Introduction
Immunoglobulin gG4-related disease (IgG4-RD) is a
recently described disease entity characterized by tumefactive
lesions, abundant tissue lymphoplasmacytic and IgG4-positive plasma
cell infiltration, storiform fibrosis and elevated serum IgG4
concentrations (1). Since autoimmune
pancreatitis, associated with high serum IgG4 expression levels,
was first reported in 2001 (2),
IgG4-RD has been described in various organ systems, including the
pancreas, kidney, biliary tree, salivary glands and the nervous
system (1,3–5). The
disease is more common in middle-aged and elderly males (6). The incidence rate of IgG4-RD has not
been established because of the relative rarity of this diagnosis
(6). IgG4-RD typically responds
significantly to treatment with immunosuppressants (7); treatment with systemic steroids is
recommended, and a longer duration of treatment with steroids may
decrease the rate of recurrence (7–9).
While optic nerve involvement is rare, a series of
recent studies have reported the presence of a mass around the
orbit (10–13). In the present study, a serologically
and histopathologically documented IgG4-RD case is reported, which
presented with rapid progressive optic neuropathy with no mass
around the orbit. The current case also responded to glucocorticoid
treatment, providing new insights into this form of IgG4-RD.
Case report
A 79-year-old woman was admitted at the China-Japan
Friendship Hospital (Beijing, China) in October 2013, complaining
of rapid loss of vision in the left eye that persisted for two
months. The study was approved by the Institutional Review Board of
the China-Japan Friendship Hospital, and written informed consent
was obtained from the patient. Approximately 30 years earlier, the
patient presented with symmetrical spot-like vitiligo of the skin
around the two ankles, which has spread upward on both legs and
lower abdomen. The patient was diagnosed with pancreatic neoplasm
and diabetes 4 years before admission for vision loss, and
treatment with oral hypoglycemic agents (30 mg acarbose, thrice
daily for 4 years; Bayer AG, Leverkusen, Germany) was initiated to
maintain normal blood glucose levels. In addition, the patient
developed enlarged right cervical and double subaxillary painless
lymph nodes 3.5 years before admission, and a right upper palpebral
mass 3 years ago. Additional features of the patient's medical
history were coronary artery disease for the past 8 years and
hypertension for the past 7 years, which were treated with oral
agents (30 mg adalat daily for 7 years; Bayer). Furthermore, the
patient had undergone bilateral cataract surgery 7 years prior to
admission; thereafter, visual acuity, as measured with a standard
logarithmic letter chart ~6 months after cataract surgery, was
maintained at 0.6–0.8/1.5 in both eyes.
During the initial evaluation in October 2013, the
patient was awake, alert and oriented to time, place and persons.
Symmetrical patchy depigmentation was identified on the legs and
the lower abdomen. Submandibular and subaxillary enlarged lymph
nodes were palpated. The results of a neurological examination
(cranial nerve, motor and sensor system examinations) were normal,
with the exception of vision loss on the left eye. At this point in
time, visual acuity was 0.03/1.5 (left eye) and 0.6/1.5 (right
eye). Dilated fundus examination by direct ophthalmoscopy revealed
optic atrophy in the left eye and arteriosclerosis in both
eyes.
Immunoglobulin G (IgG) and IgG4 serum levels were
elevated to 3,150 and 2,440 mg/dl on admission, respectively (IgG
reference range, 694–1,620 mg/dl; IgG4 reference range, 3.0–201
mg/dl). The erythrocyte sedimentation rate of the patient was 53
mm/h (reference range, 0–20 mm/h). Normal immunofixation
electrophoresis was noted in blood and urine samples. Visual evoked
potentials (VEPs; Medtronic, Minneapolis, MN, USA) were obtained to
a 26-degree square pattern of 1-degree high contrast black and
white checks that reversed at 1 Hz. VEP implicit times were
prolonged when the left eye was stimulated, but not when the right
eye was tested (Fig. 1). At
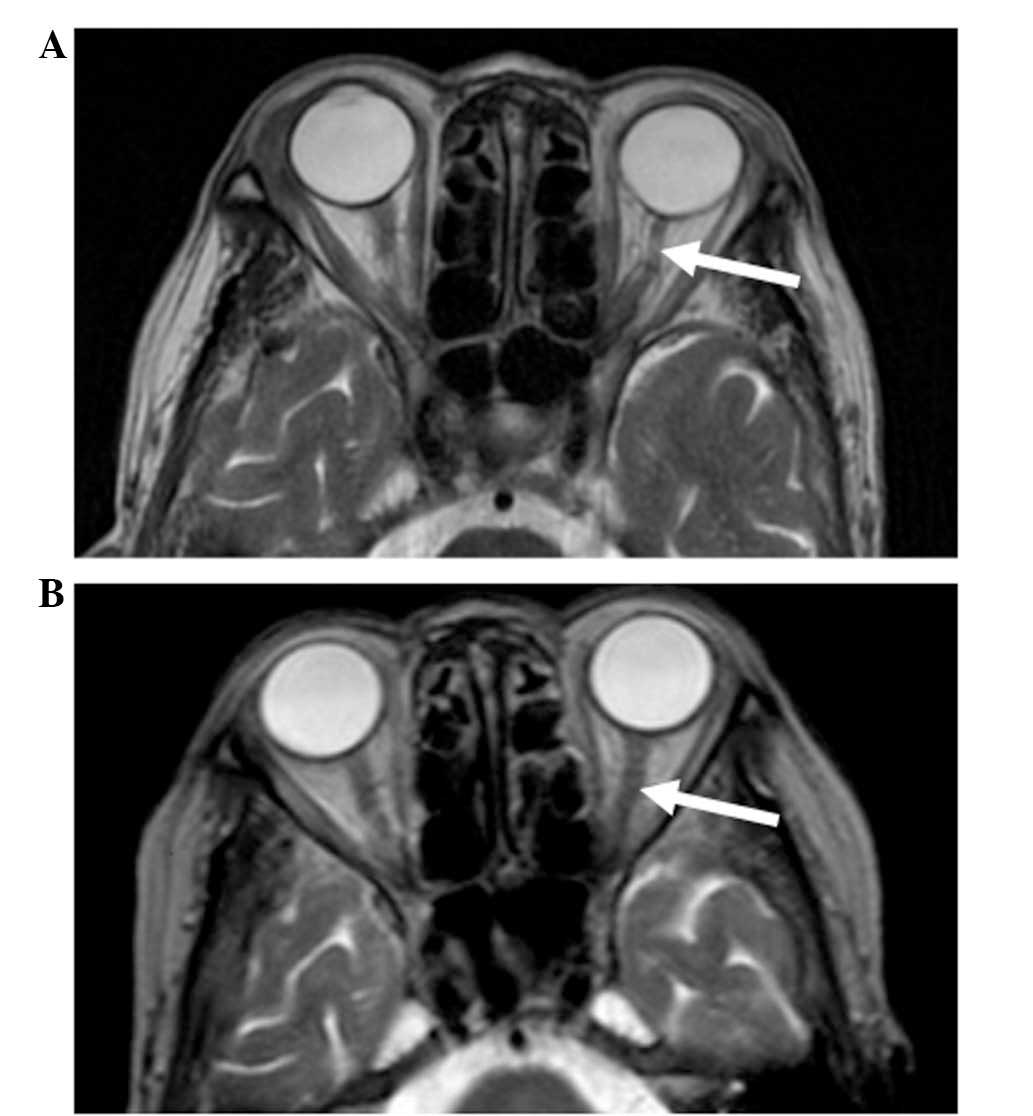
presentation, cranial magnetic resonance imaging (MRI; Gyroscan 1.5
T; Philips Healthcare, DA Best, The Netherlands) showed atrophy of
the left optic nerve with a focal hyperintense lesion, as shown in
a T2-weighted image (Fig. 2A). The
MRI analysis revealed the absence of any mass surrounding the optic
nerve or any other orbital areas. Furthermore, a thoracic computed
tomography (CT; Aquilion; Toshiba, Tokyo, Japan) scan showed
diffused interstitial pneumonia. No abnormalities were detected on
fundus fluorescein angiography, electromyography, abdominal-pelvic
CT, specific autoimmune testing, hematological examination, serum
angiotensin-converting enzyme (ACE) and other immunoglobulin (IgA,
IgM and IgE) levels.
Clinicopathological examination of cervical lymph
node by needle biopsy revealed diffuse lymphoplasmacytic
infiltration and storiform fibrosis. An immunohistochemical stain
against IgG4 demonstrated that the number of IgG4-positive plasma
cells were elevated to >50 cells per high-power field (Fig. 3A), as compared to the 0–20 cells per
high-power field observed in normal biopsy specimens (14,15). In
addition, an IgG4/IgG plasma cell ratio of >40% was observed
(Fig. 3B), which was much higher
compared with the 6% ratio typically observed in normal subjects. A
labial gland biopsy revealed lymphoplasmacytic focal infiltration
in the salivary gland stroma with mild changes in the acinus and
ducts, but was negative for IgG4. Upon analysis of the skin biopsy,
IgG4-positive plasma cells were absent from the dermis and
epidermis.
The patient was diagnosed with IgG4-RD and began
oral administration of methylprednisolone (Pfizer, Inc., New York,
NY, USA) at 40 mg/day (1 mg/kg/day), followed by gradual reduction
of the dose (~1 pill per month reduction) within a period of 24
weeks, maintaining a dose of 4 mg/day for ~1 year. This treatment
resulted in a decrease in serum IgG and IgG4 levels to 732 mg/dl
and 259 mg/dl, respectively. The visual acuity in the left eye was
stabilized and improved to 0.3/1.5, which persisted over a 1-year
post-treatment period. After 4 months of therapy, the left optic
nerve had a normal appearance on an MRI scan (Fig. 2B). The last follow-up date was
January 2014.
Discussion
The present study reported the case of a woman with
unilateral vision loss in addition to involvement of salivary
gland, lymph node, lung, pancreas, eyelid and skin. Other systemic
diseases that may cause similar vision loss include the
Vogt-Koyanagi-Harada syndrome, sarcoidosis, Sjögren's syndrome and
systemic vasculitis. In the present case, these conditions were
excluded by normal fundus fluorescein angiography, negative serum
ACE, specific autoantibody testing and labial biopsy. Lymphoma was
also excluded by normal hematological testing. The diagnosis of the
patient conformed to the 2011 diagnostic criteria for IgG4-RD based
on organs affected, enriched IgG4-positive plasma cells in involved
tissues and high serum IgG4 concentration (7). Using the key words ‘optic nerve’ and
‘IgG4-related disease’, a literature search was performed using
PubMed (http://www.ncbi.nlm.nih.gov/pubmed).
Orbital involvement in IgG4-RD has been previously
reported to affect nearly every orbital structure, including
lacrimal glands, extra-ocular muscles and the trigeminal nerve
(16,17). The term IgG4-related ophthalmic
disease (IgG4-ROD) has been suggested for IgG4-RD with ophthalmic
involvement (18). According to
Kashii (18), IgG4-ROD is
characterized by bilateral lacrimal gland enlargement accompanied
by three distinctive features, including infraorbital nerve
enlargement, extraocular myositis and compressive optic neuropathy.
Comparing the characteristics of the patient of the current study
to this classification, we note that an upper eyelid mass was
present, which was consistent with lacrimal gland enlargement;
however, this disappeared spontaneously and prior to
methylprednisolone treatment. The patient subsequently developed
rapid loss of vision only on the left eye. Cranial MRI scans
documented the presence of optic nerve atrophy with focal lesion,
but did not identify any masses infiltrating the optic nerve or
orbit, distinguishing this case from previous cases (summarized in
Table I). We hypothesize that the
underlying pathophysiologic mechanism may involve inflammatory
demyelination of the optic nerve based on the findings of MRI, VEP
implicit time delays and positive response to glucocorticoid
treatment. Although this mechanism was not confirmed through optic
nerve biopsy, this hypothesis raises a novel pathogenic feature
that should be addressed in future cases of IgG4-related optic
neuropathy.
 | Table I.Summary of recent literature
documenting optic nerve involvement in immunoglobulin G4-related
disease. |
Table I.
Summary of recent literature
documenting optic nerve involvement in immunoglobulin G4-related
disease.
| Authors | Clinical
presentation | Mechanism | Treatment | Refs. |
|---|
| Takahashi et
al | 1 case of bilateral
blurred vision with orbital apices masses and sinusitis | Compressive optic
neuropathy | Prednisolone | (10) |
| Takahira et
al | 2 cases with optic
neuropathy | Optic neuropathy
infiltrated by surrounding masses | Steroids | (11) |
| Ramirez et
al | 1 case of bilateral
vision loss with headache | Optic neuropathy
secondary to hypertrophic pachymeningitis | Steroids | (12) |
| Sogabe et
al | 6 cases of visual
disturbance due to optic nerve disturbance | Optic neuropathy
compressed by supraorbital nerve lesion in 2 patients; localized
orbital mass in 2 patients; diffuse orbital fat lesion in 1
patient; enlarged extraocular muscle; and localized orbital mass in
1 patient | N/A | (13) |
IgG4-ROD usually responds favorably to systemic
glucocorticoids (19). A recent case
has shown evident improvement to normal vision after a 3-month
course of steroid treatment (10).
By contrast, the vision of the current patient did not reach normal
levels within the 1-year follow-up period. This may reflect the
presence of an irreversible optic atrophy or reflect more severe
fundus arteriosclerosis in the left eye of the patient.
In conclusion, the current study presented a case of
IgG4-RD with ocular symptoms that were rapidly improved by
glucocorticoid treatment. The current case did not involve focal
mass infiltration of any orbital structure. By broadening the
spectrum of IgG4-ROD, the present case provides new information
regarding the underlying pathogenic mechanisms of IgG4-related
optic neuropathy.
Acknowledgements
The authors would like to thank their colleagues
from the Department of Rheumatology and Respirology (China-Japan
Friendship Hospital, Beijing, China) for their support. The present
study was sponsored by the Wu Jieping Medical Foundation (grant no.
1117) and the Yough Foundation of the China-Japan Friendship
Hospital (grant no. 2015-1-QN-12).
References
|
1
|
Stone JH, Zen Y and Deshpande V:
IgG4-related disease. N Engl J Med. 366:539–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hamano H, Kawa S, Horiuchi A, Unno H,
Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N
and Kiyosawa K: High serum IgG4 concentrations in patients with
sclerosing pancreatitis. N Engl J Med. 344:732–738. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kamisawa T and Okamoto A: IgG4-related
sclerosing disease. World J Gastroenterol. 14:3948–3955. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wallace ZS, Carruthers MN, Khosroshahi A,
Carruthers R, Shinagare S, Stemmer-Rachamimov A, Deshpande V and
Stone JH: IgG4-related disease and hypertrophic pachymeningitis.
Medicine (Baltimore). 92:206–216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohyama K, Koike H, Iijima M, Hashimoto R,
Tomita M, Kawagashira Y, Satou A, Nakamura S and Sobue G:
IgG4-related neuropathy: A case report. JAMA Neurol. 70:502–505.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wallace ZS, Deshpande V, Mattoo H, Mahajan
VS, Kulikova M, Pillai S and Stone JH: IgG4-related disease:
Clinical and laboratory features in one hundred twenty-five
patients. Arthritis Rheumatol. 67:2466–2475. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Islam AD, Selmi C, Datta-Mitra A, Sonu R,
Chen M, Gershwin ME and Raychaudhuri SP: The changing faces of
IgG4-related disease: Clinical manifestations and pathogenesis.
Autoimmun Rev. 14:914–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zen Y and Nakanuma Y: IgG4-related
disease: A cross-sectional study of 114 cases. Am J Surg Pathol.
34:1812–1819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Umehara H, Okazaki K, Masaki Y, Kawano M,
Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, et
al: Comprehensive diagnostic criteria for IgG4-related disease
(IgG4-RD), 2011. Mod Rheumatol. 22:21–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takahashi Y, Kitamura A and Kakizaki H:
Bilateral optic nerve involvement in immunoglobulin G4-related
ophthalmic disease. J Neuroophthalmol. 34:16–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takahira M, Ozawa Y, Kawano M, Zen Y,
Hamaoka S, Yamada K and Sugiyama K: Clinical aspects of
IgG4-related orbital inflammation in a case series of ocular
adnexal lymphoproliferative disorders. Int J Rheumatol.
2012:6354732012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramirez L, D'Auria A, Popalzai A and
Sanossian N: Bilateral vision loss secondary to pachymeningitis in
a patient with IgG4-related disease. Front Neurol. 5:1922014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sogabe Y, Ohshima K, Azumi A, Takahira M,
Kase S, Tsuji H, Yoshikawa H and Nakamura T: Location and frequency
of lesions in patients with IgG4-related ophthalmic diseases.
Graefes Arch Clin Exp Ophthalmol. 252:531–538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kitagawa S, Zen Y, Harada K, Sasaki M,
Sato Y, Minato H, Watanabe K, Kurumaya H, Katayanagi K, Masuda S,
et al: Abundant IgG4-positive plasma cell infiltration
characterizes chronic sclerosing sialadenitis (Küttner's tumor). Am
J Surg Pathol. 29:783–791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deshpande V, Zen Y, Chan JK, Yi EE, Sato
Y, Yoshino T, Klöppel G, Heathcote JG, Khosroshahi A, Ferry JA, et
al: Consensus statement on the pathology of IgG4-related disease.
Mod Pathol. 25:1181–1192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hagiya C, Tsuboi H, Yokosawa M, Hagiwara
S, Hirota T, Takai C, Asashima H, Miki H, Umeda N, Horikoshi M, et
al: Clinicopathological features of IgG4-related disease
complicated with orbital involvement. Mod Rheumatol. 24:471–476.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wallace ZS, Deshpande V and Stone JH:
Ophthalmic manifestations of IgG4-related disease: Single-center
experience and literature review. Semin Arthritis Rheum.
43:806–817. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kashii S: IgG4-related disease: A
neuro-ophthalmological perspective. J Neuroophthalmol. 34:400–407.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Masaki Y, Dong L, Kurose N, Kitagawa K,
Morikawa Y, Yamamoto M, Takahashi H, Shinomura Y, Imai K, Saeki T,
et al: Proposal for a new clinical entity, IgG4-positive multiorgan
lymphoproliferative syndrome: Analysis of 64 cases of IgG4-related
disorders. Ann Rheum Dis. 68:1310–1315. 2009. View Article : Google Scholar : PubMed/NCBI
|