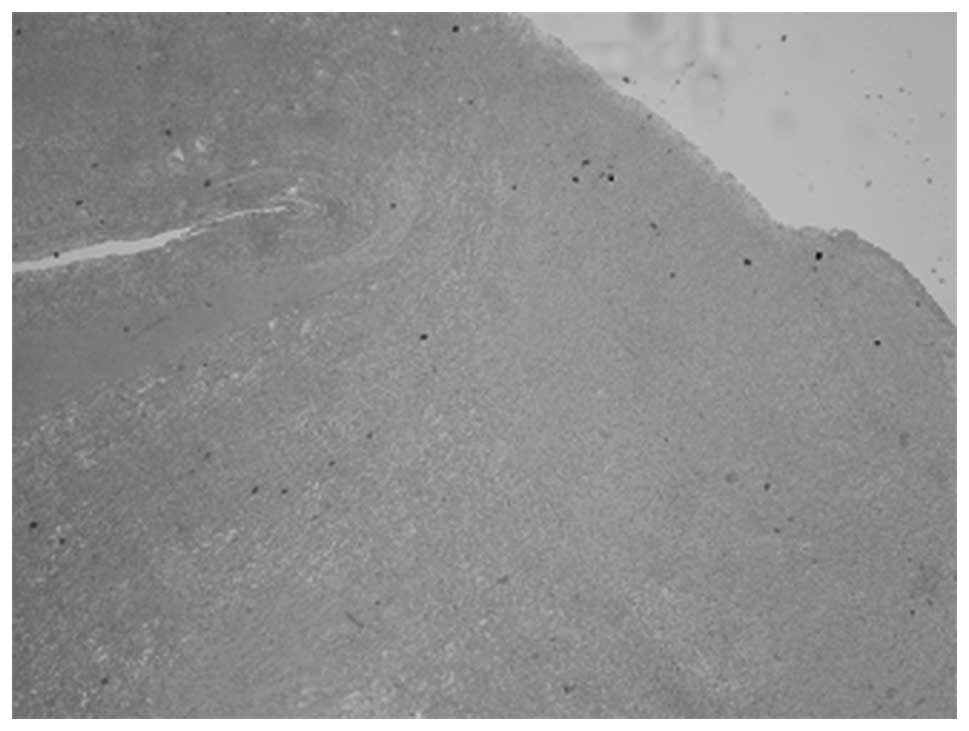
was sutured. The pathological diagnosis indicated that the primary
component of the lesion was hemorrhagic, with synovium and
hemosiderin (Figs. 6 and 7). The patient remained in good health with
no deficit as of the last follow-up visit. The patient received a
total of 2,400 units of factor VIII every 24 h for 3 days following
the second operation. The APTT was 80.6 sec following the
continuous infusion of factor VIII. The patient remained in good
health with no symptoms at a 1-year follow-up examination.
Discussion
Hemophilia A is an inherited bleeding disorder
caused by a deficiency in the synthesis of factor VIII (1). The bleeding tendency associated with
hemophilia A is proportional to the degree of factor VIII
deficiency. A genetic predisposition or congenital deficiency is
currently considered the predominant etiological factor (2).
Large hemorrhages may occur in infants with
hemophilia; the immature fragile vascular structure of lesions
leads to frequent hemorrhages in injection sites. The lesion may
also express growth factors, such as vascular endothelial growth
factor and transforming growth factor-α, which further promote the
proliferation of lesions (3,4). Without suitable treatment, these
episodes often lead to severe arthropathy.
Few non-specialist health care professionals deal
with patients with hemophilia A on a regular basis (5). For treatment of the disease,
immunosuppressive therapy is required for the eradication of
autoantibodies. Corticosteroids or combination therapy with
cyclophosphamide is considered the standard treatment (6). Early diagnosis of the disease,
communication with the patient and their parents/guardians, and
implementation of the best treatment are essential.
Doctors aim to prevent bleeding symptoms and ensure
that patients with hemophilia A have normal lifestyles; therefore,
treatment with both conservative and aggressive therapeutic
strategies is necessary. The factor VIII injection is used as the
conservative treatment, surgical hematoma clearance is used as the
aggressive treatment. The conservative treatment involves control
of bleeding and may avoid operational risks, for example, the
hemorrhage formation again, and can control bleeding by systemic
administration. By contrast, the aggressive treatment may
completely resolve the hematoma surgically. If rebreeding can be
avoided, the aggressive treatment is the better choice. For the
treatment of bleeding in patients with hemophilia, conservative
treatment with factor VIII replacement is the primary choice
(7). Surgical treatment in patients
with hemophilia remains challenging due to the high risk of intra-
or post-operative hemorrhage. In addition, fatal bleeding must be
controlled during the perioperative period.
Conservative management in bleeding patients with
severe hemophilia and factor VIII inhibitors includes the
administration of activated and non-activated prothrombin complex
concentrate (PCC) and, more frequently, the use of rFVII
concentrate (8). It is recommended
that rFVII be administered to bleeding patients, or those
undergoing surgery, at 2 h intervals, at least during the initial
24 h period of treatment; subsequently, the dosage intervals may be
increased to 3, 4 or 6 h, according to the type and extent of the
surgical procedure or bleeding (9).
Recombinant activated FVII or activated PCC should be used to
control the symptoms (10–12), and recombinant products are
increasingly regarded as the preferred choice, due to a low risk of
viral infection (5). Rituximab
monotherapy may also be considered in the case of
surgery-associated acquired hemophilia; however the effects of
immunosuppressive therapies on postoperative wound healing remain
unclear (13).
In the present case, the time between the use of
conservative (the transfusion of factor VIII) and aggressive
treatments (the surgery for evacuation of hematoma) was crucial, as
an informed decision can avoid complex or frequent operations. For
large hemorrhages, aggressive factor VIII replacement and emergency
surgical treatment is still considered at the primary stage. The
surgeon and hematologist must work together closely in order to
obtain a successful outcome. Data from a previous prospective
randomized controlled trial demonstrated that the co-treatment of a
patient by a hematologist and surgeon reduces the incidence of
joint hemorrhages and protects against the development of joint
damage (5). Blood counts and
coagulation function should be routinely monitored following
surgery. In addition, it is recommended that factor VIII be infused
regularly; the level should be raised to 80–100% for 7 days, and to
50% for a further 14 days (14).
The present case is the first, to the best of our
knowledge, to report a case of a large hemorrhage caused by
venipuncture in the elbow of a patient with hemophilia. The results
indicate that a systematic analysis of patients to determine the
appropriateness of transfusion with factor VIII should be
considered for patients with hemophilia A prior to surgery.
In the future, it seems likely that modified
molecules, which possess enhanced properties such as reduced
immunogenicity and increased half-life, will become available.
Numerous clinical trials targeting hemophilia A and B are currently
underway (15,16). In addition, developments in gene
therapy may provide an attractive model for the treatment of
hemophilia, eliminating the need for regular injections of clotting
factor VIII.
Acknowledgements
The authors wish to thank the staff of the
Department of Pathology of the West China Hospital for their
technical assistance.
Glossary
Abbreviations
Abbreviations:
|
aPTT
|
activated partial thromboplastin
time
|
References
|
1
|
Petrini P: Treatment strategies in
children with hemophilia. Pediatr Drugs. 4:427–437. 2002.
View Article : Google Scholar
|
|
2
|
Martínez-Lage JF, Torroba MA, Cuartero
Pérez B, Almagro MJ, López-Guerrero López A and de la Rosa P:
Cavernous hemangiomas of the cranial vault in infants: A case-based
update. Childs Nerv Syst. 26:861–865. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cosar M, Eser O, Aslan A, Korkmaz S,
Boyaci G and Aktepe F: Intradiploic cavernous hemangioma of the
skull in a child: A case report. Childs Nerv Syst. 24:975–977.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Politi M, Romeike BF, Papanagiotou P,
Nabhan A, Struffert T, Feiden W and Reith W: Intraosseous
hemangioma of the skull with dural tail sign: Radiologic features
with pathologic correlation. AJNR Am J Neuroradiol. 26:2049–2052.
2005.PubMed/NCBI
|
|
5
|
Giangrande PLF: Management of haemophilia.
Pead Child Healt. 21:344–347. 2011. View Article : Google Scholar
|
|
6
|
Sperr WR, Lechner K and Pabinger I:
Rituximab for the treatment of acquired antibodies to factor VIII.
Haematologica. 92:66–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhong W, Li G, Huang S, Chen H and You C:
Intradiploic hemangioma with repeated hemorrhage in a child with
hemophilia. J Neurosurg Pediatr. 10:56–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shih SL, Lin JC, Liang DC and Huang JK:
Computed tomography of spontaneous intracranial haemorrhage due to
haemostatic disorders in children. Neuroradiology. 35:619–621.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taylor CL, Selman WR and Ratcheson RA:
Brain attack. The emergent management of hypertensive hemorrhage.
Neurosurg Clin N Am. 8:237–244. 1997.PubMed/NCBI
|
|
10
|
Rangarajan S, Yee T and Wilde J:
Experience of four UK comprehensive care centres using FEIBA® for
surgeries in patients with inhibitors. Haemophilia. 17:28–34. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lauroua P, Ferrer AM and Guérin V:
Successful major and minor surgery using factor VIII inhibitor
bypassing activity in patients with haemophilia A and inhibitors.
Haemophilia. 15:1300–1307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lak M, Sharifian RA, Karimi K and
Mansouritorghabeh H: Acquired hemophilia. A. Clinical features,
surgery and treatment of 34 cases, and experience of using
recombinant factor VIIa. Clin Appl Thromb Hemost. 16:294–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kam G, Lee YS, Tan TT, Chow P and Ng HJ:
Surgery-associated acquired haemophilia and response to combined
rituximab and cyclosporine treatment. Haemophilia. 17:715–716.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahlangu JN and Gilham A: Medical and
Scientific Advisory Council of the South African Haemophilia
Foundation: Guideline for treatment of haemophilia in South Africa.
S Afr Med J. 98:126–140. 2008.PubMed/NCBI
|
|
15
|
Lak M, Sharifian RA, Karimi K and
Mansouritorghabeh H: Acquired Hemophilia A. Clinical features,
surgery and treatment of 34 Cases, and experience of using
recombinant factor VIIa. Clin Appl Thromb Hemost. 16:294–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huth-Kühne A, Baudo F, Collins P,
Ingerslev J, Kessler CM, Lévesque H, Castellano ME, Shima M and
St-Louis J: International recommendations on the diagnosis and
treatment of patients with acquired hemophilia A. Haematologica.
94:566–575. 2009. View Article : Google Scholar : PubMed/NCBI
|