Introduction
Chronic obstructive pulmonary disease (COPD), which
is characterised by poor reversible airflow limitation, is a major
chronic lung disease and a global health problem (1,2). The
alveolar response to noxious substances, including cigarette smoke,
is considered to be a risk factor for the development of COPD
(3); however, the underlying
molecular mechanisms remain to be elucidated. Airway fibrosis has
been reported to contribute to physiological airway dysfunction in
COPD (4). Previous studies have
suggested that epithelial-mesenchymal transition (EMT), a feature
of lung fibrosis, may be important for the development of COPD
(5–8). De-differentiation of the respiratory
epithelium occurs via active EMT, however the underlying mechanism
and the association between EMT and peribronchial fibrosis remain
unclear (9). It was therefore
hypothesised that de-differentiation of the COPD respiratory
epithelium via EMT may serve a role in airway fibrosis and thereby
in airway obstruction.
It has been reported that the airway epithelium
provides frontline innate defence mechanisms as a physical barrier
and via the secretion of protective factors. Interleukin (IL)-17A
is an important cytokine that serves a vital role in a number of
chronic inflammatory diseases (10,11).
Previous studies have reported a greater number of
IL-17A+ cells and increased IL-17A secretion in the
submucosa of patients with COPD (12–14) and
have also investigated the expression of IL-17A and IL-17F in the
lung tissues of patients with stable COPD (15). Increased levels of growth
differentiation factor 15 (GDF15) have been reported in the airway
epithelium of smokers with COPD and in human airway epithelial
cells exposed to cigarette smoke; as such, elevated GDF15
expression may contribute to the progression of COPD.
The aim of the present study was to elucidate the
role of IL-17A and GDF15 in the pathogenesis of COPD. The results
demonstrated that IL-17A and GDF15 are upregulated in patients with
COPD, particularly in those with a history of smoking. IL-17A and
GDF15 expression is negatively correlated with epithelial
(E)-cadherin levels and positively correlated with the mesenchymal
marker vimentin in clinical specimens. The results also revealed
that treatment with cigarette smoke extract (CSE) and IL-17A was
able to induce GDF15 expression, while increased IL-17A and GDF15
expression caused a corresponding increase in EMT in human small
epithelial HSAEpiC cells in vitro. The results of the
present study demonstrate that IL-17A and GDF15-induced EMT serves
an important role in the pathology of COPD.
Materials and methods
Patients
A total of 143 patients with COPD (57 non-smokers,
86 smokers) and 75 patients without COPD (45 non-smokers, 30
smokers) were recruited from Hunan Provincial People's Hospital
during (Changsha, Hunan Province, China) from March 2014 to
September 2016. Patients were excluded from the present study if
they exhibited acute and chronic pulmonary diseases, including
bronchial asthma, bronchiectasis, interstitial lung disease, heart
disease and autoimmune disease. Patients were also excluded if they
possessed other systemic disorders, including orthopedic,
neurologic or unstable cardiac diseases, which may interfere with
results. Patients were recruited into the present study if they had
not received glucocorticoid and antibiotics within the last 3
months. A total of 99 male and 44 female patients with COPD and
average age of 56.4 years (range, 39–74 years) were recruited. In
addition, 43 male and 32 female patients without COPD were
recruited (average age, 52.5 years; range, 33–72 years). Lung
tissue samples were obtained following lobectomy or pneumonectomy
for various medical reasons, including solitary pulmonary nodules,
peripheral space-occupying lesions and peripheral lung cancer. COPD
was diagnosed according to the guidelines of the Global Initiative
for Chronic Obstructive Lung Disease (16) as follows: Non-COPD, forced expiratory
volume in 1 sec (FEV1)/forced vital capacity (FVC) ≥70% and FEV1
≥70%; COPD, FEV1/FVC <70% and FEV1 <70%. Patients with a
history of any other significant respiratory diseases were
excluded. Non-smokers were defined as those who had smoked on
average <1 cigarette/day for <1 year or had never smoked.
Smokers were defined as patients who had a smoking history of ≥1
year and had smoked on average ≥300 cigarettes/year. None of the
patients received corticosteroids prior to surgery. All experiments
were approved by the Ethics Committee of Hunan Provincial People's
Hospital and informed consent was obtained from all patients prior
to specimen collection.
Immunohistochemistry
Immunohistochemical (IHC) staining for IL-17A and
GDF15 proteins was performed using the labeled streptavidin-biotin
method with an LSAB kit (Dako; Agilent Technologies, Inc., Santa
Clara, CA, USA) according to the manufacturer's protocol. Surgical
specimens were fixed using 10% neutral-buffered formalin. Following
fixation for 24–48 h at room temperature, an initial 3–5 mm thick
section was routinely processed, paraffin embedded, deparaffinized
and rehydrated. 5-µm sections were then cut, placed on charged
slides and dried in an oven for 1 h at 56–60°C. Sections were then
stored in the dark at 2–8°C and used for the IHC assay. Sections
were then immersed in 0.01 Mcitric buffer (pH 6.0) and preheated to
97°C, for 30 min. The slides were incubated with 3% hydrogen
peroxide for 10 min at room temperature, followed by incubation in
normal goat serum (cat. no. ab7481; Abcam, Cambridge, UK) for 10
min at room temperature. The slides were incubated with antibodies
against IL-17A (ab217359; 1:200) and GDF15 (ab206414; 1:100; both
Abcam) at 37°C for 1 h, followed by incubation in Biotin-SP (long
spacer) AffiniPure Goat Anti-Rabbit Immunoglobulin G (IgG; 1:2,000;
cat. no. 111-065-008; Jackson ImmunoResearch Laboratories, Inc.,
West Grove, PA, USA) for 10 min at room temperature and
subsequently with streptavidin peroxidase for 10 min at room
temperature. Diaminobenzidene was employed as the chromogen for 5
min at room temperature. Samples were then counterstained with
hematoxylin at room temperature for 30 sec and coverslipped for
microscopic examination (50I; light microscope; Nikon Corporation,
Tokyo, Japan).
Cell culture
Human small airway epithelial cells (HSAEpiCs) were
obtained from ScienCellResearch Laboratories, Inc. (San Diego, CA,
USA; 3230) and cultured at 37°C in Small Airway Epithelial Cell
Medium (cat. no. 3131; ScienCell Research Laboratories, Inc.) in a
humidified atmosphere containing 5% carbon dioxide. To induce GDF15
overexpression, the recombinant expression vector pReceiver-Lv154
which contains the coding sequence of GDF15 (NM_004864) was bought
from GeneCopoeia, Inc. (Rockville, MD, USA). Lipofectamine™ 3000
(Thermo Fisher Scientific, Inc., Waltham, MA, USA; cat. no.
L3000015) was used to transfect cells with DNA complexes according
to the manufacturer's protocol. Cells were harvested for RNA and
protein extraction 72 h following transfection.
CSE preparation
CSE was prepared according to a previously published
protocol (17). CSE was made from
one brand of commercial cigarette (Baisha; Hunan Changsha Tobacco
Industrial Co., Ltd., Changsha, China) bubbled through 12.5 ml of
bronchial epithelium basal medium (cat. no. CC-3171; Lonza Group
Ltd., Basel, Switzerland), which was filtered through a 0.2-mm pore
filter. To ensure standardization between experiments and batches
of CSE, the absorbance was measured at 320 nm on a
spectrophotometer. An optical density of 1 was defined as 100%. CSE
was frozen in single use aliquots at −20°C. According to previous
reports, 1% CSE is the equivalent of 5 cigarettes/day, 3% is 15
cigarettes/day, 6% is 30 cigarettes/day and 10% is equal to 50
cigarettes/day, which corresponds to human daily exposure to
cigarette smoke (18,19). The cytotoxicity of CSE on primary
basal cells was measured using a Cytotoxicity Detection kit
1644793001; Roche Diagnostics GmbH, Mannheim, Germany).
Cell stimulation
Cells were seeded at a density of 1×106
onto 6-well plates and grown in bronchial epithelial growth medium
(BEGM; Lonza Group Ltd.) supplemented with a BEGM™ BulletKit™ (cat.
no. CC-3170; Lonza Group Ltd.) containing bovine pituitary extract,
insulin, hydrocortisone, gentamycin/amphotericin, retinoic acid,
transferrin, epinephrine and human epidermal growth factor (Lonza
Group Ltd.). The medium was supplemented with heat-inactivated
foetal bovine serum (cat. no. 10099141; Gibco; Thermo Fisher
Scientific, Inc.; 10% in the growth medium or 1% in starvation
medium). At confluence, cells were starved for 24 h (BEGM + 1%
FBS), then treated daily with IL-17A 00 ng/ml) and CSE (10%) for 4
days, or treated with varying concentrations of IL7A (10, 25, 50,
100 ng/ml) or CSE (1, 2.5, 5, 10, 15%) for 3 days.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. First strand cDNA was
synthesised using Superscript II reverse transcriptase (Invitrogen;
Thermo Fisher Scientific, Inc.). The reverse transcription
temperature protocol was 42°C for 50 min, followed by 70°C for 15
min. Real-time PCR reactions were performed using the SYBR-Green
Real time PCR Master Mix (Invitrogen; Thermo Fisher Scientific,
Inc.) and PCR was performed using an ABI PRISM 7900 HT Sequence
Detection System (Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: 95°C for 5 min, followed
by 95°C for 30 sec, 58°C for 30 sec and 40 cycles of 72°C for 10
sec. All reactions were performed in triplicate. Primers used for
the amplification of the indicated genes are listed in Table I. Results were normalised to
endogenous GAPDH and quantified using the 2−∆∆Cq method
(20).
 | Table I.Primers used in reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used in reverse
transcription-quantitative polymerase chain reaction.
| Genes | Direction | Primer sequences
(3′-5′) |
|---|
| Interleukin 17A | Forward |
ACAACCGATCCACCTCACCTT |
|
| Reverse |
CCCACGGACACCAGTATCTTCT |
|
Growth/differentiation factor 15 | Forward |
GTTGCGGAAACGCTACGAGGA |
|
| Reverse |
CGGAACAGAGCCCGGTGAAG |
|
Epithelial-cadherin | Forward |
CGCATTGCCACATACACTCTCT |
|
| Reverse |
GAGCACCTTCCATGACAGACC |
|
Neural-cadherin | Forward |
TCAGTGGCGGAGATCCTACTG |
|
| Reverse |
TTGACTGAGGCGGGTGCTGAA |
| Vimentin | Forward |
AACTAATCTGGATTCACTCCCTCTGC |
|
| Reverse |
GAGAAGTTTCGTTGATAACCTGTC |
| β-actin | Forward |
AGGGGCCGGACTCGTCATACT |
|
| Reverse |
GGCGGCACCACCATGTACCCT |
Western blot analysis
Cells were washed with PBS and lysed with RIPA lysis
buffer (cat. no. 89900; Invitrogen; Thermo Fisher Scientific)
containing a 10% protease inhibitor cocktail (cat. no. 78429;
Thermo Fisher Scientific, Inc.) on ice for 30 min. Protein
concentrations were determined using a BCA Protein Assay Reagent
kit (Thermo Fisher Scientific, Inc.). Aliquots of cell lysates
containing 20 µg protein were separated using 10%
SDS-polyacrylamide gel and transferred to polyvinylidene difluoride
membranes (Invitrogen; Thermo Fisher Scientific, Inc.). Subsequent
to blocking with bovine serum albumin (Sigma-Aldrich; Merck KGaA;
Darmstadt, Germany) at room temperature for 1 h, membranes were
incubated at 4°C overnight with primary antibodies against the
following: E-cadherin (1:500; cat. no. ab15148), N-cadherin
(1:1,000; cat. no. ab18203), Vimentin (1:1,500; cat. no. ab137321).
The anti-E-cadherin, N-cadherin and Vimentin antibodies were
obtained from Abcam. Subsequently, membranes were washed with
Tris-buffered saline and Tween-20 (Sigma-Aldrich; Merck KGaA) and
incubated with horseradish peroxidase-conjugated anti-Rabbit IgG
secondary antibodies (1:1,000; cat. no. A21253; Thermo Fisher
Scientific, Inc.). Immunoreactive proteins were detected using an
ECL kit (Pierce; Thermo Fisher Scientific, Inc.). The relative
protein expression was analyzed using Image-Pro plus software 6.0
(Media Cybernetics Inc., Rockville, MD, USA). The expression of
β-actin (1:5,000; cat. no. ab227387; Abcam) was used as an internal
control to normalize the expressions of other proteins.
Statistical analysis
Data were analysed using SPSS v.17.0 (SPSS, Inc.,
Chicago, IL, USA) and are presented as the mean ± standard
deviation. The non-parametric Mann Whitney U test was used for
comparisons-between two groups. One-way analysis of variance with a
post-hoc Tukey's test was used for comparisons between more than
two groups. The correlation between gene expression levels was
analysed using Pearson's correlation. P<0.05 was considered to
indicate a statistically significant difference.
Results
IL-17A and GDF15 are upregulated in
the lung tissues of patients with COPD and a history of
smoking
IHC staining and RT-qPCR were performed in 143
samples from patients with COPD and 75 samples from patients
without COPD. The results of IHC staining demonstrated that IL-17A
and GDF15 were markedly upregulated in lung tissues from patients
with COPD compared with non-COPD tissues (Fig. 1A). RT-qPCR revealed that IL-17A and
GDF15 expression was significantly increased in patients with COPD
compared with patients who did not have COPD (Fig. 1B and C). Furthermore, the expression
of IL-17A and GDF15 was significantly increased in smokers compared
with non-smokers in the COPD and non-COPD groups (Fig. 1B and C). A significant positive
correlation was identified between IL-17A and GDF15 expression
(Fig. 1D). These results suggest
that upregulated IL-17A and GDF15 expression may contribute to COPD
development, particularly in patients with a history of
smoking.
Correlation between IL-17A, GDF15 and
EMT markers
A correlation analysis was performed to assess the
association between IL-17A, GDF15 and EMT markers [E-cadherin,
neural (N)-cadherin and vimentin] in clinical specimens (Fig. 2). IL-17A expression was significantly
negatively correlated with E-cadherin (Fig. 2A), whereas it was significantly
positively correlated with N-cadherin (Fig. 2C) and vimentin (Fig. 2E). GDF15 expressionwas significantly
negatively correlated with E-cadherin (Fig. 2B) and significantly positively
correlated with vimentin (Fig. 2F).
No significant correlation was observed between GDF15 and
N-cadherin expression (Fig. 2D).
These results suggest a possible link between increased IL-17A and
GDF15 expression and EMT during the development of COPD.
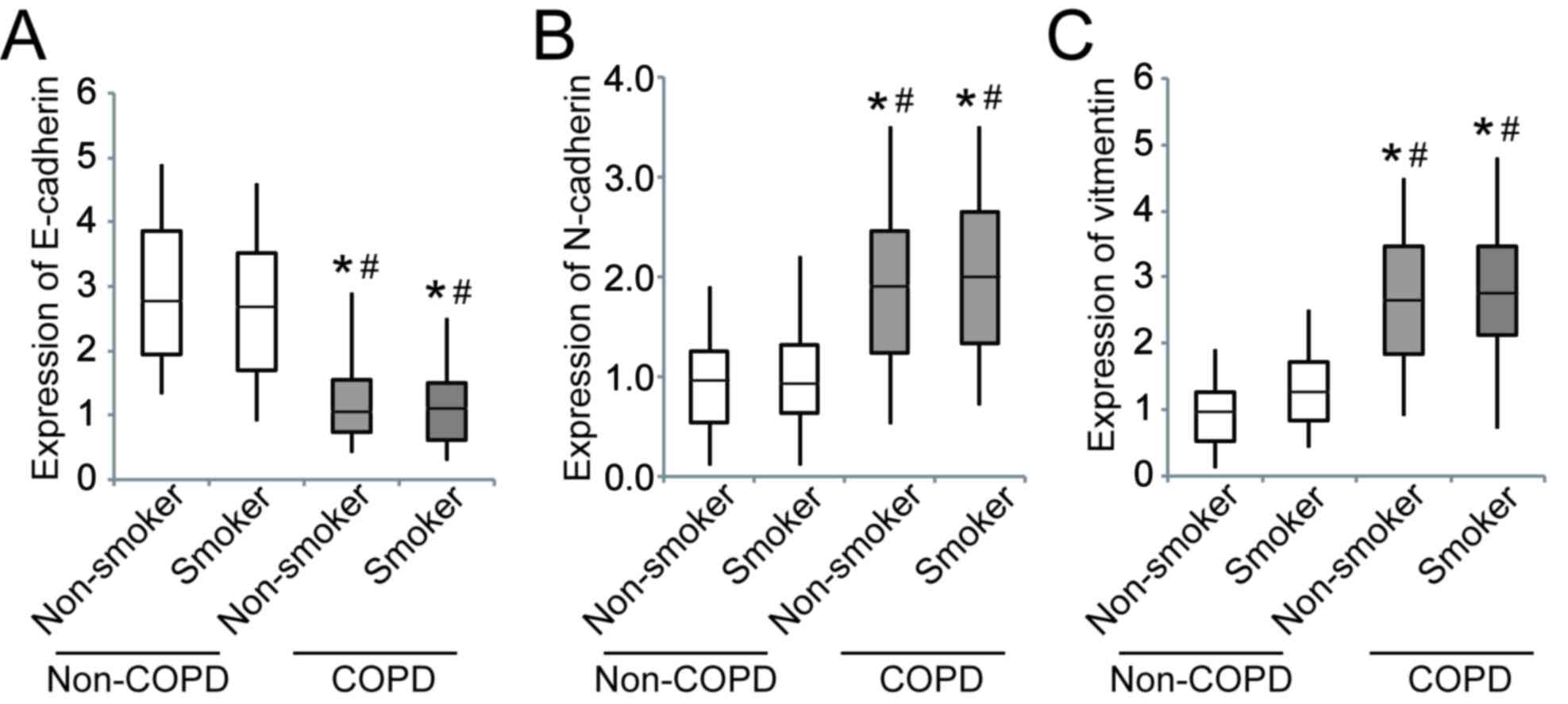
The expression of E-cadherin was significantly
downregulated in patients with COPD compared to those without COPD,
whereas N-cadherin and vimentin were significantly upregulated
(Fig. 3).
GDF15 is upregulated in CSE- and
IL-17A-treated HSAEpiC cells
To address whether smoking affects GDF15 expression,
HSAEpiCs were stimulated with CSE and assessed. The results
demonstrated that the expression GDF15 mRNA significantly increased
in a dose-dependent manner following CSE exposure for 3 days
(Fig. 4A). Additionally, GDF15
expression increased significantly with exposure to 10% CSE in a
time-dependent manner, reaching a peak following 3 days of exposure
(Fig. 4B). It was also demonstrated
that GDF15 expression was significantly upregulated by IL-17A in a
dose- and time-dependent manner (Fig. 4C
and D), suggesting that GDF15 expression is affected by both
cigarette smoke exposure and IL-17A treatment in HSAEpiC cells.
 | Figure 4.IL-17A and CSE induce GDF15
expression. (A) Cells were treated with 1, 2.5, 5, 10 or 15% CSE
for 3 days. (B) Cells were treated with 10% CSE for 1, 2, 3 or 4
days. (C) Cells were treated with 10, 25, 50 or 100 ng/ml IL-17A
for 3 days. (D) Cells were treated with 100 ng/ml IL-17A for 1, 2,
3 or 4 days. *P<0.05 vs. control. IL, interleukin; CSE,
cigarette smoke extract; GDF, growth/differentiation factor. |
IL-17A and GDF15 induce EMT in HSAEpiC
cells
To investigate the role of IL-17A and GDF15 in EMT,
HSAEpiC cells were transfected with a GDF15-overexpressing
lentivirus and treated with IL-17A (100 ng/ml) for 3 days. The
results revealed that HSAEpiC cells lost their honeycomb-like
epithelial cell morphology and became spindle shape, which is
indicative of EMT (Fig. 5A). RT-qPCR
and western blotting were performed to assess the expression of EMT
markers and the results demonstrated that E-cadherin was
downregulated while N-cadherin and vimentin were upregulated in
cells GDF15-overexpressing cells with or without IL-17A treatment
compared with control cells (Fig. 5B and
C). Furthermore, the combination of IL-17A treatment and GDF15
overexpression had a significantly greater effect on E-cadherin,
N-cadherin and vimentin expression compared with GDF15
overexpression alone (Fig. 5B and
C). These data suggest that IL-17A, in conjunction with GDF15,
is able to induce EMT in HSAEpiC cells.
Discussion
The results of the present study demonstrate that
IL-17A and GDF15 are upregulated in patients with COPD,
particularly in those with a history of smoking, and that IL-17A
and GDF15 expression levels are positively correlated with EMT
progression. Furthermore, IL-17A treatment in conjunction with
CSE-induced GDF15 overexpression triggered EMT in HSAEpiC cells
in vitro.
It has been reported that cigarette smoking
contributes to lung remodelling and peribronchiolar fibrosis in the
small airways, leading to airway obstruction (5). The formation of peribronchiolar
fibrosis is associated with EMT (8),
while CSE exposure is able to induce EMT in human bronchial
epithelial cells (6). COPD has a
complex aetiology and is characterised by a combination of airway
and lung parenchymal damage (21).
The earliest changes in the small airways appear to be due to
active EMT, which is associated with a cascade of changes in the
expression of regulators, including Smads, Twist and β-catenin
(7). Proinflammatory cytokines,
including tumour necrosis factor-α, interferon-γ and IL-17, serve
an important role in the pathobiology of COPD (22). There is also evidence that high
levels of the aforementioned cytokines are responsible for the
clinical manifestations of COPD (22,23).
Elevated IL-17 expression has been reported in the lungs of
patients with COPD; this elevation was attributed to cigarette
smoke exposure-dependent effects on the nuclear factor-κB and the
phosphoinositide3-kinase pathways (24). Additionally, a combination of IL-17A
and CSE stimulation has been reported to affect the proliferation
of human epithelial cells and cigarette smoke increases T helper-17
immunity in the lung tissue of patients with COPD (15). In IL-17A knockout mice exposed to
cigarette smoke, lymphoid neogenesis was attenuated and chemokine
C-X-C motif ligand (CXCL)12 expression was reduced, suggesting that
IL-17A contributes to COPD disease progression and the development
of lymphoid follicles by activating CXCL12 (25).
It has been reported that the number of
CD4+IL-17+ cells is higher in patients with
COPD compared with non-smokers, as well as in healthy smokers
compared with non-smokers (26). The
increase in CD4+IL-17+ cells was positively
correlated with pathological changes and airflow limitations
(27,28). However, Freeman et al
(23) reported a decrease in
CD4+ and CD8+ T cells in the peripheral blood
during acute exacerbations of COPD, indicating T cell extravasation
into inflammatory sites. They also reported that GDF15 is a
sensitive marker of cardiopulmonary stress that greatly increases
during acute exacerbations of COPD (29). Elevated serum GDF15 levels have been
demonstrated to independently predict adverse outcomes in patients
with exacerbated COPD (30);
furthermore, circulating GDF15 is inversely correlated with rectus
femora's cross-sectional area and exercise capacity in patients
with COPD (31). This suggests that
GDF15 overexpression may be associated with a higher frequency of
exacerbations and increased mortality in patients with COPD
(32). Importantly, a GDF15
deficiency attenuated cigarette smoke-induced pulmonary
inflammation (33) and disruption of
GDF15 expression significantly inhibited CSE-induced airway
epithelial senescence via activation of the Smad1 pathway (34). The results of the present study
revealed that GDF15 expression in HSAEpiC cells was significantly
upregulated by IL-17A in a dose- and time-dependent manner.
Furthermore, IL-17A in conjunction with GDF15 activated EMT in
HSAEpiC cells, which may explain the correlation between IL-17A,
GDF15 and EMT markers in clinical specimens. In future studies,
knocking out GDF15 and IL-17A is necessary to further determine
whether GDF15 and IL-17A are effectors of smoking on COPD.
Furthermore, whether monoclonal antibodies for GDF15 and IL-17A
diminish the effects of tobacco on airway and lung parenchymal
damage in vivo should be assessed in further studies.
In summary, the results of the present study
highlight the role of IL-17A and GDF15 in the development of COPD
following exposure to cigarette smoke. IL-17A and GDF15-induced EMT
was demonstrated to serve an important role in the etiopathology of
COPD, which suggests that IL-17A and GDF15 suppression may be an
effective therapeutic treatment for COPD. However, more in
vivo studies should be conducted in the future.
Acknowledgements
The authors would like to thank Mrs Merissa E.
Garvey for language modifications.
Funding
The present study was supported by the Hunan
Provincial Natural Science Foundation of China (grant no.
2015JJ2091), the Scientific Research Foundation from Ministry
Education of Hunan Province of China (grant no. 15C0832) and the
Hunan Provincial Health and Family planning commission of China
(grant no. B2017078).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GJ conceived and designed the experiments and wrote
the manuscript; CTL performed the experiments and analyzed the
data; WDZ contributed in collecting clinical tissue samples,
performing the IHC assay and revising the manuscript.
Ethics approval and consent to
participate
Ethical approval for the study was granted from the
Hunan Provincial People's Hospital Ethics Committee and all
patients gave written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4422006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sohal SS, Ward C, Danial W, Wood-Baker R
and Walters EH: Recent advances in understanding inflammation and
remodeling in the airways in chronic obstructive pulmonary disease.
Expert Rev Respir Med. 7:275–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hogg JC, Chu F, Utokaparch S, Woods R,
Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson
HO and Paré PD: The nature of small-airway obstruction in chronic
obstructive pulmonary disease. N Engl J Med. 350:2645–2653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sohal SS and Walters EH: Role of
epithelial mesenchymal transition (EMT) in chronic obstructive
pulmonary disease (COPD). Respir Res. 14:1202013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milara J, Peiró T, Serrano A and Cortijo
J: Epithelial to mesenchymal transition is increased in patients
with COPD and induced by cigarette smoke. Thorax. 68:410–420. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Q, Wang Y, Zhang Y, Zhang Y and Xiao
W: The role of uPAR in epithelial-mesenchymal transition in small
airway epithelium of patients with chronic obstructive pulmonary
disease. Respir Res. 14:672013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nowrin K, Sohal SS, Peterson G, Patel R
and Walters EH: Epithelial-mesenchymal transition as a fundamental
underlying pathogenic process in COPD airways: Fibrosis, remodeling
and cancer. Expert Rev Respir Med. 8:547–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sohal SS, Mahmood MQ and Walters EH:
Clinical significance of epithelial mesenchymal transition (EMT) in
chronic obstructive pulmonary disease (COPD): Potential target for
prevention of airway fibrosis and lung cancer. Clin Transl Med.
3:332014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gohy ST, Hupin C, Fregimilicka C, Detry
BR, Bouzin C, Chevronay Gaide H, Lecocq M, Weynand B, Ladjemi MZ,
Pierreux CE, et al: Imprinting of the COPD airway epithelium for
dedifferentiation and mesenchymal transition. Eur Respir J.
45:1258–1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holgate ST: The sentinel role of the
airway epithelium in asthma pathogenesis. Immunol Rev. 242:205–219.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Isailovic N, Daigo K, Mantovani A and
Selmi C: Interleukin-17 and innate immunity in infections and
chronic inflammation. J Autoimmun. 60:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Stefano A, Caramori G, Gnemmi I,
Contoli M, Vicari C, Capelli A, Magno F, D'Anna SE, Zanini A, Brun
P, et al: T helper type 17-related cytokine expression is increased
in the bronchial mucosa of stable chronic obstructive pulmonary
disease patients. Clin Exp Immunol. 157:316–324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Doe C, Bafadhel M, Siddiqui S, Desai D,
Mistry V, Rugman P, McCormick M, Woods J, May R, Sleeman MA, et al:
Expression of the T helper 17-associated cytokines IL-17A and
IL-17F in asthma and COPD. Chest. 138:1140–1147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eustace A, Smyth LJC, Mitchell L,
Williamson K, Plumb J and Singh D: Identification of cells
expressing IL-17A and IL-17F in the lungs of patients with COPD.
Chest. 139:1089–1100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montalbano AM, Riccobono L, Siena L,
Chiappara G, Di Sano C, Anzalone G, Gagliardo R, Ricciardolo FLM,
Sorbello V, Pipitone L, et al: Cigarette smoke affects IL-17A,
IL-17F and IL-17 receptor expression in the lung tissue: Ex vivo
and in vitro studies. Cytokine. 76:391–402. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baker CL, Zou KH and Su J: Long-acting
bronchodilator use after hospitalization for COPD: An observational
study of health insurance claims data. Int J Chron Obstruct Pulmon
Dis. 9:431–439. 2014.PubMed/NCBI
|
|
17
|
Wang G, Zhou H, Strulovici-Barel Y,
Al-Hijji M, Ou X, Salit J, Walters MS, Staudt MR, Kaner RJ and
Crystal RG: Role of OSGIN1 in mediating smoking-induced autophagy
in the human airway epithelium. Autophagy. 13:1205–1220. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hogan AE, Corrigan MA, O'Reilly V,
Gaoatswe G, O'Connell J, Doherty DG, Lynch L and O'Shea D:
Cigarette smoke alters the invariant natural killer T cell function
and may inhibit anti-tumor responses. Clin Immunol. 140:229–235.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kosmider B, Messier EM, Chu HW and Mason
RJ: Human alveolar epithelial cell injury induced by cigarette
smoke. PLoS One. 6:e260592011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nowrin K, Sohal SS, Peterson G, Patel R
and Walters EH: Epithelial-mesenchymal transition as a fundamental
underlying pathogenic process in COPD airways: Fibrosis, remodeling
and cancer. Expert Rev Respir Med. 8:547–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Caramori G, Adcock IM, Di Stefano A and
Chung KF: Cytokine inhibition in the treatment of COPD. Int J Chron
Obstruct Pulmon Dis. 9:397–412. 2014.PubMed/NCBI
|
|
23
|
Zhang X, Zheng H, Zhang H, Ma W, Wang F,
Liu C and He S: Increased interleukin (IL)-8 and decreased IL-17
production in chronic obstructive pulmonary disease (COPD) provoked
by cigarette smoke. Cytokine. 56:717–725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chang Y, Al-Alwan L, Alshakfa S, Audusseau
S, Mogas AK, Chouiali F, Nair P, Baglole CJ, Hamid Q and Eidelman
DH: Upregulation of IL-17A/F from human lung tissue explants with
cigarette smoke exposure: Implications for COPD. Respir Res.
15:1452014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roos AB, Sandén C, Mori M, Bjermer L,
Stampfli MR and Erjefält JS: IL-17A is elevated in End-stage
chronic obstructive pulmonary disease and contributes to cigarette
smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med.
191:1232–1241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang J, Chu S, Zhong X, Lao Q, He Z and
Liang Y: Increased expression of CD4+IL-17+ cells in the lung
tissue of patients with stable chronic obstructive pulmonary
disease (COPD) and smokers. Int Immunopharmacol. 15:58–66. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Chu S, Zhong X, Lao Q, He Z and
Liang Y: Increased expression of CD4+IL-17+ cells in the lung
tissue of patients with stable chronic obstructive pulmonary
disease (COPD) and smokers. Int Immunopharmacol. 15:58–66. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin Y, Wan Y, Chen G, Chen L, Zhang MQ,
Deng L, Zhang JC, Xiong XZ and Xin JB: Treg/IL-17 ratio and Treg
differentiation in patients with COPD. PLoS One. 9:e1110442014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Q, Jiang D and Chu HW: Cigarette smoke
induces growth differentiation factor 15 production in human lung
epithelial cells: Implication in mucin over-expression. Innate
Immun. 18:617–626. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim M, Cha SI, Choi KJ, Shin KM, Lim JK,
Yoo SS, Lee J, Lee SY, Kim CH, Park JY, et al: Prognostic value of
serum growth differentiation factor-15 in patients with chronic
obstructive pulmonary disease exacerbation. Tuberc Respir Dis
(Seoul). 77:243–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patel MS, Lee J, Baz M, Wells CE, Bloch S,
Lewis A, Donaldson AV, Garfield BE, Hopkinson NS, Natanek A, et al:
Growth differentiation factor-15 is associated with muscle mass in
chronic obstructive pulmonary disease and promotes muscle wasting
in vivo. J Cachexia Sarcopenia Muscle. 7:436–448. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Husebo GR, Grønseth R, Lerner L, Gyuris J,
Hardie JA, Bakke PS and Eagan TM: Growth differentiation factor-15
is a predictor of important disease outcomes in patients with COPD.
Eur Respir J. 49:pii: 1601298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Verhamme FM, Seys LJ, De Smet EG, Provoost
S, Janssens W, Elewaut D, Joos GF, Brusselle GG and Bracke KR:
Elevated GDF-15 contributes to pulmonary inflammation upon
cigarette smoke exposure. Mucosal Immunol. 10:1400–1411. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Freeman CM, Martinez CH, Todt JC, Martinez
FJ, Han MK, Thompson DL, McCloskey L and Curtis JL: Acute
exacerbations of chronic obstructive pulmonary disease are
associated with decreased CD4+ &; CD8+ T cells and increased
growth &; differentiation factor-15 (GDF-15) in peripheral
blood. Respir Res. 16:942015. View Article : Google Scholar : PubMed/NCBI
|