Introduction
Multiple myeloma (MM) is a B-cell malignancy
characterized by the aberrant clonal expansion of plasma cells
(PCs) within the bone marrow, and, as a consequence, osteolytic
bone destruction with hypercalcemia, anemia, immunosuppression and
end organ damage frequently occurs (1). Recent advances in molecular and genetic
research into MM have led to the discovery that although MM is
defined histologically as a single entity, it encompasses a wide
range of genomic abnormalities, including numerical and structural
chromosomal abnormalities, gene mutations and epigenetic
alterations (2–4), which differ in their molecular
pathogenesis and prognostic significance (5).
MicroRNAs (miRNAs/miRs) are small non-coding
single-stranded RNAs of ~22 nucleotides in length, which control
gene expression at a post-transcriptional level by degrading or
repressing target mRNAs, resulting in translational repression or
mRNA degradation. miRNAs serve roles in essential biological
processes, including cellular growth, differentiation and
proliferation. In addition, miRNAs regulate the expression of
>30% of protein-coding genes, and >50% of miRNA target genes
are located in cancer-associated genomic regions, suggesting that
miRNAs serve an important role in the pathogenesis of human cancer
(6,7). It is well known that the dysregulation
of miRNAs is associated with the pathogenesis of cancer, and that
miRNA expression profiles have prognostic implications in numerous
types of cancer. Thus, inhibiting specific miRNAs is a therapeutic
strategy for the treatment of cancer (8).
Numerous previous studies have detected miRNA
expression in MM via microarray profiling and reverse
transcription-quantitative polymerase chain reaction analysis
(9–11), with results suggesting that miRNAs
serve an important role in the molecular pathogenesis, progression
and prognosis of MM. Lionetti et al (9) evaluated the influence of allelic
imbalances on miRNA expression in MM, and identified that
differential miRNA expression patterns were associated with the
cytogenetic abnormalities in MM, particularly with immunoglobulin
heavy locus translocations. Furthermore, Wu et al (10) was able to develop an ‘outcome
classifier’ in patients newly diagnosed with myeloma based on their
expression of specific miRNAs.
The miR-17-92 cluster, located in an intron of
miR-17-92a-1 cluster host gene on chromosome 13q31.3, was
originally reported to be implicated in B-cell neoplasms, including
MM (11). Later, the miR-17-92
cluster was identified as an oncomiR due to its oncogenic activity
in several types of cancer (12).
Mendell (11) identified that the
deletion of miR-17-92 inhibited B-cell proliferation and
development, whereas its overexpression induced B-cell
hyperproliferation and autoimmune diseases. Another study revealed
that members of the miR-17-92 cluster, particularly miR-19a and b,
were upregulated in MM, but not in healthy cases or monoclonal
gammopathy of undetermined significance (MGUS), suggesting a
potential role of the cluster in the progression from MGUS to MM,
likely representing MM-specific genetic changes (2).
miR-19a, a key component of miR-17-92 cluster, has
been directly implicated in myeloma pathogenesis (13,14). In
addition, miR-19a has been demonstrated to be upregulated in
patients with MM, and in MM cell lines compared with normal plasma
cells (13,15). Furthermore, miR-19a was more highly
expressed in patients with MM with 13q14 deletions compared with
those without these deletions (5,16,17).
Additionally, miR-19a antagonists have been revealed to suppress MM
tumor growth in nude mice (14).
miR-19a can modulate the expression of proteins that are essential
in myeloma pathogenesis, including suppressors of cytokine
signaling (SOCS), a gene that is frequently silenced in MM,
releasing inhibition of interleukin 6 and leading to pro-growth
signaling (14). These results
highlight the contribution of miR-19a to the pathogenesis of MM and
its potential application as a molecular biomarker for MM.
Although the function of miR-19a has been relatively
well studied, its exact role in the development and progression of
MM remains unclear. Systematic analyses of miR-19a-associated
malignant cell behavior is required. Since miR-19a's function is
mediated through its target genes, the exploration of its target
genes is also essential. The inverse correlation between miRNA-mRNA
interactions may aid in the identification of target genes
regulated by miR-19a in the pathogenesis of MM. Combined with the
MM gene expression profiling data generated by high-throughput
technology in a previous study (18), the present study performed a
systematic analysis of miR-19a predicted target genes associated
with the carcinogenesis, prognosis and chemoresistance of MM in
order to further investigate the potential involvement of miR-19a
in MM.
Materials and methods
Prediction of miRNA target genes
miR-19a target prediction was performed using the
online tool miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk)
(19) with a combination of three
currently available independent target prediction programs,
including PicTar (version 2005; http://pictar.mdc-berlin.de/cgi-bin/PicTar_vertebrate.cgi),
miRanda (version 5; http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5)
and TargetScan (version 5.1; http://www.targetscan.org). Only the targets genes
that were confirmed by all of the above prediction programs or
experimentally validated according to the miRWalk database were
considered putative targets of miR-19a.
Identifying gene expression
profiles
The Gene Expression Omnibus database (GEO,
http://www.ncbi.nlm.nih.gov/geo), a
public repository for high-throughput gene expression datasets, was
searched for MM gene expression profiling studies. Expression
profiling studies of peripheral blood mononuclear cells from
patients with MM were obtained from previous studies (20–22).
Differential analysis of genes in
MM
The raw gene expression data of each study was
downloaded from the GEO database, and preprocessed for background
correction and Z-score normalization. The Bioconductor limma
package (version 1.9.6) in R (23)
was used to perform differential analysis of genes between MM and
controls using a two-tailed Student's t-test. The p-value of
individual microarray studies were combined using Fisher's exact
test. Differently expressed genes with a false discovery rate (FDR)
<0.01 were selected.
Functional classification
GeneCodis (http://genecodis.cnb.csic.es/) was used to perform
Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analysis, in order to annotate the
function of the selected genes. The functional GO terms were
classified into three groups, biological processes, cellular
components and molecular functions. Genes were mapped to
corresponding signaling pathways according to KEGG signaling
pathway database, and the enrichment FDR was calculated for each
pathway, and the criteria of FDR <0.05 was used as the threshold
for significance.
Protein-protein interaction (PPI)
network analysis
To explore the function of genes at the protein
level, PPI analysis was performed as previously described (24). The Biological General Repository for
Interaction Datasets (BioGRID; http://thebiogrid.org) was used to construct the PPI
network. BIOGRID is an online interaction repository, confirmed by
existing high-throughput experiments. Cytoscape version 3.3.0
software was used to display the PPI network, as previously
described (25). In the PPI network
produced, nodes indicate proteins and edges indicate interactions
between these nodes. The nodes that contain the most connectivity
degrees are defined as significant hub proteins.
Natural language processing (NLP)
analysis of MM
Document searching and formatting were performed in
PubMed (http://www.ncbi.nlm.nih.gov/pubmed) using the keywords
‘multiple myeloma’ and ‘resistance or prognosis or carcinogenesis
or tumorigenesis’. All of the genes and proteins associated with
these keywords were extracted, followed by gene mention tagging
using A Biomedical Named Entity Recognizer software, version 1.5
(http://pages.cs.wisc.edu/~bsettles/abner). For the
conditions, multiple genes were described in a word, such as
‘STAT3/5 gene’, and these were translate manually to ‘STAT3 gene’,
and ‘STAT5 gene’. Gene names were normalized based on the Entrez
database (https://www.ncbi.nlm.nih.gov/gene). Gene names were
normalized based on the Entrez database (https://www.ncbi.nlm.nih.gov/gene).
The frequency of the occurrence of each gene was
calculated. The higher the frequency of the gene, the greater the
likelihood of the association between MM and the gene. The total
number of studies in PubMed database was recorded as ‘N’. The
frequency of the genes and diseases associated with these in the
PubMed database were denoted as ‘m’ and ‘n’, respectively. It was
hypothesized that subtracting the disease co-occurrent from the
actual frequency of the gene would equal ‘k’. Then, by using
hypergeometric distribution, the probability of a frequency greater
than k co-citation at completely random conditions was calculated
as follows:
p=1–∑i=0k–1p(i|n,m,N)p(i|n,m,N)=n!(N–n)!m!(N–m)!(n–i)!i!(n–m)!(N–n–m+i)!N!
Results
Predicted target genes of miR-19a
Target genes of miR-19a were predicted using three
target prediction programs. A total of 715 putative targets of
miR-19a were identified using these three programs, among which 40
were experimentally validated in miRWalk (data not shown).
Differentially expressed genes in
MM
Following searching the GEO database, three gene
expression profiling studies of MM were collected (GSE23832,
GSE21942 and GSE17048; Table I). The
raw data was downloaded and processed, and 121 genes were
identified to be differentially expressed in MM with an FDR
<0.01, including 80 upregulated genes and 41 downregulated genes
(Fig. 1). The top 10 most
significantly upregulated or downregulated genes are listed in
Table II. Interestingly, four
putative targets of miR-19a, ras homolog family member B (RHOB),
clathrin heavy chain (CLTC), prosaposin (PSAP) and protein
phosphatase 6 regulatory subunit 2 (PPP6R2), were identified to be
differentially expressed.
 | Table I.Characteristics of the three gene
expression profiling datasets for multiple myeloma downloaded for
integrated analysis. |
Table I.
Characteristics of the three gene
expression profiling datasets for multiple myeloma downloaded for
integrated analysis.
| Author, year | GEO dataset ID | Platform for
detection | Samples (N:M) | (Refs.) |
|---|
| Zhang et al,
2011 | GSE23832 | GPL6244
[HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array [transcript
(gene) version] | 4:8 | (20) |
| Kemppinen et
al, 2011 | GSE21942 | GPL570
[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 15:12 | (21) |
| Gandhi et
al, 2010 | GSE17048 | GPL6947 Illumina
HumanHT-12 V3.0 expression beadchip | 45:99 | (22) |
| Table II.Top 10 significantly upregulated and
downregulated DEGs identified in multiple myeloma. All gene ID's
were taken from the Entrez database on NCBI. |
Table II.
Top 10 significantly upregulated and
downregulated DEGs identified in multiple myeloma. All gene ID's
were taken from the Entrez database on NCBI.
| A, Upregulated
DEGs |
|---|
|
|---|
| Entrez gene ID | Abbreviation | Name | FDR |
|---|
| 84265 | POLR3GL | Polymerase (RNA)
III (DNA directed) polypeptide G (32kD)-like | 0.0001793 |
| 8364 | HIST1H4C | Histone cluster 1,
H4c | 0.0002646 |
| 6170 | RPL39 | Ribosomal protein
L39 | 0.0013368 |
| 5880 | RAC2 | Ras-related C3
botulinum toxin substrate 2 (rho family, small GTP binding protein
Rac2) | 0.0013368 |
| 29080 | CCDC59 | Coiled-coil domain
containing 59 | 0.0013368 |
| 521 | ATP5I | ATP synthase, H+
transporting, mitochondrial Fo complex, subunit E | 0.0013773 |
| 9991 | PTBP3 | Polypyrimidine
tract binding protein 3 | 0.001458 |
| 79023 | NUP37 | Nucleoporin
37kDa | 0.001458 |
| 64801 | ARV1 | ARV1 homolog, fatty
acid homeostasis modulator | 0.001458 |
| 3700 | ITIH4 | Inter-α-trypsin
inhibitor heavy chain family, member 4 | 0.001458 |
|
| B, Downregulated
DEGs |
|
| Entrez gene ID | Abbreviation | Name | FDR |
|
| 387 | RHOA | Ras homolog family
member A | 0.0004738 |
| 23256 | SCFD1 | Sec1 family domain
containing 1 | 0.0013368 |
| 92241 | RCSD1 | RCSD domain
containing 1 | 0.0014580 |
| 126364 | LRRC25 | Leucine rich repeat
containing 25 | 0.0017804 |
| 5226 | PGD | Phosphogluconate
dehydrogenase | 0.0026507 |
| 129531 | MITD1 | MIT, microtubule
interacting and transport, domain containing 1 | 0.0027447 |
| 10023 | FRAT1 | Frequently
rearranged in advanced T-cell lymphomas 1 | 0.0027447 |
| 8883 | NAE1 | NEDD8 activating
enzyme E1 subunit 1 | 0.0028332 |
| 81631 | MAP1LC3B |
Microtubule-associated protein 1 light
chain 3 β | 0.0028998 |
| 4904 | YBX1 | Y box binding
protein 1 | 0.0028998 |
Functional classification of
differentially expressed genes
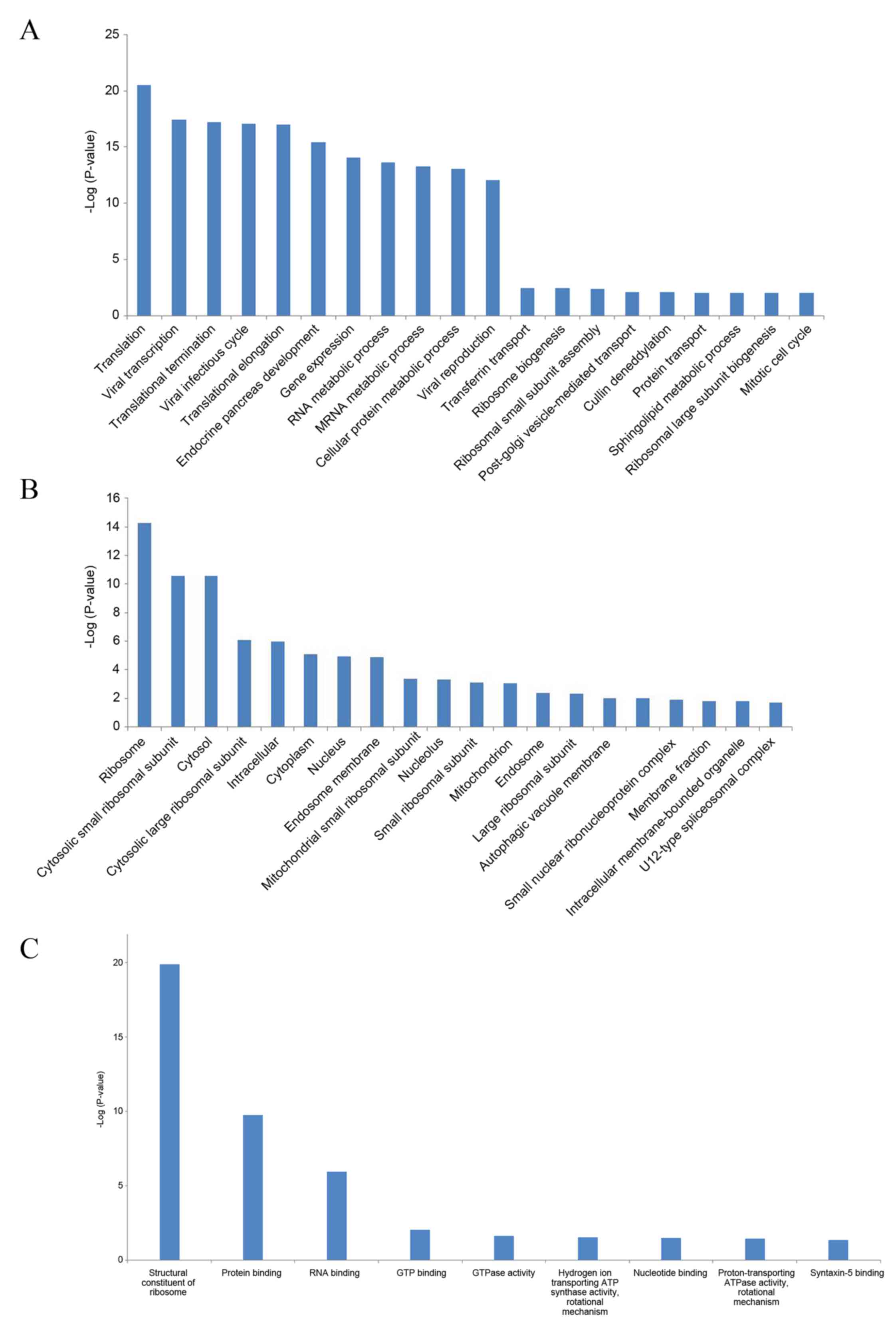
For the differentially expressed genes in MM, GO
term enrichment analysis was performed. For biological processes,
38 GO terms were significantly enriched, and the most significantly
enriched GO terms were translation (GO:0006412; FDR,
3.14×10−21) and viral transcription (GO:0019083; FDR,
3.59×10−18) (Fig. 2A).
For cellular components, 29 GO terms were significantly enriched,
and the most significantly enriched GO terms were ribosome
(GO:0005840; FDR, 5.12×10−15) and cytosolic small
ribosomal subunit (GO:0022627; FDR, 2.60×10−11)
(Fig. 2B). For molecular functions,
9 GO terms were significantly enriched, and the most significantly
enriched GO terms were structural constituent of ribosome
(GO:0003735; FDR, 1.40×10−20) and protein binding
(GO:0005515; FDR, 1.82×10−10) (Fig. 2C).
When performing the KEGG pathway enrichment
analysis, 7 signaling pathways were significantly enriched with the
criteria of FDR <0.05 (Table
III). The most significant pathway was ribosome (FDR,
2.25×10−16). Furthermore, oxidative phosphorylation
(FDR, 0.0277), bacterial invasion of epithelial cells (FDR,
0.0286), lysosome (FDR, 0.0318), the Wnt signaling pathway (FDR,
0.0343), tuberculosis (FDR, 0.0385) and collecting duct acid
secretion (FDR, 0.0407) were also significantly enriched.
| Table III.Significantly enriched KEGG signaling
pathways of the differentially expressed genes identified in
multiple myeloma. |
Table III.
Significantly enriched KEGG signaling
pathways of the differentially expressed genes identified in
multiple myeloma.
| KEGG ID | KEGG term | No. of enriched
genes | FDR | Genes |
|---|
| hsa03010 | Ribosome | 13 |
2.25×10−16 | RPS15A, RPS27A,
RPS25, RPS13, RPL26,RPL39, RPL21, RPS29, RPS14, RPS27, RPL11,
RPL27, RPS10 |
| hsa00190 | Oxidative
phosphorylation | 4 |
2.77×10−02 | NDUFS4, ATP6V1A,
ATP5I, ATP6V0C |
| hsa05100 | Bacterial invasion
of epithelial cells | 3 |
2.86×10−02 | CLTC, RHOA,
CRKL |
| hsa04142 | Lysosome | 4 |
3.18×10−02 | CLTC, PSAP,
ATP6V0C, LAMP2 |
| hsa04310 | Wnt signaling
pathway | 4 |
3.43×10−02 | SIAH1, RAC2, RHOA,
FRAT1 |
| hsa05152 | Tuberculosis | 4 |
3.85×10−02 | APAF1, RHOA,
ATP6V0C, LAMP2 |
| hsa04966 | Collecting duct
acid secretion | 2 |
4.07×10−02 | ATP6V1A,
ATP6V0C |
PPI network
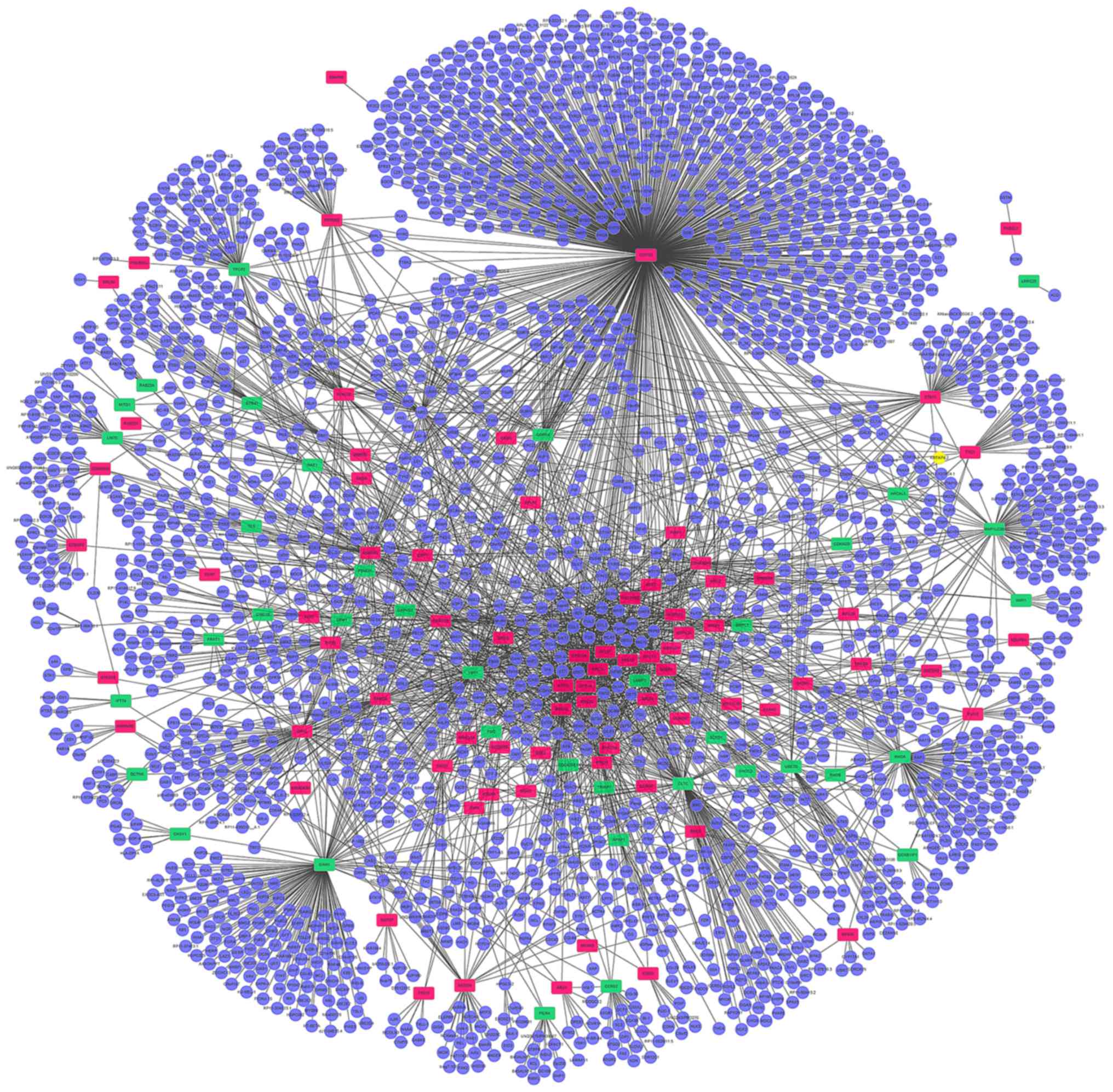
A PPI network including all of the differentially
expressed genes identified was constructed. The PPI network
produced included 2,355 nodes and 3,707 edges (Fig. 3). Highly connected proteins in the
network are called hub proteins, which are the core of regulation
and serve an important role in the stability of the network. The
significant hub proteins were identified, including COP9
signalosome complex subunit 5 (COPS5; connectivity degree, 791),
CLTC (connectivity degree, 172) and 60S ribosomal protein L11
(connectivity degree, 167).
NLP results
The abstracts of 6,795 primary studies were
identified using the aforementioned search strategy and a total of
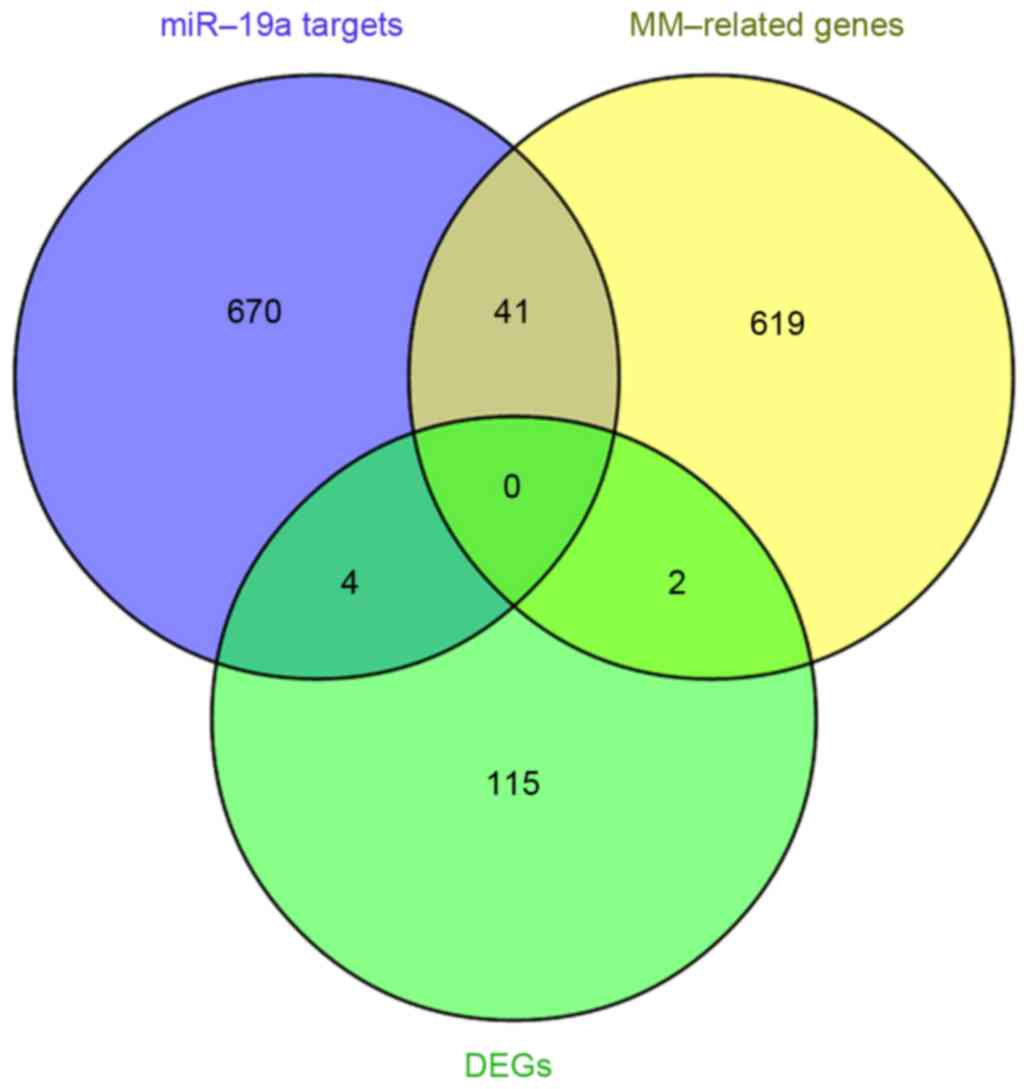
662 MM-associated genes were obtained (data not shown). Integrated
analysis was performed to identify the overlap between the 715
miR-19a target genes previously identified and the 662
MM-associated genes obtained from NLP analysis. This revealed that
there were 41 overlapping genes (Table
IV and Fig. 4), which were
associated with the development and progression of MM, and were
putative miR-19a target genes. In addition, 2 of the differentially
expressed genes, Y-box binding protein 1 (YBX1) and TP53 regulated
inhibitor of apoptosis 1 (TRIAP1) were identified to be associated
with MM (Fig. 4).
| Table IV.Overlapping genes that were
associated with the development and progression of multiple
myeloma, and were putative target genes of microRNA-19a (n=41). |
Table IV.
Overlapping genes that were
associated with the development and progression of multiple
myeloma, and were putative target genes of microRNA-19a (n=41).
| Gene
abbreviation | Gene name | Count | P-value |
|---|
| CCND1 | Cyclin D1 | 78 | <0.0001 |
| CCNA2 | Cyclin A2 | 1 |
0.2128 |
| CCND2 | Cyclin D2 | 16 | <0.0001 |
| CD69 | CD69 molecule | 1 |
0.0818 |
| CTGF | Connective tissue
growth factor | 1 |
0.2861 |
| S1PR1 |
Sphingosine-1-phosphate receptor 1 | 1 |
0.1044 |
| EREG | Epiregulin | 1 |
0.0388 |
| ESR1 | Estrogen receptor
1 | 3 |
0.4207 |
| F3 | Coagulation factor
III (thromboplastin, tissue factor) | 2 |
0.0853 |
| GJA1 | Gap junction
protein, α 1, 43kDa | 1 |
0.2950 |
| GRK6 | G protein-coupled
receptor kinase 6 | 1 |
0.0625 |
| ID2 | Inhibitor of DNA
binding 2, dominant negative helix-loop-helix protein | 1 |
0.1265 |
| IL6ST | Interleukin 6
signal transducer (gp130, oncostatin M receptor) | 2 |
0.0231 |
| ITGA6 | Integrin, α 6 | 1 |
0.1979 |
| KIT | V-kit
Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | 10 | <0.0001 |
| KRAS | V-Ki-ras2 Kirsten
rat sarcoma viral oncogene homolog | 1 |
0.5071 |
| LIF | leukemia inhibitory
factor (cholinergic differentiation factor) | 1 |
0.1516 |
| SMAD4 | SMAD family member
4 | 1 |
0.3981 |
| MDM4 | Mdm4 p53 binding
protein homolog (mouse) | 1 |
0.1155 |
| PTEN | Phosphatase and
tensin homolog | 5 |
0.0038 |
| ATXN1 | Ataxin 1 | 1 |
0.1155 |
| SDC1 | Syndecan 1 | 46 | <0.0001 |
| TGFBR2 | Transforming growth
factor, β receptor II (70/80kDa) | 1 |
0.3081 |
| THBS1 | Thrombospondin
1 | 1 |
0.3418 |
| KLF10 | Kruppel-like factor
10 | 1 |
0.0487 |
| TNFAIP3 | Tumor necrosis
factor, α-induced protein 3 | 2 |
0.0064 |
| SOCS1 | Suppressor of
cytokine signaling 1 | 8 | <0.0001 |
| SOCS3 | Suppressor of
cytokine signaling 3 | 3 |
0.0022 |
| HDAC4 | Histone deacetylase
4 | 1 |
0.1463 |
| FOXP1 | Forkhead box
P1 | 2 |
0.0016 |
| MIB1 | Mindbomb homolog 1
(Drosophila) | 1 |
0.0267 |
| PCDH10 | Protocadherin
10 | 1 |
0.0226 |
| CYLD | Cylindromatosis
(turban tumor syndrome) | 1 |
0.1007 |
| PTK2B | PTK2B protein
tyrosine kinase 2 β | 1 |
0.2757 |
| IGF1 | Insulin-like growth
factor 1 (somatomedin C) | 14 | <0.0001 |
| MAPK10 | Mitogen-activated
protein kinase 10 | 3 |
0.0001 |
| RAF1 | V-raf-1 murine
leukemia viral oncogene homolog 1 | 1 |
0.4154 |
| SGK1 |
Serum/glucocorticoid regulated kinase
1 | 2 |
0.0173 |
| TSC1 | Tuberous sclerosis
1 | 1 |
0.1861 |
| BCL2L11 | BCL2-like 11
(apoptosis facilitator) | 4 |
0.0002 |
| TLR2 | Toll-like receptor
2 | 1 |
0.5613 |
Discussion
Several previous studies (2,13–15) have
demonstrated that miR-19a is deregulated in MM as an oncomiR,
suggesting it serves an important role in MM. Considering that the
biological significance of miRNA deregulation relies on the effect
upon target protein-coding genes, predicted target genes of miR-19a
that were associated with the carcinogenesis, prognosis and
chemoresistance of MM were systematically analyzed in the present
study in order to further investigate the potential involvement of
miR-19a in MM. Strategies to determine miRNA targets include
bioinformatical prediction and experimental assays. The present
study utilized three common computational algorithms, miRanda,
PicTar and TargetScan, to identify 715 putative target genes of
miR-19a, among which 40 were experimentally validated in miRWalk.
In addition, NLP analysis was performed in the current study, which
identified 662 MM-associated genes. Then, integrated analysis
revealed 41 predicted target genes of miR-19a that were associated
with the development and progression of MM, including Kirsten rat
sarcoma viral oncogene homolog (KRAS), SOCS and CCND1. Several of
these MM-associated putative miR-19a targets, including SOCS and
CCND1, have already been verified by miRNA functional experiments
(14). Previous studies have
demonstrated that oncogenic mutations of RAS occur in 30–40% of
patients with MM and are rarely found in MGUS (26–28). The
occurrence of RAS mutation appears independent of clinical stage,
but is associated with disease progression, an aggressive
phenotype, resistance to therapy and poor patient survival
(26,27,29).
Steinbrunn et al (27) also
reported that the ectopic overexpression of oncogenic RAS induces
MM cell proliferation and lowers drug efficacy.
Given that the altered expression of miR-19a in MM
would cause changes in target gene expression, differentially
expressed genes between MM and normal controls were assessed in
current study using gene expression data. This revealed that 121
genes were differentially expressed in MM, including 80 upregulated
genes and 41 downregulated genes. In addition, 2 of the
differentially expressed genes, YBX1 and TRIAP1, were identified to
be associated with MM in the present study. YBX1, a member of the
cold-shock domain protein superfamily, is involved in a wide range
of cellular functions, including DNA transcription, replication and
repair, and environmental stress and chromatin remodeling, in
addition to pre-mRNA splicing (30).
Chatterjee et al (31)
demonstrated that YBX1 was overexpressed in immature and anaplastic
MM cells, but not expressed in normal PCs, MGUS PCs or the majority
of MM specimens, suggesting it serves a role in dedifferentiation
as part of the malignant transformation process. Furthermore, other
studies have reported that the aberrant expression of YBX1 is
associated with tumorigenesis, and cancer cell proliferation,
survival and drug resistance (32,33).
Interestingly, four putative targets of miR-19a,
RHOB, CLTC, PSAP and PPP6R2, were identified to be differentially
expressed in MM in the present study. The tumor suppressor RHOB has
been demonstrated to downregulated in various types of cancer
(34,35), which is in accord with the findings
of the present study. Notably, Tan et al (36) revealed that RHOB induced apoptosis,
and inhibited proliferation and migration in pancreatic cancer as a
direct target of miR-19a. Chromosomal and genomic analyses have
revealed that the ALK receptor tyrosine kinase gene is fused to
CLTC in inflammatory myofibroblastic tumors and B-cell lymphoma
(37,38). However, the exact role of CLTC in MM
has not yet been reported. PSAP, a highly conserved glycoprotein,
is overexpressed in prostate cancer and esophageal squamous cell
carcinoma (39,40). A similar expression trend for PSAP
was observed in the present study, indicating that it may be a
candidate biomarker for MM.
In the PPI network of differentially expressed genes
produced in the present study, COPS5 had the highest connectivity
degree, suggesting that it serves an important role in MM
progression. COPS5, one of the eight subunits of the COP9
signalosome, is overexpressed in a variety of types of human cancer
(41). COPS5was identified to be
overexpressed in MM in the present study. A previous study
demonstrated that the specific knockdown of COPS5 inhibits the
proliferation of colorectal cancer cells (42), and that COPS5-transgenic mice
developed a phenotype similar to that of myeloproliferative
disorders (43). In addition, COPS5
is involved in Ras-mediated cell transformation by inhibiting
premature senescence (44).
All of the differentially expressed genes in MM
identified in the present study underwent GO term and signaling
pathway enrichment analysis, in addition to PPI network
construction, in order to understand their function. This revealed
functions in viral transcription, the viral infectious cycle, viral
reproduction and the bacterial invasion of epithelial cells. A
previous clinical investigation revealed that the risk of bacterial
and viral infections was seven times higher in MM patients compared
with matched controls due to MM-associated immunodeficiency
resulting from PC disorders, including B-cell dysfunction, and
T-cell, dendritic cell and NK cell abnormalities (45).
In conclusion, the present study identified and
systematically analyzed predicted MM-associated target genes of
miR-19a. A total of 121 differentially expressed genes in MM were
identified, including 80 upregulated genes and 41 downregulated
genes. Among these differentially expressed genes, RHOB, CLTC, PSAP
and PPP6R2, were predicted target genes of miR-19a. The results of
NLP analysis revealed that 2 of the differentially expressed genes,
YBX1 and TRIAP1, were associated with MM. In addition, 41 target
genes of miR-19a were associated with the development and
progression of MM. The combined examination of gene expression and
bioinformatical prediction for miR-19a target genes may provide new
insights into carcinogenic mechanisms of MM, in addition to
highlighting potential areas for the development of novel
personalized therapies. Further studies are required to confirm the
results of the present study in patients with MM.
References
|
1
|
Bommert K, Bargou RC and Stühmer T:
Signalling and survival pathways in multiple myeloma. Eur J Cancer.
42:1574–1580. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Benetatos L and Vartholomatos G:
Deregulated microRNAs in multiple myeloma. Cancer. 118:878–887.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raab MS, Podar K, Breitkreutz I,
Richardson PG and Anderson KC: Multiple myeloma. Lancet.
374:324–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dolloff NG and Talamo G: Targeted therapy
of multiple myeloma. Adv Exp Med Biol. 779:197–221. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chi J, Ballabio E, Chen XH, Kušec R,
Taylor S, Hay D, Tramonti D, Saunders NJ, Littlewood T, Pezzella F,
et al: MicroRNA expression in multiple myeloma is associated with
genetic subtype, isotype and survival. Biol Direct. 6:232011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carthew RW: Gene regulation by microRNAs.
Curr Opin Genet Dev. 16:203–208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rushworth SA, Murray MY, Barrera LN,
Heasman SA, Zaitseva L and Macewan DJ: Understanding the role of
miRNA in regulating NF-kB in blood cancer. Am J Cancer Res.
2:65–74. 2012.PubMed/NCBI
|
|
8
|
Zhou Y, Chen L, Barlogie B, Stephens O, Wu
X, Williams DR, Cartron MA, van Rhee F, Nair B, Waheed S, et al:
High-risk myeloma is associated with global elevation of miRNAs and
overexpression of EIF2C2/AGO2. Proc Natl Acad Sci USA. 107:pp.
7904–7909. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lionetti M, Biasiolo M, Agnelli L,
Todoerti K, Mosca L, Fabris S, Sales G, Deliliers GL, Bicciato S,
Lombardi L, et al: Identification of microRNA expression patterns
and definition of a microRNA/mRNA regulatory network in distinct
molecular groups of multiple myeloma. Blood. 114:e20–e26. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu P, Agnelli L, Walker BA, Todoerti K,
Lionetti M, Johnson DC, Kaiser M, Mirabella F, Wardell C, Gregory
WM, et al: Improved risk stratification in myeloma using a
microRNA-based classifier. Br J Haematol. 162:348–359. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fuziwara CS and Kimura ET: Insights into
regulation of the miR-17-92 cluster of miRNAs in cancer. Front Med
(Lausanne). 2:642015.PubMed/NCBI
|
|
13
|
Todoerti K, Barbui V, Pedrini O, Lionetti
M, Fossati G, Mascagni P, Rambaldi A, Neri A, Introna M, Lombardi L
and Golay J: Pleiotropic anti-myeloma activity of ITF2357:
Inhibition of interleukin-6 receptor signaling and repression of
miR-19a and miR-19b. Haematologica. 95:260–269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pichiorri F, Suh SS, Ladetto M, Kuehl M,
Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, et
al: MicroRNAs regulate critical genes associated with multiple
myeloma pathogenesis. Proc Natl Acad Sci USA. 105:pp. 12885–12890.
2008; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Corthals SL, Sun SM, Kuiper R, de Knegt Y,
Broyl A, Van der Holt B, Beverloo HB, Peeters JK, el Jarari L,
Lokhorst HM, et al: MicroRNA signatures characterize multiple
myeloma patients. Leukemia. 25:1784–1789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen L, Li C, Zhang R, Gao X, Qu X, Zhao
M, Qiao C, Xu J and Li J: miR-17-92 cluster microRNAs confers
tumorigenicity in multiple myeloma. Cancer Lett. 309:62–70. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gutierrez NC, Sarasquete ME,
Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV,
Fermiñán E, Martín-Jiménez P, Chillón C, Risueño A, et al:
Deregulation of microRNA expression in the different genetic
subtypes of multiple myeloma and correlation with gene expression
profiling. Leukemia. 24:629–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Claudio JO, Masih-Khan E, Tang H,
Goncalves J, Voralia M, Li ZH, Nadeem V, Cukerman E,
Francisco-Pabalan O, Liew CC, et al: A molecular compendium of
genes expressed in multiple myeloma. Blood. 100:2175–2186. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang F, Shi Y, Wang L and Sriram S: Role
of HDAC3 on p53 expression and apoptosis in T cells of patients
with multiple sclerosis. PLoS One. 6:e167952011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kemppinen AK, Kaprio J, Palotie A and
Saarela J: Systematic review of genome-wide expression studies in
multiple sclerosis. BMJ Open. 1:e0000532011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gandhi KS, McKay FC, Cox M, Riveros C,
Armstrong N, Heard RN, Vucic S, Williams DW, Stankovich J, Brown M,
et al: The multiple sclerosis whole blood mRNA transcriptome and
genetic associations indicate dysregulation of specific T cell
pathways in pathogenesis. Hum Mol Genet. 19:2134–2143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Giot L, Bader JS, Brouwer C, Chaudhuri A,
Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al: A protein
interaction map of Drosophila melanogaster. Science. 302:1727–1736.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoang B, Zhu L, Shi Y, Frost P, Yan H,
Sharma S, Sharma S, Goodglick L, Dubinett S and Lichtenstein A:
Oncogenic RAS mutations in myeloma cells selectively induce cox-2
expression, which participates in enhanced adhesion to fibronectin
and chemoresistance. Blood. 107:4484–4490. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Steinbrunn T, Stühmer T, Gattenlöhner S,
Rosenwald A, Mottok A, Unzicker C, Einsele H, Chatterjee M and
Bargou RC: Mutated RAS and constitutively activated Akt delineate
distinct oncogenic pathways, which independently contribute to
multiple myeloma cell survival. Blood. 117:1998–2004. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rasmussen T, Kuehl M, Lodahl M, Johnsen HE
and Dahl IM: Possible roles for activating RAS mutations in the
MGUS to MM transition and in the intramedullary to extramedullary
transition in some plasma cell tumors. Blood. 105:317–323. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chng WJ, Gonzalez-Paz N, Price-Troska T,
Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S,
Greipp P, et al: Clinical and biological significance of RAS
mutations in multiple myeloma. Leukemia. 22:2280–2284. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eliseeva IA, Kim ER, Guryanov SG,
Ovchinnikov LP and Lyabin DN: Y-box-binding protein 1 (YB-1) and
its functions. Biochemistry (Mosc). 76:1402–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chatterjee M, Rancso C, Stuhmer T,
Eckstein N, Andrulis M, Gerecke C, Lorentz H, Royer HD and Bargou
RC: The Y-box binding protein YB-1 is associated with progressive
disease and mediates survival and drug resistance in multiple
myeloma. Blood. 111:3714–3722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vaiman AV, Stromskaya TP, Rybalkina EY,
Sorokin AV, Ovchinnikov LP and Stavrovskaya AA: Development of drug
resistance in the population of colon cancer cells under the effect
of multifunctional protein YB-1. Bull Exp Biol Med. 143:463–466.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lasham A, Print CG, Woolley AG, Dunn SE
and Braithwaite AW: YB-1: Oncoprotein, prognostic marker and
therapeutic target? Biochem J. 449:11–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou J, Zhu Y, Zhang G, Liu N, Sun L, Liu
M, Qiu M, Luo D, Tang Q, Liao Z, et al: A distinct role of RhoB in
gastric cancer suppression. Int J Cancer. 128:1057–1068. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim DM, Chung KS, Choi SJ, Jung YJ, Park
SK, Han GH, Ha JS, Song KB, Choi NS, Kim HM, et al: RhoB induces
apoptosis via direct interaction with TNFAIP1 in HeLa cells. Int J
Cancer. 125:2520–2527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tan Y, Yin H, Zhang H, Fang J, Zheng W, Li
D, Li Y, Cao W, Sun C, Liang Y, et al: Sp1-driven up-regulation of
miR-19a decreases RHOB and promotes pancreatic cancer. Oncotarget.
6:17391–17403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chikatsu N, Kojima H, Suzukawa K,
Shinagawa A, Nagasawa T, Ozawa H, Yamashita Y and Mori N: ALK+,
CD30-, CD20- large B-cell lymphoma containing anaplastic lymphoma
kinase (ALK) fused to clathrin heavy chain gene (CLTC). Mod Pathol.
16:828–832. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cerchietti L, Damm-Welk C, Vater I,
Klapper W, Harder L, Pott C, Yang SN, Reiter A, Siebert R, Melnick
A and Woessmann W: Inhibition of anaplastic lymphoma kinase
(ALK)activity provides a therapeutic approach for CLTC-ALK-positive
human diffuse large B cell lymphomas. PLoS One. 6:e184362011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gunia S, Koch S, May M, Dietel M and
Erbersdobler A: Expression of prostatic acid phosphatase (PSAP) in
transurethral resection specimens of the prostate is predictive of
histopathologic tumor stage in subsequent radical prostatectomies.
Virchows Arch. 454:573–579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pawar H, Kashyap MK, Sahasrabuddhe NA,
Renuse S, Harsha HC, Kumar P, Sharma J, Kandasamy K, Marimuthu A,
Nair B, et al: Quantitative tissue proteomics of esophageal
squamous cell carcinoma for novel biomarker discovery. Cancer Biol
Ther. 12:510–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kato JY and Yoneda-Kato N: Mammalian COP9
signalosome. Genes Cells. 14:1209–1225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schütz AK, Hennes T, Jumpertz S, Fuchs S
and Bernhagen J: Role of CSN5/JAB1 in Wnt/β-catenin activation in
colorectal cancer cells. FEBS Lett. 586:1645–1651. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mori M, Yoneda-Kato N, Yoshida A and Kato
JY: Stable form of JAB1 enhances proliferation and maintenance of
hematopoietic progenitors. J Biol Chem. 283:29011–29021. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsujimoto I, Yoshida A, Yoneda-Kato N and
Kato JY: Depletion of CSN5 inhibits Ras-mediated tumorigenesis by
inducing premature senescence in p53-null cells. FEBS Lett.
586:4326–4331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Blimark C, Holmberg E, Mellqvist UH,
Landgren O, Björkholm M, Hultcrantz M, Kjellander C, Turesson I and
Kristinsson SY: Multiple myeloma and infections: A population-based
study on 9253 multiple myeloma patients. Haematologica.
100:107–113. 2015. View Article : Google Scholar : PubMed/NCBI
|