Case report
The current report presents the case of a
45-year-old female patient who was previously diagnosed in January
2010 at the Dermatological University Clinic (Cluj, Romania) with
terbinafine-induced subacute lupus erythematosus (SCLE). The
patient received terbinafine (250 mg/day) for distal subungual
onychomycosis of the toenail for 6 months and after 2 months of
treatment she developed specific lesions for SCLE. The patient
received systemic treatment with antimalarial drugs
(hydroxychloroquine; 250 mg/day), corticosteroids (prednisone; 50
mg/day) and immunomodulators (azathioprine; 100 mg/day), and
complete remission of lesions was achieved within a year. Although,
the course of the disease varied after this. The present study was
approved by the Ethics Committee of Grigore T. Popa University of
Medicine and Pharmacy (Iași, Romania), and the patient provided
written informed consent.
In October 2015, the patient presented, for the
first time at our clinic (Clinic of Dermatovenereology, St.
Spiridon Emergency Hospital, Iași, Romania), with a skin rash
consisting of erythematous-papular erythema multiforme-like lesions
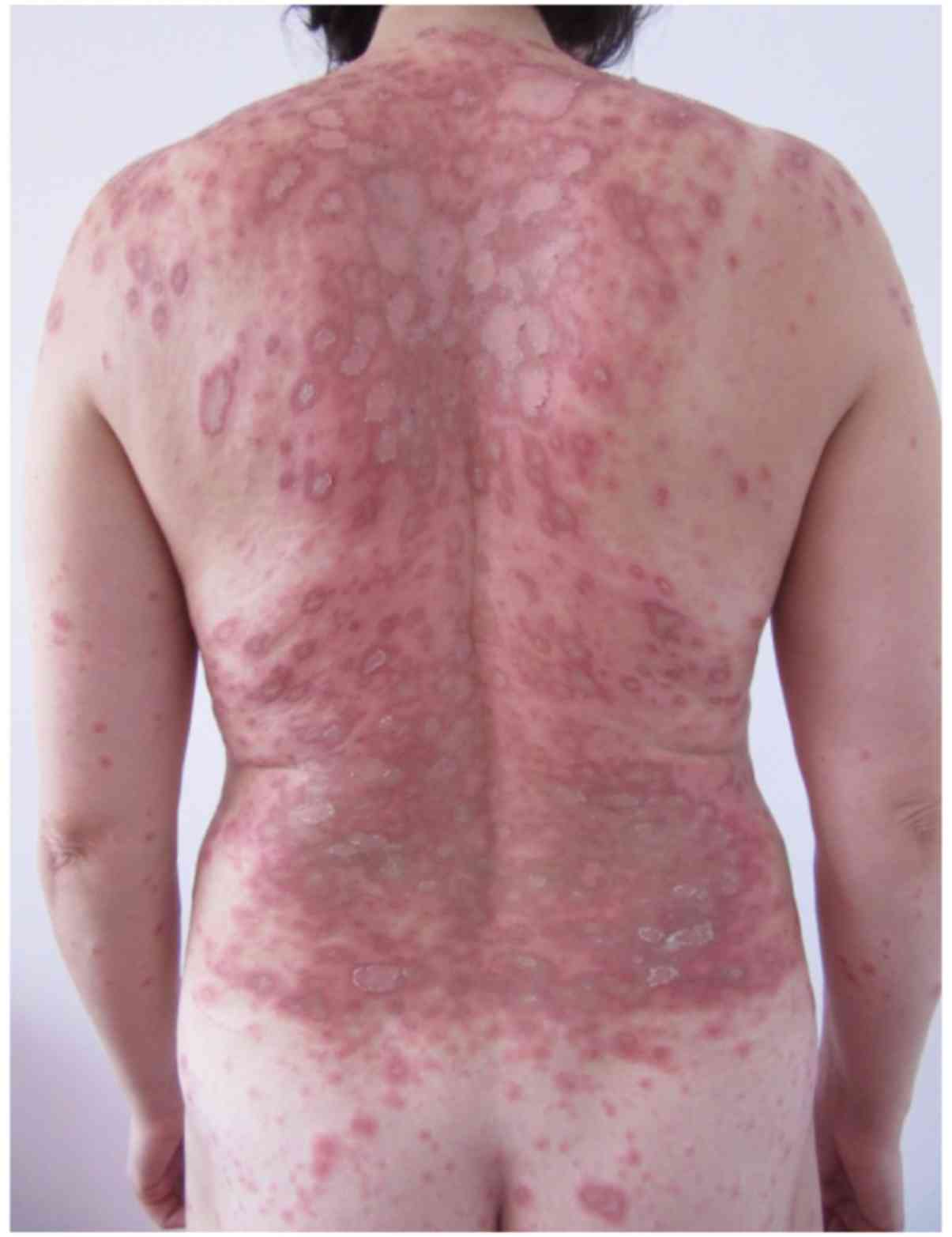
on the skin, primarily located on the trunk and limbs in (Fig. 1). Onset of illness occurred ~1 week
after the initiation of a Helicobacter pylori (H.
pylori) eradication regimen (80 mg/day esomeprasolum and 1 g/12
h amoxicillin) and subsequently this treatment for H. pylori
eradication was terminated.
Clinical dermatological examination (Fig. 1) revealed erythematous-edematous
macules and plaques, which were well-defined, round and arranged en
cocarde, with a tendency to expand and evolve into placards.
Certain lesions were solitary, while others evolved into placards.
The specific lesions were covered by scales that were only adherent
to the skin in the center of the lesions and exhibited a pale
center. These lesions were accompanied by intense itching and local
inflammatory phenomena. In addition, several petechiae were present
on the hard palate mucosa. The associated symptoms consisted of
fatigability, myalgia and gonalgia. In addition, tests for anti-Ro
antibodies and for total antinuclear antibodies using fluorometric
enzyme immunoassay (1), and
rheumatoid factor using latex-immunoturbidimetry (2) were positive. Serology for herpes
infection was performed using the electro-chemiluminescence
immunoassay for immunoglobulin G and an IMMULITE 2000 Systems
Analyzer (L2KHVG2; Siemens Healthieers, Erlangen Germany.) and
ELISA for immunoglobulin M using a microplate Reader (HSVM.CE, lot
0416; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Both tests
were negative.
A skin biopsy was taken for pathological examination
and direct immunofluorescence microscopy. The skin biopsy for
histopathology was fixed in a buffered 10% formalin solution for
~12 h, at room temperature. Subsequently, the tissue was embedded
in paraffin, cut into 4-µm sections, placed on glass microscope
slide and stained with hematoxylin and eosin. The slides were
analyzed with a light microscope, using a magnification of ×40,
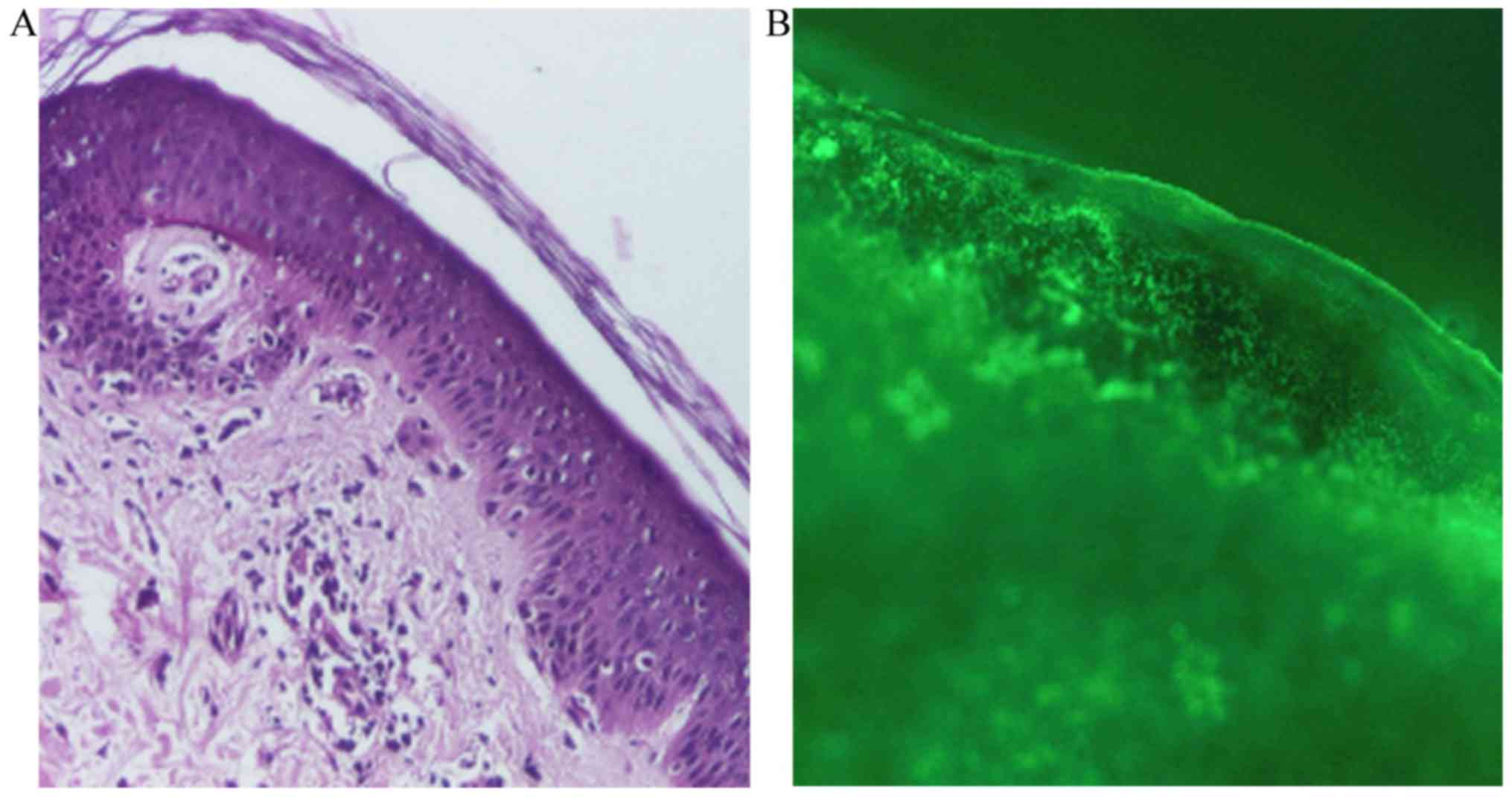
×100 and ×200. Histopathologically, the following features were
identified: Areas of atrophied epidermis with exocytosis,
vacuolization and foci of necrotic keratinocytes, and a moderate
inflammatory perivascular and peridnexal mononuclear infiltrate
(Fig. 2A). For the direct
immunofluorescence examination, the skin biopsy was immediately
transported in cold physiological saline. Section of 4-µm thick
obtained at cryotome were fixed with acetone for 15 min at room
temperature. The following antibodies were used: IgA (F031601;
dilution 1:20), IgG (F031501; dilution 1:20), IgM (F031701;
dilution 1:20), C1q (F025402; dilution 1:4), C3 (F020102; dilution
1:10), Fibrinogen (F011102; dilution 1:60; all from Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA). Fluorescence-labeled
antibodies were applied to sections and allowed to incubate at room
temperature for 30 min. Mounting medium (Dako; Agilent
Technologies, Inc.) was applied. Subsequently, using a fluorescence
microscope Nikon Eclipse E600, the sites of attachment of the
labeled antibodies in the skin were identified at a magnification
of ×40, ×100 and ×200. Direct immunofluorescence revealed
complement component 3 (C3; Fig. 2B)
and immunoglobulin M in the basement membrane (data not shown).
The patient was monitored periodically (every 6
months) to enable the early detection of potential eye concerns,
including orbital and external eye disease, proptosis,
enophthalmos, orbital pain, blurred vision, chemosis, restriction
of extraocular motility, keratoconjunctivitis sicca, corneal
erosion, peripheral corneal infiltration, ulcerative keratitis,
interstitial keratitis, endotheliitis, lupus retinopathy, optic
neuritis, ischemic optic neuropathy and papilledema, induced by the
disease. Furthermore, the potential manifestation of chloroquine
maculopathy in response to the administered antimalarial therapy
(hydroxychloroquine therapy 200 mg/day; initiated in June 2016 and
continued; last follow-up, May 2017) was also monitored.
Based on the patient's history, clinical
manifestations (erythema multiforme-like lesions), the antibody
detection, histopathological analysis and direct immunofluorescence
findings, a diagnosis of Rowell syndrome was made. Therapy with
systemic corticosteroids (methylprednisolone; 0.5 mg/kg) in
combination with immunomodulators (azathioprine; 50 mg/day) was
initiated in October 2015, which together with the associated
medication (omeprazole, 20 mg/day; KCl, 1 g/day) and topical
dermocorticoids (fluticasone propionate 0.05% cream; 1
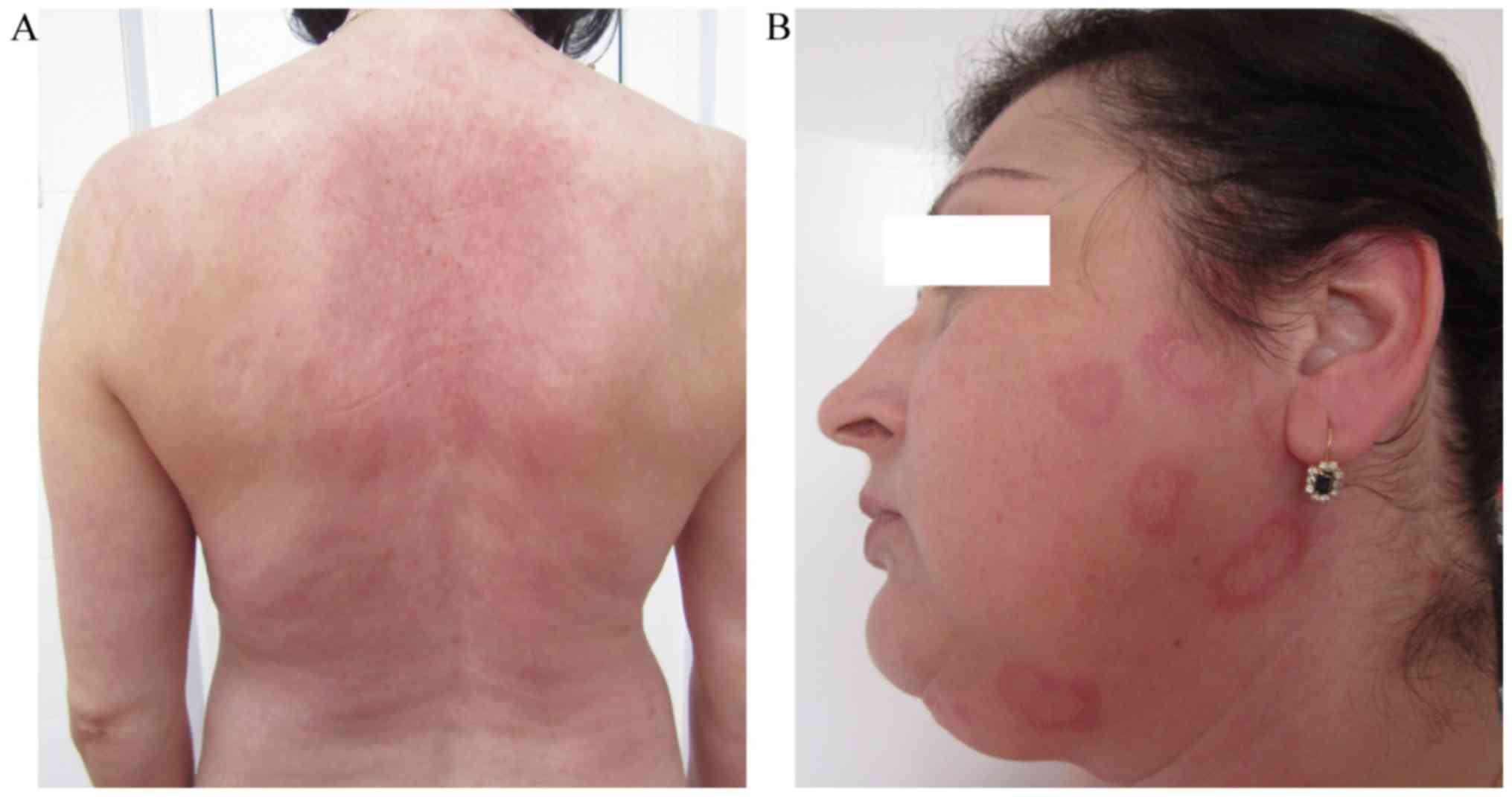
application/day) markedly reduced the patient's symptoms (Fig. 3A). The levels of anti-Ro antibodies,
rheumatoid factor and total antinuclear antibodies were monitored
periodically, every 3 months.
Given the favorable outcome, the dose of
methylprednisolone was gradually reduced with a reduction of 4
mg/month while maintaining the dose of 100 mg/day of azathioprine.
When a dose of 8 mg/day methylprednisolone was reached in May 2016,
the patient noticed the reoccurrence of erythema multiforme-like
lesions on their trunk, upper limbs and face (Fig. 3B). Therefore, the dose of
methylprednisolone was increased to 12 mg/day in June 2016. The
course of the disease varied after this, without total control of
the skin lesions; however, the patients remained in good general
health. Furthermore, hydroxychloroquine (200 mg/day) was added to
the treatment regime (June 2016), which reduced the lesions, but
did not achieve complete remission. The treatment with dapsone (100
mg/day) was initiated in October 2016 in association with
azathioprine (50 mg/day) and hydroxychloroquine (200 mg/day). The
patient decided to terminate all treatments in December 2016
because she developed notable digestive manifestations (nausea and
vomiting). At that time (December 2016), all cutaneous lesions were
in complete remission until today (last follow-up, May 2017), but
arthalgia, myalgia and immunological abnormalities persisted. From
the onset of the disease (October 2015) until present (May 2017),
the levels of anti-Ro antibodies varied from 44.8 to 46.8 UI/ml,
the levels of rheumatoid factor varied from 17 to 16.32 UI/ml and
the total antinuclear antibodies varied from 5.53 to 1.6. In
February 2017, the patient persisted with arthalgia, myalgia and
asthenia and therefore the hydroxycholoroquine (200 mg/day) was
reinitiated. Furtherore hydroxycholoroquine was also administered
for photoprotection reasons and for the prevention of relapse. The
patient remains under clinical surveillance, with follow-ups every
6 months.
Discussion
Rowell syndrome is characterized by the coexistence
of lupus erythematosus, erythema multiforme-like lesions and
characteristic immunological changes (positive tests for
antinuclear antibodies, rheumatoid factor and anti-Ro/anti-La
antibodies) (3–11). Until 2014, 37 cases of Rowell
syndrome were reported in the literature according to the data
published by Bhat et al (4).
The first description of this association was made by Scholtz in
1922 (6). In 1963, Rowell et
al (7) defined a syndrome
consisting of discoid lupus erythematosus, erythema multiforme-like
lesions and immunological abnormalities, including the presence of
rheumatoid factor and antinuclear antibodies. In 1995, Lee et
al (9) suggested the inclusion
of chilblains (pernio) to the diagnostic criteria for Rowell
syndrome.
In 2000, Zeitouni et al (10) proposed major and minor criteria for
the diagnosis of Rowell syndrome. The major criteria were as
follows: The simultaneous presence of lupus erythematosus (acute,
subacute or systemic), erythema multiforme-like lesions and
antinuclear antibodies. The minor criteria were the presence of
chilblains, anti-Ro/anti-La antibodies and rheumatoid factor. A
positive diagnosis of Rowell syndrome is made in the presence of
all major criteria and at least one minor criterion.
An analysis of the 18 case reports of Rowell
syndrome diagnosed from 1963–2000 (11) revealed that total antinuclear
antibody positivity is the most important feature for the diagnosis
of Rowell syndrome, occurring in 88% of cases, of which 53% were
anti-Ro/anti-La antibodies. Rheumatoid factor was present in 41% of
cases (4,11). The immunologic abnormalities observed
in Rowell syndrome are also observed in subacute cutaneous lupus
erythematosus (SCLE) (4).
In recent years, the definition of Rowell syndrome
has become controversial, with certain researchers suggesting that
the association of lupus erythematosus and erythema multiforme may
be a coincidence (12), an
overlapping syndrome (4,12), a different variant of cutaneous lupus
erythematosus, a subtype of chronic lupus erythematosus or an
independent subtype of lupus erythematosus (4,12).
Classic erythema multiforme is triggered by various
factors, including infectious agents (Mycoplasma pneumonia and
herpes simplex virus), medicines (antibiotics, anticonvulsants,
nonsteroidal anti-inflammatory drugs and tuberculostatic drugs),
malignancies and diseases of the connective tissue (6,13).
Erythema multiforme is not associated with specific autoimmune
abnormalities (3,6).
Subacute lupus erythematosus (SCLE) is a specific
form of lupus erythematosus, in which patients primarily exhibit
cutaneous manifestations and typically have a good prognosis
(14,15). SCLE is an autoimmune disease
(16) characterized by the excessive
production of autoantibodies by activated B cells and autoreactive
T cells (17,18). SCLE is characterized by annular or
psoriasiform cutaneous lesions located above the waist (neck, trunk
and outer aspect of the arms) (14).
The following etiologic factors are associated with SCLE: Genetic
predisposition (human leukocyte antigen-DR3); immunological factors
(60% of cases are positive for total antinuclear antibodies, of
which 80% of cases are associated with the presence of anti-Ro
antibodies; environmental factors (exposure to ultraviolet
radiation, radiotherapy, psoralen combined with ultraviolet A
treatment and certain drugs, including thiazide diuretics, calcium
blockers, statins, inhibitors of angiotensin converting enzyme,
terbinafine and griseofulvin) (15,19–22); and
an epigenetics study has reported that autoreactive T cells and B
cells in patients with SCLE have evidence of altered patterns of
DNA methylation, modifications of histones and microRNA) (23).
The clinical and histological differentiation of
discoid lupus erythematosus from erythema multiforme is
challenging. The early skin lesions observed in SCLE are annular
and polycyclic, and may resemble erythema multiforme. Also, the
necrotic keratinocytes that are frequently present in erythema
multiforme can be present in SCLE (3,6). In the
early stages of erythema multiforme, there may be a swelling of the
endothelial cells and perivascular mononuclear infiltration,
followed by hydropic degeneration, keratinocyte necrosis and
subepidermal bubbling (12,13). SCLE is characterized by the presence
of hyperparakeratosis with follicular plugs, vacuolar changes at
the dermoepidermal junction, apoptotic keratinocytes, thinning of
the basal membrane, and perivascular and periadnexal lymphocytic
infiltrate (15,24).
Anatomopathologically, Rowell syndrome and erythema
multiforme are similar, with the presence of necrotic keratinocytes
(12). In terms of direct
immunofluorescence, Rowell syndrome and erythema multiforme exhibit
similar finding, including deposits of immunoglobulin M and C3 in
the dermal capillaries in the early stage, followed by granular C3
deposits along the dermoepidermal junction (12). Deposits of immunoglobulin A, G and M,
and C3, at the dermoepidermal junction have been identified in 60%
of patients with SCLE (12). Lupus
erythematosus and Rowell syndrome respond to the same therapeutic
regimen, including azathioprine, antimalarial drugs (particularly
hydroxychloroquine), prednisone, dapsone and cyclosporine (3,12).
Response to treatment is variable and frequent recurrences have
been reported (12).
The case discussed in the present reports meets all
the major and minor diagnostic criteria for Rowell syndrome,
excluding the presence of chilblains. The patient discusse in the
present study did not present with Raynaud syndrome, erythema
multiforme-like palmar-plantar involvement or bullous lupus
erythematosus lesions, as other cases of Rowell syndrome reported
in the literature have (11,24). The present case was not preceded by
signs of respiratory infection or herpes simplex virus type I or II
lesions, and serology for herpes infection was negative. Notably,
in the present case a drug was the triggering factor for SCLE and
Rowell syndrome lesions. The first SCLE episode was triggered by
terbinafine, for which adverse effects have previously been
reported in the literature (25).
The course of the disease in the present study was favorable under
treatment, with complete remission after ~1 year. However, after a
4-year-long interval, the SCLE reoccurred with the erythema
multiforme-like lesions, this time shortly after the introduction
of treatment to eradicate H. pylori infection.
The cases of Rowell syndrome reported in the
literature had a good prognosis with complete remission of skin
lesions within 1 year. In the present study, although the patient
entered remission for ~1 year initially, reoccurrence of the
disease occurred after a 4-year-long interval and the interval to
complete remission of lesions was double in the present study (~2
years). In addition, in order to avoid the fluctuating course with
relapse of the Rowell syndrome, proactive treatment with
hydroxychloriquine 200 mg/day was introduced throughout the summer
months of the year. Furthermore, in the reported case, the Rowell
syndrome was drug-induced. In addition, the typical Rowel syndrome
skin lesions, the patient continued to present with high levels of
anti-Ro antibodies, rheumatoid factor and total antinuclear
antibodies, though at much lower levels compared with at onset. The
high levels of these antibodies whilst under therapy highlights the
requirement for surveillance in Rowell syndrome, in addition to the
long-term and unpredictable course of the disease. Different
therapies studied for Rowell syndrome have produced variable
results (4,11,12),
highlighting the importance of developing novel and more effective
treatments. However, a more accurate understanding of the
pathophysiology of Rowell syndrome, in addition to a consensus
classification of Rowell syndrome as a distinct entity or a
particular form of lupus erythematosus/erythema multiforme is
required.
References
|
1
|
Kumar Y, Bhatia A and Minz RW: Antinuclear
antibodies and their detection methods in diagnosis of connective
tissue diseases: A journey revisited. Diagn Pathol. 4:12009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Borque L, Barozzi D and Ferrari L: The
determination of rhematoid factors by an immunoturbidimetric assay
on boehringer mannheim/hitachi analysis systems. Klin Lab.
40:445–453. 1994.
|
|
3
|
Yachoui R and Cronin P: Systemic lupus
erythematosus associated with erythema multiforme-like lesions.
Case Rep Rheumatol. 2013:2121452013.PubMed/NCBI
|
|
4
|
Bhat RY, Varna C, Bhatt S and Balachandran
C: Rowell syndrome. Indian Dermatol Online J. 5 Suppl 1:S33–S35.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aydin F, Senturk N, Yuksel EP, Yildiz L,
Canturk T and Turanli AY: Systemic lupus erythematosus with an
erythema multiforme-like lesions. Indian J Dermatol. 52:56–58.
2007. View Article : Google Scholar
|
|
6
|
Solanki D, Dalal E and Darji N: Case
report of Rowell's Syndrome. Int J Sci Res. 3:7–8. 2014.
|
|
7
|
Rowell NR, Beck JS and Anderson JR: Lupus
eythematosus and erythema multiforme-like lesions. A syndrome with
characteristic immunological abnormalities. Arch Dermatol.
88:176–180. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Antiga E, Caproni M, Bonciani D,
Bonciolini V and Fabbri P: The last word on the so-called ‘Rowell's
syndrome’? Lupus. 21:577–585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee S, Schloss E and Kowichi J: Rowell's
syndrome: A case report with subacute cutaneous lupus erythematosus
and erythema multiforme. Can J Dermatol. 7:807–810. 1995.
|
|
10
|
Zeitouni NC, Funaro D, Cloutier RA, Gagné
E and Claveau L: Redefining Rowell's syndrome. Brit J Dermatol.
142:343–346. 2000. View Article : Google Scholar
|
|
11
|
Lee A, Batra P, Furer V, Cheung W, Wang N
and Franks A Jr: Rowell syndrome (systemic lupus
erythematosus+erythema multiforme). Dermatol Online J.
15:12009.PubMed/NCBI
|
|
12
|
Andronache IT, Suta C, Ionescu C,
Calistrat A and Suta M: Rowell syndrome-a controversial clinical
entity. Rom J Rhematol. 24:230–234. 2015.
|
|
13
|
Creţu A, Dimitriu A, Brănişteanu D and
Brinişteanu DE: Erythema multiforme-etiopathogenic, clinical and
therapeutic aspects. Rev Med Chir Soc Med Nat Iasi. 119:55–61.
2015.PubMed/NCBI
|
|
14
|
Wolff K, Goldsmith LA, Katz S, Gilchrest
BA, Paller AS and Leffell D: Fitzpatrik's Dermatology in General
Medicine. 7th. USA: pp. 376–384. 2008
|
|
15
|
Branisteanu DE, Labontu A, Ciobanu D,
Stoleriu G, Branisteanu DC and Oanta A: Possible progression of
subacute lupus erythematosus-case report. Rev Med Chir Soc Med Nat.
118:381–386. 2014.
|
|
16
|
Gu Y, Zhu T, Wang Y and Xu H: Systemic
lupus erythematosus with intestinal perforation: A case report. Exp
Ther Med. 10:1234–1238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang B, Shi Y and Lei TC: Detection of
active P-glucoprotein in systemic lupus erythematosus patients with
poor disease control. Exp Ther Med. 4:705–710. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rao L, Liu G, Li C, Li Y, Wang Z, Zhou Z,
Tong S and Wu X: Specificity of anti-SSB as a diagnostic marker for
the classification of systemic lupus erythematosus. Exp Ther Med.
5:1710–1714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saurat JH, Lachapelle JM, Lipsker D and
Thomas D: Dermatologie et infections sexuellement transmissibles.
5th. Ed Tsunami; France: pp. 346–353. 2009
|
|
20
|
Burns T, Breathnach S, Copz N and
Griffiths C: Rook's Textbook of Dermatology. eighth. Blackwell
Publishing; USA: 51. pp. 22010
|
|
21
|
Zhu X, Li F, Yang B, Liang J, Qin H and Xu
J: Effects of ultraviolet B exposure on DNA methylation in patients
with systemic lupus erythematosus. Exp Ther Med. 5:1219–1225. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brănişteanu DE, Molodoi AD, Stătescu L,
Petrescu Z, Vasiluţ D, Anisiei E, Ferariu D and Brănişteanu D:
Chilblain lupus in an adolescent. Rev Med Chir Soc Med Nat Iasi.
112:646–651. 2008.(In Romanian). PubMed/NCBI
|
|
23
|
Xiao B and Zuo X: Epigenetics in systemic
lupus erythematosus. Biomed Rep. 4:135–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kempf W, Hantschke M, Kutzner H and
Burgdorf WHC: Dermatopathology. Steinkopff Verlag; Germany: pp.
662008
|
|
25
|
Bonsmann G, Schiller M, Luger TA and
Ständer S: Terbinafine-induced subacute cutaneous lupus
erythematosus. J Am Acad Dermatol. 44:925–931. 2001. View Article : Google Scholar : PubMed/NCBI
|