Introduction
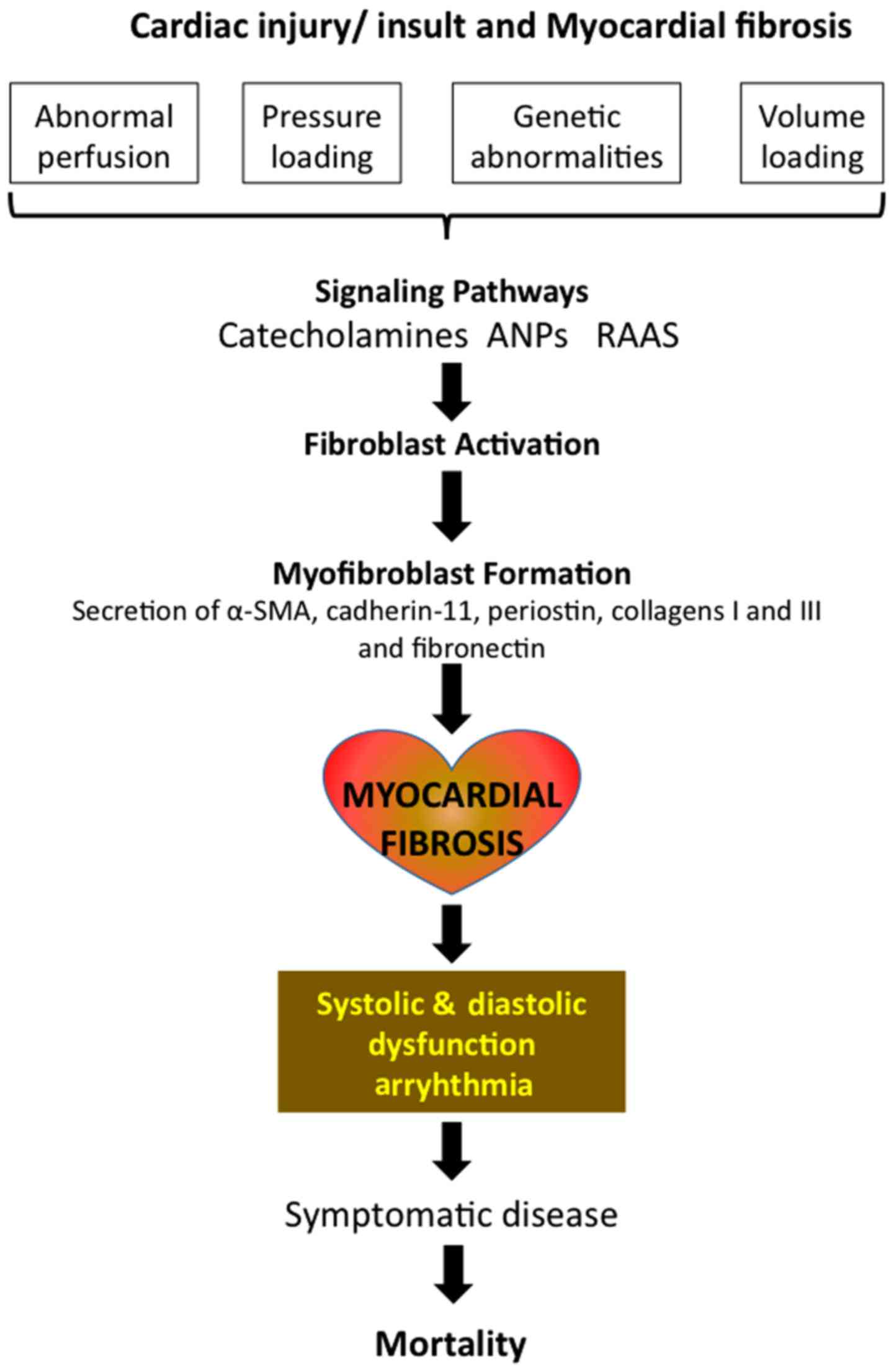
Cardiac fibrosis is a common histopathological
occurrence with many types of heart diseases, including ischemic
heart disease, inherited cardiomyopathy mutations, diabetes, and
ageing and is associated with morbidity and mortality. While there
are several mechanisms of myocardial fibrosis with acute and
chronic etiologies, increased accumulation of extracellular matrix
(ECM) that impacts cardiac function, is the underlying cause of
fibrotic heart disease. Besides the disturbances in the mechanical
functions of heart, fibrosis mediated scarring and electrical
dysfunction often initiates ventricular arrhythmias, which in turn
accelerates events towards heart failure and then sudden cardiac
death (1–3). Myocardial fibrosis presents one of the
major challenges clinically not just to improve the survival rate
and also the quality of life. Cardiac magnetic resonance (CMR) has
been useful in measuring the extent of diffuse myocardial fibrosis,
which is considered as a common pathological pathway that leads to
the loss of myocardial function (4,5).
Inasmuch as fibrosis can be reversible (6), its quantification using CMR can be
useful in changing the way the cardiac fibrosis patients are
monitored and treated (7).
Myocardial fibrosis has been demonstrated in the pressure-loaded
left ventricle of infants and children with aortic stenosis and
coarctation (8). Histological
studies revealed right ventricular myofiber disorganization and
interstitial fibrosis in patients with tetralogy of Fallot
(9). Right ventricular fibrosis is
not just found in late adult survivors, but actually is already
present in infants suffering with this condition (10). Alterations in myocardial architecture
associated with fibrotic remodeling lead to deranged heart function
in different ways. Thus, in patients after tetralogy of Fallot
repair, fibrous endocardial thickening of the right ventricular
infundibulum is a predictor of poor right ventricular function
(5). On the other hand, myocardial
fibrosis is associated with systolic ventricular dysfunction early
in life in patients with tricuspid atresia (11).
Healthy myocardium consists of mostly cardiomyocytes
(~75%), with the remaining partition, the interstitium regions
comprising of fibroblasts, endothelial cells, and coronary arteries
(12). The fibroblasts participate
in the continuous formation and degradation of ECM, which is
composed of types I and III collagen fibers, that act as the
scaffold for the myocardium. Disturbances in the balance between
collagen production and degradation in the ECM, that led to
expansion of collagens, cause myocardial fibrosis. Such derangement
occurs either due to the death of cardiomyocytes, or because of
stimuli that trigger elevated collagen synthesis (13).
Types of cardiac fibrosis
There are multiple types of cardiac fibrosis
depending upon location and causes. i) Reactive interstitial
fibrosis (RIF): This is characterized by an increase in collagen
synthesis without much effect on the viability of myocardium
(14). RIF is considered as a type
of myocardial remodeling and leads to an increased interstitial
compartment volume without any associated changes in myocyte
volume. This type of fibrosis is associated with diffused
deposition of collagen and occurs in response to increased pressure
and/or volume loads as in the cases of hypertension, aortic
stenosis, chronic regurgitation, and shunt, ischemia,
hyperglycemia, or ageing. RIF, which generally presents with a
progressive chronic course, is possibly reversible by curtailing
the damage causing stimuli or even by targeted therapy. ii)
Replacement fibrosis: An increased type I collagen deposition and
expansion of ECM, following cardiomyocyte death contributes to this
type of fibrosis (14). The elevated
levels of collagen fibers replace the dead cardiomyocytes, but lack
the contractile and electric functionality. Acute or chronic
conditions that trigger cardiomyocyte death such as myocardial
ischemia and events that damage the cardiomyocyte membrane
integrity causing cell death, lead to diffused or focal replacement
fibrosis. Unlike the RIF, in replacement fibrosis, the affected
myocardium is not viable and thus is unable to recover contractile
properties following revascularization or by blocking the damaging
process. iii) Infiltrative interstitial fibrosis: This fibrosis is
seen in conditions such as amyloidosis or Anderson-Fabry disease
(14). Inflammatory cell
infiltration was proposed to be an important factor underlying the
interstitial fibrosis seen in right ventricles of systemic
sclerosis-associated pulmonary arterial hypertension (15). iv) Endomyocardial fibrosis (EMF): EMF
affects children under age of 2 years in tropical and subtropical
regions, and involves the apical endocardium of either or both
right and left ventricles. It has been suggested that EMF as one of
the primary causes of pediatric congestive heart failure, which is
often overlooked clinically (16).
There is no clear etiology for EMF, but hypereosinophilia,
infections, autoimmunity, genetic factors and nutritional
deficiencies may play a role. EMF has been described to present in
three phases: Initial phase of acute carditis with febrile illness
and with heart failure in severe cases; an intermediary subacute
phase followed by a chronic burnt-out phase (17). Notably, EMF is identified mostly at
late stages of the disease and is not observed in the early phase
of the disease. However, following diagnosis, there is rapid onset
of associated complications such as atrial fibrillation,
thromboembolism and progressive atrioventricular valve
regurgitation. EMF patients display clinical features of heart
failure, with enlargement of both atria with normally functioning
ventricles during echocardiography (18). Currently, the approach to treat EMF
patients is by surgery, involving targeted endocardial resection
combined with valve repair or replacement, as there is no specific
medical treatment (16).
Fibroblasts, myofibroblasts and
fibrosis
Although the biology of cardiomyocyte and its
apoptotic or necrotic death have been the focus of several
investigations addressing cardiac diseases, increasing evidence
suggests cardiac fibroblasts in the pathogenic mechanisms (19,20).
Fibroblasts, which are abundant in the normal myocardium, have a
structural function by providing support (21). However, when these normally quiescent
fibroblasts are activated, they transdifferentiate into
myofibroblasts, which appear similar to a hybrid of fibroblast and
smooth muscle cells. Myofibroblasts are able to effectively secrete
and myofibroblasts are absent in healthy heart, they appear within
days following a cardiac injury (22). Myofibroblasts appear as
spindle-shaped and possess dendritic-like processes (23) that protrude from the cell body.
Characteristic elongated and serrated nuclei, extensive areas of
rough endoplasmic reticulum and irregular non-sarcomeric
myofibrillar structures are morphological features of
myofibroblasts as observed in electron micrographs of
pressure-overloaded rat heart cross sections (24). Specific protein markers of
myofibroblasts are mainly the ECM proteins, such as periostin,
collagens I and III and fibronectin (25). Besides these ECM proteins,
myofibroblasts also express smooth muscle α-actin, SM22 and
caldesmon (26,27). Of note, most of the myofibroblasts
present in the border zone and approximately half of those in the
fibrotic lesions of damaged myocardium, express embryonic smooth
muscle myosin and α-SMA but not other smooth muscle markers, which
is a major difference from smooth muscle cells (28).
Transformation of fibroblasts to
myofibroblasts
It has been suggested that there are two stages in
the transformation of myofibroblast: in the first stage, there is
development of ‘proto-myofibroblasts’ from fibroblasts, with the
characteristic formation of cytoplasmic actin stress fibers and
small adhesion complexes, which facilitate the migration of these
cells to the injured myocardium, where these cells secrete
collagens and fibronectin and assume physical orientation in order
to align themselves with the primary stress axes of the injured
tissue (25). In the second stage,
which starts approximately 20–30 h following cardiac injury, the
cytokines and mechanical stress convert these proto-myofibroblasts
to mature myofibroblasts, which express α-SMA, which forms the
stress fiber network (25). With
further maturation of the myofibroblast, besides the α-SMA,
cadherin-11 is also secreted and adds to the strength of the stress
fiber network (29) (Fig. 1). Normally myofibroblasts have a
protective function in the heart by participating acutely in injury
repair response. Although uninterrupted fibrosis can be dangerous
to heart, the function of myofibroblasts is important to maintain
functional myocardium following any stress insult. Thus, following
myocardial infarction, when cardiomyocytes die over several days
due to ischemia, myofibroblasts play a major role in the remodeling
of a fibrotic scar in the affected myocardium, in order to prevent
wall rupture (30,31). Myofibroblasts eventually disappear,
through less understood mechanisms, in many situations of wound
healing. One possible mechanism is that following the formation of
permanent scar and stabilization of cardiac injury, which may take
up to 2 weeks, a significant proportion of the myofibroblasts
undergo senescence (32) and/or
apoptosis (33). It has also been
proposed that some of the myofibroblasts may dedifferentiate back
to fibroblast phenotype, even though strong evidence for this is
lacking in myocardium. Critical for the pathogenesis of cardiac
fibrosis is the persistent presence of myofibroblasts in the
myocardium, long after the resolution of stress insult (Fig. 1). This is generally either due to
concurrent dysregulation of neuroendocrine factors or even
mechanical stress, both of which lead to and aggravate chronic
cardiac fibrosis and/or hypertrophic scarring (34,35). A
common example for this is the intermittent ischemia, chronically
affecting the heart, creating constant wound healing milieu, which
leads to persistent activation and formation of the myofibroblasts
and the eventual fibrosis (30). It
has been proposed that the function of resident fibroblasts is to
maintain normal tissue integrity and structure, whereas, acute
injury or altered neuroendocrine signaling stimulates fibroblasts
from other sources, which ultimately contribute to pathophysiologic
fibrosis (36,37). A better understanding of the
molecular mechanisms involved in cardiac fibrosis and the
regulation of the players involved such as myofibroblast formation
and activation, is essential for developing effective
therapeutics.
Assessment of myocardial fibrosis
The most commonly employed techniques in patients
with congenital heart disease to assess the fibrotic features of
myocardium include stress echocardiography and CMR and also
positron emission tomography (PET), which is the reference standard
for monitoring myocardial viability. Stress echocardiography allows
for the assessment of myocardial contractile reserve in patients
with adequate acoustic windows. Because of this, the main
limitation of stress echocardiography is decreased diagnostic
accuracy in patients with poor acoustic windows (38). PET scan is a nuclear scintigraphy and
employs 13N-labeled ammonia, which a marker for coronary
perfusion and 18F-fluorodeoxyglucose, a glucose
surrogate, for monitoring myocardial metabolism. A mismatch between
the imaging of coronary perfusion and metabolism is indicative of
nonviable tissue with the possibility of replacement fibrosis.
However, PET scanner availability is limited and its usage requires
an on-site cyclotron, and PET is not sensitive enough to detect
interstitial fibrosis (39). CMR is
the major technique used to collect data on cardiac fibrosis. CMR
is able to provide much better spatial resolution and visualization
of all myocardial segments, in the absence of any
contaminating/interfering metallic particles, and this technique is
used extensively for the quantitative assessment of ventricular
size and systolic function. There are two major CMR approaches for
assessing myocardial fibrosis: Late gadolinium enhancement (LGE)
imaging and T1 mapping/ECV fraction calculation. LGE identifies
areas of discrete replacement fibrosis, with high sensitivity and
specificity (13).
Therapeutic approaches to treat cardiac
fibrosis
In the normal healthy heart, collagen network, which
contributes to ECM and maintains myocardial architecture as well as
chamber geometry, also provides tensile strength and elasticity,
the necessary factors for normal systolic and diastolic function of
ventricles (40). Several local and
hormonal systems, such as renin-angiotensin-aldosterone system
(RAAS), regulate the dynamic collagen turnover, and disturbances in
these systems lead to an imbalance between collagen synthesis and
degradation with the resultant accumulation of collagen, the
underlying factor of cardiac fibrosis, followed by deranged
myocardial architecture (41). Thus
it has been shown that pharmacological intervention of RAAS could
prevent this process of fibrosis and at times even reverse the
damage caused (42). Thus,
aldosterone antagonism with spironolactone has been found to lower
collagen levels in hearts, ex vivo (43), improve diastolic function (44), decrease left ventricular size and
ejection fraction (45), and also
mortality (Fig. 2) (46).
There is ample evidence indicating increased
expression of transforming growth factor-β (TGF-β) in response to
injury and TGF-β is known to play an important role during
fibroblast activation and myofibroblast differentiation, and also
fibrosis (Fig. 2) (47,48).
TGF-β acts via binding to its cell surface receptor, activin linked
kinase 5 (ALK5), which phosphorylates Smad2 and −3. Phospho-Smad2
and −3 bind to Smad4, which translocates into the nucleus, leading
to the activation of target gene transcription. It has been shown
that inhibitors of ALK5 signaling block certain steps of fibrosis
process and thus there is a potential that ALK5 can be a
therapeutic target for cardiac fibrosis (49). Even though neutralizing antibodies
against TGF-β have been tried in some studies (50), because of involvement of TGF-β in
several different cellular functions, such approach proved to be
not appropriate (51) and ALK5
antagonists are much better choice (52). Angiotensin II has been shown to
function in concert with TGF-β in the signaling pathways that lead
to cardiac fibrosis (53,54). Blockade of angiotensin receptor with
inhibitors like losartan is effective in decreasing cardiac
fibrosis in animal models and also in humans and this approach of
angiotensin-II inhibitors seems to be better than blocking TGF-β
(55).
Similarly, antagonism of endothelin-1, which induces
ECM production and myofibroblast transformation (56) was found to reduce cardiac fibrosis.
Other possible therapeutic targets include platelet derived growth
factor and connective tissue growth factor, CCN2, but further work
is needed to ascertain this possibility (57).
Conclusion
Cardiac fibrosis, which is seen in many types of
heart diseases, involves increased accumulation of ECM, mediated by
the activated myofibroblasts, arising from cardiac fibroblasts.
Myocardial fibrosis include stress echocardiography and CMR and
also PET. Renin-angiotensin-II-aldosterone system, TGF-β signaling
and ALK5 have been found useful as therapeutic targets. However,
because of the importance of these pathways in many other
physiological functions, their therapeutic targeting needs to be
approached with caution.
References
|
1
|
Harris KM, Spirito P, Maron MS, Zenovich
AG, Formisano F, Lesser JR, Mackey-Bojack S, Manning WJ, Udelson JE
and Maron BJ: Prevalence, clinical profile, and significance of
left ventricular remodeling in the end-stage phase of hypertrophic
cardiomyopathy. Circulation. 114:216–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
O'Hanlon R, Grasso A, Roughton M, Moon JC,
Clark S, Wage R, Webb J, Kulkarni M, Dawson D, Sulaibeekh L, et al:
Prognostic significance of myocardial fibrosis in hypertrophic
cardiomyopathy. J Am Coll Cardiol. 56:867–874. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vasquez C and Morley GE: The origin and
arrhythmogenic potential of fibroblasts in cardiac disease. J
Cardiovasc Transl Res. 5:760–767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ho SY, Jackson M, Kilpatrick L, Smith A
and Gerlis LM: Fibrous matrix of ventricular myocardium in
tricuspid atresia compared with normal heart. A quantitative
analysis. Circulation. 94:1642–1646. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farah MC, Castro CR, Moreira VM, Binotto
MA, Guerra VC, Riso AA, Marcial MB, Lopes AA, Mathias W Jr and
Aiello VD: The impact of preexisting myocardial remodeling on
ventricular function early after tetralogy of Fallot repair. J Am
Soc Echocardiogr. 23:912–918. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burns KM, Byrne BJ, Gelb BD, Kühn B,
Leinwand LA, Mital S, Pearson GD, Rodefeld M, Rossano JW, Stauffer
BL, et al: New mechanistic and therapeutic targets for pediatric
heart failure: Report from a National Heart, Lung, and Blood
Institute working group. Circulation. 130:79–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moon JC, Messroghli DR, Kellman P,
Piechnik SK, Robson MD, Ugander M, Gatehouse PD, Arai AE, Friedrich
MG, Neubauer S, et al: Society for Cardiovascular Magnetic
Resonance Imaging; Cardiovascular Magnetic Resonance Working Group
of the European Society of Cardiology: Myocardial T1 mapping and
extracellular volume quantification: A Society for Cardiovascular
Magnetic Resonance (SCMR) and CMR Working Group of the European
Society of Cardiology consensus statement. J Cardiovasc Magn Reson.
15:922013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheitlin MD, Robinowitz M, McAllister H,
Hoffman JI, Bharati S and Lev M: The distribution of fibrosis in
the left ventricle in congenital aortic stenosis and coarctation of
the aorta. Circulation. 62:823–830. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chowdhury UK, Sathia S, Ray R, Singh R,
Pradeep KK and Venugopal P: Histopathology of the right ventricular
outflow tract and its relationship to clinical outcomes and
arrhythmias in patients with tetralogy of Fallot. J Thorac
Cardiovasc Surg. 132:270–277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peters TH, Sharma HS, Yilmaz E and Bogers
AJ: Quantitative analysis of collagens and fibronectin expression
in human right ventricular hypertrophy. Ann N Y Acad Sci.
874:278–285. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanchez-Quintana D, Climent V, Ho SY and
Anderson RH: Myoarchitecture and connective tissue in hearts with
tricuspid atresia. Heart. 81:182–191. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baum J and Duffy HS: Fibroblasts and
myofibroblasts: What are we talking about? J Cardiovasc Pharmacol.
57:376–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rathod RH, Powell AJ and Geva T:
Myocardial fibrosis in congenital heart disease. Circ J.
80:1300–1307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mewton N, Liu CY, Croisille P, Bluemke D
and Lima JA: Assessment of myocardial fibrosis with cardiovascular
magnetic resonance. J Am Coll Cardiol. 57:891–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Overbeek MJ, Mouchaers KT, Niessen HM,
Hadi AM, Kupreishvili K, Boonstra A, Voskuyl AE, Belien JA, Smit
EF, Dijkmans BC, et al: Characteristics of interstitial fibrosis
and inflammatory cell infiltration in right ventricles of systemic
sclerosis-associated pulmonary arterial hypertension. Int J
Rheumatol. 2010:6046152010.PubMed/NCBI
|
|
16
|
Rohit M, Gupta A and Talwar KK: Heart
failure in children in tropical regions. Curr Heart Fail Rep.
10:277–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ball JD, Williams AW and Davies JN:
Endomyocardial fibrosis. Lancet. 266:1049–1054. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ammash NM, Seward JB, Bailey KR, Edwards
WD and Tajik AJ: Clinical profile and outcome of idiopathic
restrictive cardiomyopathy. Circulation. 101:2490–2496. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krenning G, Zeisberg EM and Kalluri R: The
origin of fibroblasts and mechanism of cardiac fibrosis. J Cell
Physiol. 225:631–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Turner NA and Porter KE: Function and fate
of myofibroblasts after myocardial infarction. Fibrogenesis Tissue
Repair. 6:52013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Banerjee I, Yekkala K, Borg TK and Baudino
TA: Dynamic interactions between myocytes, fibroblasts, and
extracellular matrix. Ann N Y Acad Sci. 1080:76–84. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun Y and Weber KT: Infarct scar: A
dynamic tissue. Cardiovasc Res. 46:250–256. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eyden B: The myofibroblast: Phenotypic
characterization as a prerequisite to understanding its functions
in translational medicine. J Cell Mol Med. 12:22–37. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shiojima I, Aikawa M, Suzuki J, Yazaki Y
and Nagai R: Embryonic smooth muscle myosin heavy chain SMemb is
expressed in pressure-overloaded cardiac fibroblasts. Jpn Heart J.
40:803–818. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier
C and Brown RA: Myofibroblasts and mechano-regulation of connective
tissue remodelling. Nat Rev Mol Cell Biol. 3:349–363. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hinz B: Formation and function of the
myofibroblast during tissue repair. J Invest Dermatol. 127:526–537.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hinz B: The myofibroblast: Paradigm for a
mechanically active cell. J Biomech. 43:146–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frangogiannis NG, Michael LH and Entman
ML: Myofibroblasts in reperfused myocardial infarcts express the
embryonic form of smooth muscle myosin heavy chain (SMemb).
Cardiovasc Res. 48:89–100. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hinz B, Pittet P, Smith-Clerc J,
Chaponnier C and Meister JJ: Myofibroblast development is
characterized by specific cell-cell adherens junctions. Mol Biol
Cell. 15:4310–4320. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weber KT, Sun Y, Bhattacharya SK, Ahokas
RA and Gerling IC: Myofibroblast-mediated mechanisms of
pathological remodelling of the heart. Nat Rev Cardiol. 10:15–26.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van den Borne SW, Isobe S, Verjans JW,
Petrov A, Lovhaug D, Li P, Zandbergen HR, Ni Y, Frederik P, Zhou J,
et al: Molecular imaging of interstitial alterations in remodeling
myocardium after myocardial infarction. J Am Coll Cardiol.
52:2017–2028. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Krizhanovsky V, Yon M, Dickins RA, Hearn
S, Simon J, Miething C, Yee H, Zender L and Lowe SW: Senescence of
activated stellate cells limits liver fibrosis. Cell. 134:657–667.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kissin E and Korn JH: Apoptosis and
myofibroblasts in the pathogenesis of systemic sclerosis. Curr
Rheumatol Rep. 4:129–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scotton CJ and Chambers RC: Molecular
targets in pulmonary fibrosis: The myofibroblast in focus. Chest.
132:1311–1321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Crawford JR, Haudek SB, Cieslik KA, Trial
J and Entman ML: Origin of developmental precursors dictates the
pathophysiologic role of cardiac fibroblasts. J Cardiovasc Transl
Res. 5:749–759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Biernacka A and Frangogiannis NG: Aging
and cardiac fibrosis. Aging Dis. 2:158–173. 2011.PubMed/NCBI
|
|
38
|
Camici PG, Prasad SK and Rimoldi OE:
Stunning, hibernation, and assessment of myocardial viability.
Circulation. 117:103–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Auerbach MA, Schöder H, Hoh C, Gambhir SS,
Yaghoubi S, Sayre JW, Silverman D, Phelps ME, Schelbert HR and
Czernin J: Prevalence of myocardial viability as detected by
positron emission tomography in patients with ischemic
cardiomyopathy. Circulation. 99:2921–2926. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Delcayre C and Swynghedauw B: Molecular
mechanisms of myocardial remodeling. The role of aldosterone. J Mol
Cell Cardiol. 34:1577–1584. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Weber KT and Brilla CG: Pathological
hypertrophy and cardiac interstitium. Fibrosis and
renin-angiotensin-aldosterone system. Circulation. 83:1849–1865.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cohn JN: Myocardial structural effects of
aldosterone receptor antagonism in heart failure. J Am Coll
Cardiol. 50:597–599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suzuki G, Morita H, Mishima T, Sharov VG,
Todor A, Tanhehco EJ, Rudolph AE, McMahon EG, Goldstein S and
Sabbah HN: Effects of long-term monotherapy with eplerenone, a
novel aldosterone blocker, on progression of left ventricular
dysfunction and remodeling in dogs with heart failure. Circulation.
106:2967–2972. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Izawa H, Murohara T, Nagata K, Isobe S,
Asano H, Amano T, Ichihara S, Kato T, Ohshima S, Murase Y, et al:
Mineralocorticoid receptor antagonism ameliorates left ventricular
diastolic dysfunction and myocardial fibrosis in mildly symptomatic
patients with idiopathic dilated cardiomyopathy: A pilot study.
Circulation. 112:2940–2945. 2005.PubMed/NCBI
|
|
45
|
Cicoira M, Zanolla L, Rossi A, Golia G,
Franceschini L, Brighetti G, Marino P and Zardini P: Long-term,
dose-dependent effects of spironolactone on left ventricular
function and exercise tolerance in patients with chronic heart
failure. J Am Coll Cardiol. 40:304–310. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pitt B, Zannad F, Remme WJ, Cody R,
Castaigne A, Perez A, Palensky J and Wittes J: Randomized Aldactone
Evaluation Study Investigators: The effect of spironolactone on
morbidity and mortality in patients with severe heart failure. N
Engl J Med. 341:709–717. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Massagué J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Y, Xu SW, Eastwood M, Black CM,
Denton CP, Leask A and Abraham DJ: Contribution of activin
receptor-like kinase 5 (transforming growth factor beta receptor
type I) signaling to the fibrotic phenotype of scleroderma
fibroblasts. Arthritis Rheum. 54:1309–1316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kuwahara F, Kai H, Tokuda K, Kai M,
Takeshita A, Egashira K and Imaizumi T: Transforming growth
factor-beta function blocking prevents myocardial fibrosis and
diastolic dysfunction in pressure-overloaded rats. Circulation.
106:130–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Frantz S, Hu K, Adamek A, Wolf J, Sallam
A, Maier SK, Lonning S, Ling H, Ertl G and Bauersachs J:
Transforming growth factor beta inhibition increases mortality and
left ventricular dilatation after myocardial infarction. Basic Res
Cardiol. 103:485–492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tan SM, Zhang Y, Connelly KA, Gilbert RE
and Kelly DJ: Targeted inhibition of activin receptor-like kinase 5
signaling attenuates cardiac dysfunction following myocardial
infarction. Am J Physiol Heart Circ Physiol. 298:H1415–H1425. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Campbell SE and Katwa LC: Angiotensin II
stimulated expression of transforming growth factor-beta1 in
cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol.
29:1947–1958. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schultz JJ, Witt SA, Glascock BJ, Nieman
ML, Reiser PJ, Nix SL, Kimball TR and Doetschman T: TGF-beta1
mediates the hypertrophic cardiomyocyte growth induced by
angiotensin II. J Clin Invest. 109:787–796. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
De Mello WC and Specht P: Chronic blockade
of angiotensin II AT1-receptors increased cell-to-cell
communication, reduced fibrosis and improved impulse propagation in
the failing heart. J Renin Angiotensin Aldosterone Syst. 7:201–205.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Leask A: Targeting the TGFbeta,
endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell
Signal. 20:1409–1414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Leask A: Potential therapeutic targets for
cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|