Introduction
Infection by pathogenic microorganisms and viruses
presents a significant threat to human life worldwide; their
constant variation, evolution and spread render it difficult to
prevent and control infection. HBV, one of the most infectious
diseases worldwide, often varies due to the pressures of host
immunity, natural selection and the use of antiviral agents. Such
variations may cause changes in HBV pathogenicity, including the
development of tolerance and immune escape, and have greatly
hindered clinical diagnosis and treatment (1). A number of new HIV GAG subtypes have
been reported due to its constant variation and accumulation
(2). Continual variation in the
influenza virus hemagglutinin (HA) antigen gene is the main cause
of influenza outbreaks (3). This
poses challenges for immunology, virology and immunopharmacology
research, and for the development of vaccines against influenza and
other pathogenic microorganisms.
Epitopes, also known as antigenic determinants,
represent the material base of immunogen antigenicity, and is the
part of an antigen recognized by the immune system. Epitopes can be
classified as either conformational epitopes or linear epitopes,
based on their structure and interaction with the paratope
(4). The linear epitope is a section
of the continual amino acid sequence of the antigen, and its
interaction with the paratope predominantly depends on its primary
structure. Variations in any area of the linear epitopes may lead
to structural changes, a reduced antibody binding ability, and the
ability to escape recognition by existing antibodies and vaccines
(5).
Different subtypes of a pathogen may have a variety
of antigens; thus, it is challenging to distinguish the subtype of
pathogenic microorganisms, to establish immunodetection
technologies, and to clarify the mechanisms of disease spread.
Consequently, epitope prediction and utilization are of value in
differential diagnosis, the prediction of variation trends,
determining the mechanisms of pathogenic microorganism infection,
and in the design of multi-epitope vaccines (6).
Recently, several methods of epitope prediction have
been in use, the majority of which are limited to one antigen,
although they still provide a satisfactory predictive capacity
(7–9). X-ray diffraction requires more time and
energy to identify epitope structures. To elucidate the biological
profile of the epitope, multiple factors should be considered,
including its location on the surface of the antigen, the
flexibility, and the accessibility, although it showed a growing
acceptance among this field (10–14). In
addition to α-helices and β-pleated sheets, glycosylation sites are
also important for prediction (15).
However, the predictive accuracy of these methods is just ~60%
(16). Larger protein libraries are
required for phage display technology, and certain peptides have
strong hydrophobicity, which influences their structure on the
surface of phages. Furthermore, the predictions obtained via this
method still require further verification (17). Thus, a single optimal approach is
required, which is capable of predicting the epitope sequences of
microorganisms comprehensively and in one pass, establishing a
biological profile with the characteristics and functions of the
epitopes, and modeling the behavior of these epitopes during
changes to virus antigenicity. This will have an important and
direct role in the design of biologically active drugs, research
into pathogenic mechanisms, and the prediction of variation in
certain pathogenic microorganisms.
Monoclonal antibodies (mAbs) are a subset of
antibodies generated by identical immune cells with a strong
monovalent affinity, in that they bind to the same epitope, with
high specificity and sensitivity, and define the structure and
character of epitopes (18). Such
specificity can also be used as a tool to analyze the epitopes of
viruses and their subtypes, provide information on the main
functions of the epitopes and on genetic variations involved in
changes to the epitopes, and assist research into epitope variation
and improvements in vaccine design (19,20).
In the present study, mAbs from 40 previously
developed anti-H1N1 influenza virus HA split vaccines had been
developed and characterized (21),
which were used as experimental tools to predict the epitopes of
influenza virus HA proteins, after which their distribution and
expression were investigated using synthesized peptides. The
present study aimed to illuminate the association between variation
in the influenza virus and its immunogenicity, and to develop a
useful method for predicting the variable epitopes of other
pathogenic microorganisms. In the present study, we just
preliminary report a new method for predicting the variability
epitope of influenza virus. Next, we will carry out biological
functional studies on predicted different epitopes one by one,
which can help us to develop epitope vaccines of influenza virus,
further contribute to the diagnosis and prevention of influenza
virus.
Materials and methods
Antigens
H1N1 influenza virus split vaccine (2009; SFDA
Approval no.: S20090015) was obtained from Hualan Biological
Bacterin Co., Ltd., (Henan, China); seasonal A1 and A3 influenza
[2009; Veterinary Drug Production Approval no.: 150132145], and
H9N2 (SD696) strains were purchased from Qingdao Yebio
Bioengineering Co., Ltd., (Shandong, China).
Antibodies
mAbs against the anti-H1N1 A influenza virus HA
protein were prepared in our laboratory, and HRP-conjugated goat
anti-mouse antibodies were provided by Beijing Zhongshan Golden
Bridge Biotechnology Co., Ltd., (Beijing, China).
HA protein synthetic peptides
Part of the continuous amino acid sequence of
influenza virus HA was determined using DNAMAN software, and
peptides were synthesized by ChinaPeptides Co., Ltd., (Shanghai,
China).
ELISA analysis and classification
Indirect ELISA analyses were performed using the
following: Hybridoma culture supernatant; H1N1 influenza virus
split vaccine (2009); seasonal influenza viruses A1 and A3; and
avian influenza viruses H5N1 and H9N2. Briefly, the 96-well plate
was pre-coated with 100 µl of each vaccine (2–5 µg/ml). After
washing three times with PBST (including 8 g of NaCl, 0.2 g of KCl,
1.44 g of Na2HPO4, 0.24 g of
KH2PO4, 2 ml of Tween-20, pH 7.2, volume
adjusted to 1L with additional distilled H2O), the
plates were blocked with 200 µl skim milk (dilution, 1:20) and
incubated for 1 h at 37°C. Subsequently, 100 µl/well supernatant
aspirated from the hybridoma cell cultures for 40 mAbs was added,
including the supernatant of SP2/0 as a negative control, which was
incubated for 1 h at 37°C. After washing a further three times, the
concentration (dilution, 1:2,500) of the HRP-labeled
goat-anti-mouse IgG mAb (100 µl/well) was added and incubated for 1
h at 37°C. Next, 100 µl TMB-H2O2 chromogenic
solution was added to each well and incubated for 10 min at 37°C in
the dark, and terminated with H2SO4 solution
(2 M, 50 µl/well). Finally, the proportion of bound antibodies,
which is correlated with the color intensity, was measured with an
ELISA reader via absorbance at 450 nm. The ratio of each test
sample (OD450: Control OD450) was calculated.
Samples with a ratio of ≥2.1 were classified as exhibiting a
positive reaction. Considering each test sample reaction with the
five subtypes of the influenza virus, the antibodies were
categorized into different groups.
Epitopes of influenza A virus HA
protein prediction
In the NCBI database (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html),
the amino acid sequences of various influenza virus subtypes were
accessed and downloaded with their GenBank IDs (Table I). Consequently, a multiple sequence
alignment analysis was performed using DNAMAN software, following
which the common continuous amino acid sequence (5–7 aa) between
the antigens of the different groups were defined, and used to
predict the epitopes of influenza A virus HA proteins. Overall, 27
candidate epitope fragments were selected, and complementary
peptides were synthesized, each with a >85% purity as measured
by HPLC and MS methods; these peptides were stored as freeze-dried
powders at −20°C.
 | Table I.Information about the amino acid
sequences of subtype influenza virus. |
Table I.
Information about the amino acid
sequences of subtype influenza virus.
| Name of
antigens | Source of HA amino
acid sequence | GenBank ID |
|---|
| 2009 H1N1-HA |
(A/reassortant/NYMCX-179A
(California/07/2009×NYMC X-157)(H1N1)) | ACR47014.1 |
| H3N2-HA | Influenza A virus
(A/Victoria/210/2009(H3N2)) | CY121077.1 |
| Seasonal
H1N1-HA | Influenza A virus
(A/Brisbane/59/2007(H1N1)) | CY163864.1 |
| H5N1-HA | Influenza A virus
(A/Goose/Guangdong/1/96(H5N1)) | AF144305.1 |
| H9N2-HA | Influenza A virus
(A/chicken/Shandong/6/96(H9N2)) | AAY52514.1 |
Localization of predicted epitopes
with anti-influenza virus HA mAbs
To investigate the positions of the predicted
epitopes of influenza A virus HA, 27 candidate-epitope peptides
were used and screened using mAbs against influenza virus HA. The
process was as follows: The synthesized peptides were mixed with 40
HA mAbs, and incubated for 1 h at 37°C. A total of 100 µl mixed
reagent was placed into each well of an ELISA plate pre-coated with
H1N1 influenza virus HA antigens (2 µg/ml), according to standard
ELISA protocols. After a 1 h incubation and three washes, the goat
anti-mouse antibodies (dilution, 1:2,500) were added, and the steps
of a conventional ELISA were performed. The OD450 values
for all wells were calculated from TMB coloration and an inhibition
rate (IR) was calculated. The formula used to calculate the IR was
as follows:
IR=(ODCTL-ODTEST)/ODCTL.
Correlations between the antigens and the antibody binding sites
were defined according to the following criteria: No correlation
(IR ≤0.4); correlation (0.4≤ IR ≤0.8); and strong correlation (IR
≥0.8).
Distribution of predicted epitopes in
the HA crystal structure
The PyMOL Molecular Graphics System (http://www.PyMOL.org) and Protein Database (PDB) were
used to analyze the distribution of predicted epitopes in the HA
crystal structure. Peptides recognized by mAbs against influenza
virus HA proteins in the ELISA experiments were selected and
analyzed. First, the PDB database was used to search for and
generate a model of the HA protein X-ray crystal structure by
referring to the 3LZG structure, which was produced from the
A/California/04/2009 H1N1 virus HA and had a similar structure to
that of the antigen in the present study. Secondly, the selected
peptides' distributions were determined using PyMOL software
according to the manufacturer's protocol.
Results
Specificity and cross reactivity of
mAbs
ELISA reactions between 40 influenza virus HA
antigen mAbs and five different influenza virus subtype vaccines
were evaluated using the OD450 ratio, and classified as
positive (OD450 ≥2.1) or negative (OD450
<2.1) reactions. According to the cross-ELISA results, all the
assessed influenza virus HA antigens can be classified into three
groups. Approximately half (20/40) were recognized by all five
antigens, ~35% (14/40) were recognized by the antigens of 2009 H1N1
virus A, or seasonal influenza virus A1 and A3, and 6 mAbs only
reacted with the antigens of H1N1 virus A and seasonal A1 (Table II).
| Table II.mAb cross-reactivity with various
subtypes of influenza virus. |
Table II.
mAb cross-reactivity with various
subtypes of influenza virus.
| mAb group | No. of cell
lines |
|---|
| Common antigens of
influenza virus | 20 |
| (2009 H1N1 and
seasonal A1, A3 and avian influenza H5N1 and H9N2) |
|
| Common antigens of
2009 H1N1 influenza virus and seasonal influenza virus | 14 |
| (2009 H1N1 and
seasonal A1, A3) |
|
| Specific H1
subtype | 6 |
| (2009 H1N1and
seasonal A1) |
|
| Total | 40 |
Detection of conserved peptides in
influenza virus A HA
Twenty seven common continuous amino acid sequences
of influenza HA antigens detected through multiple sequence
alignment analysis of the three groups using DNAMAN software
(Table III). There were 9 peptides
located in the conserved sequences of vaccines in group 1, 7
peptides in the conserved sequences of group 2, and 11 in group 3
(Table III).
| Table III.Peptide fragments in influenza virus
HA identified subtype influenza virus mAbs. |
Table III.
Peptide fragments in influenza virus
HA identified subtype influenza virus mAbs.
| Groups and peptides
no. | Sequence of
peptides | Position |
|---|
| Group 1:
(9)a |
|
|
|
Peptide | LVLWGIHHP | 191aa-199aa |
| Peptide
2 | LPFQNI | 307aa-312aa |
| Peptide
3 | LATGLRN | 331aa-337aa |
| Peptide
4 |
RGLFGAIAGFIEGGW | 344aa-358aa |
| Peptide
5 | GWYGYHH | 364aa-370aa |
| Peptide
6 | STQNAID | 384aa-390aa |
| Peptide
7 | YNAELLVL | 438aa-445aa |
| Peptide
8 | ENERTLD | 447aa-453aa |
| Peptide
9 | WSYIVE | 93aa-98aa |
| Group 2:
(7)b |
|
|
| Peptide
10 |
DTLCIGYHANNSTDT | 17aa-32aa |
| Peptide
11 | MNYYWTLVEPGD | 244aa-255aa |
| Peptide
12 | ATGNLVVPR | 261aa-269aa |
| Peptide
13 |
GYAADLKSTQNAIDEI | 377aa-392aa |
| Peptide
14 | EIGNGCF | 476aa-482aa |
| Peptide
15 | FYHKCDNT | 484aa-491aa |
| Peptide
16 | SVKNGTYD | 495aa-502aa |
| Group 3:
(11)c |
|
|
| Peptide
17 | KAILVVLLYTFA | 2aa-13aa |
| Peptide
18 | SVNLLEDK | 46aa-53aa |
| Peptide
19 | KLRGVAPLHLGK | 60aa-71aa |
| Peptide
20 | ESLSTASS | 85aa-92aa |
| Peptide
21 | TSSSDNGT | 99aa-106aa |
| Peptide
22 | PNHDSNKGVTA | 141aa-151aa |
| Peptide
23 | PHAGAKSFYKNLI | 154aa-166aa |
| Peptide
24 | KLSKSYINDKGKEV | 177aa-190aa |
| Peptide
25 | GSSRYSKKFKPE | 219aa-230aa |
| Peptide
26 | RYAFAMERNAGSG | 269aa-281aa |
| Peptide
27 | VVSLGAISF | 544aa-552aa |
Locations of predicted epitopes
determined using anti-influenza virus HA mAbs
The ELISA results demonstrated that 9/27 peptides
were recognized by 13/40 mAbs, considering their IRs calculated
with OD450 values (Figs.
1–3). In group 1, 5 mAbs were
identified by 3 peptides, designated peptides 1, 2 and 9 (Fig. 1); in group 2, 6 mAbs reacted with 4
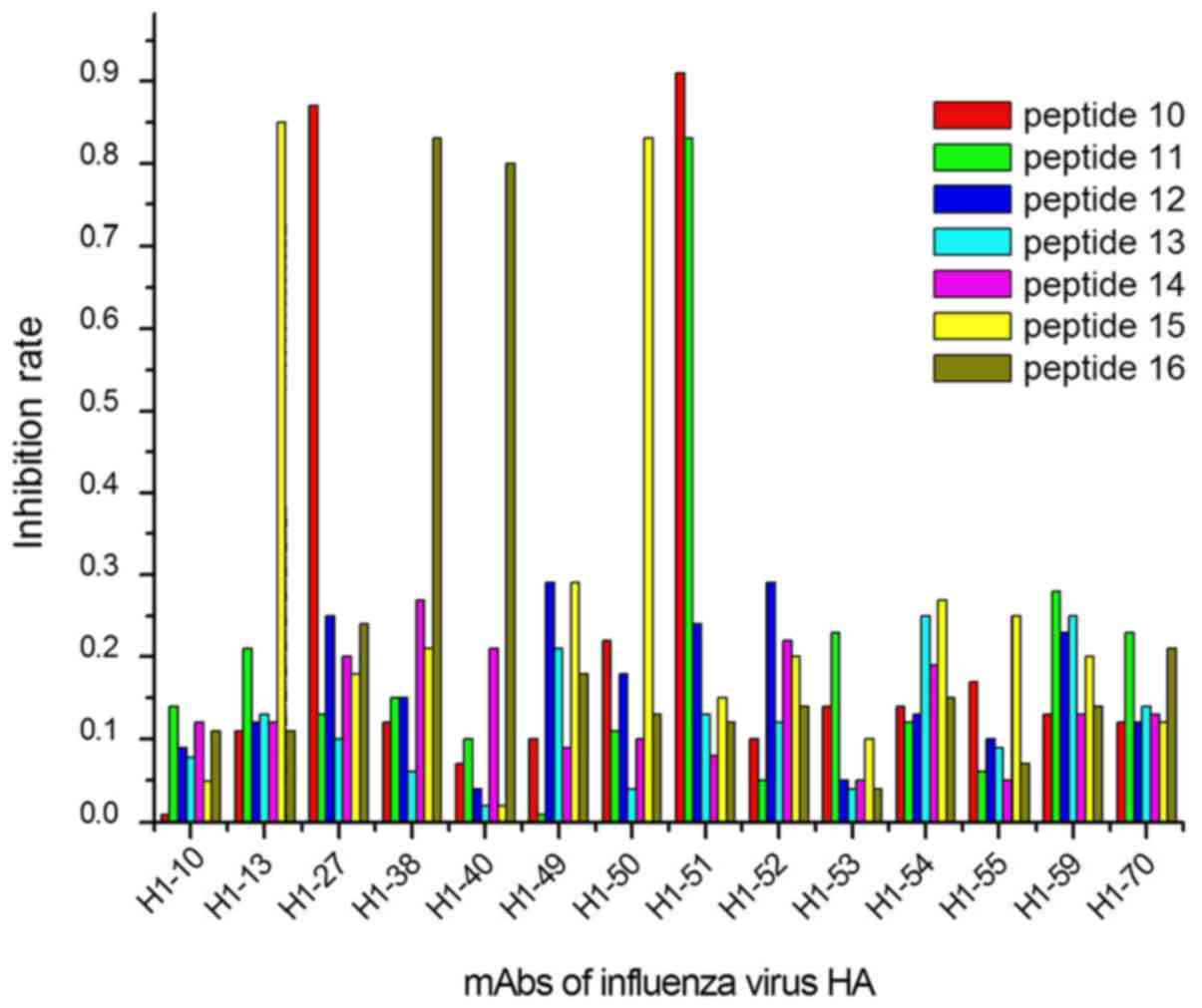
peptides (peptides 10, 11, 15 and 16; Fig. 2); and 2 mAbs in group 3 were
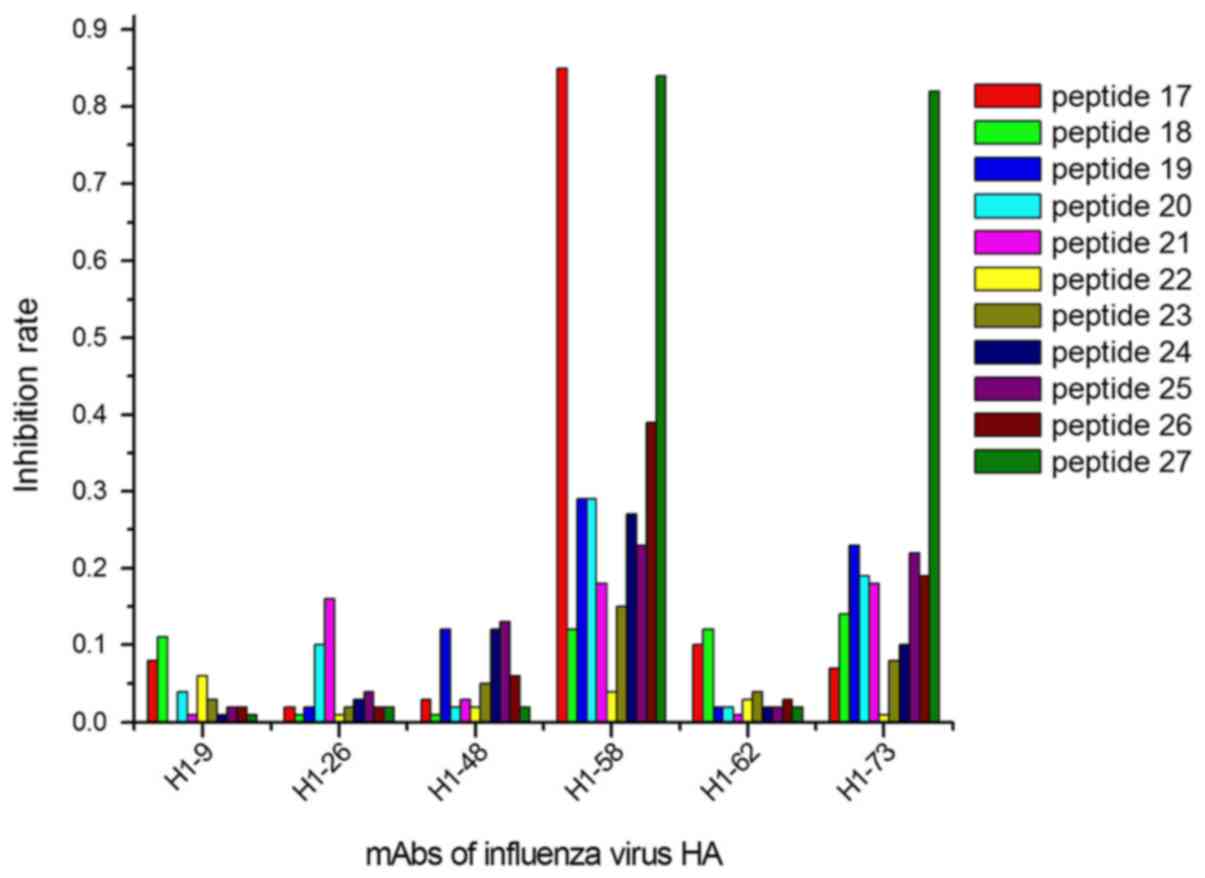
identified by 2 peptides (peptides 17 and 27; Fig. 3).
Distribution of predicted epitopes in
the HA crystal structure
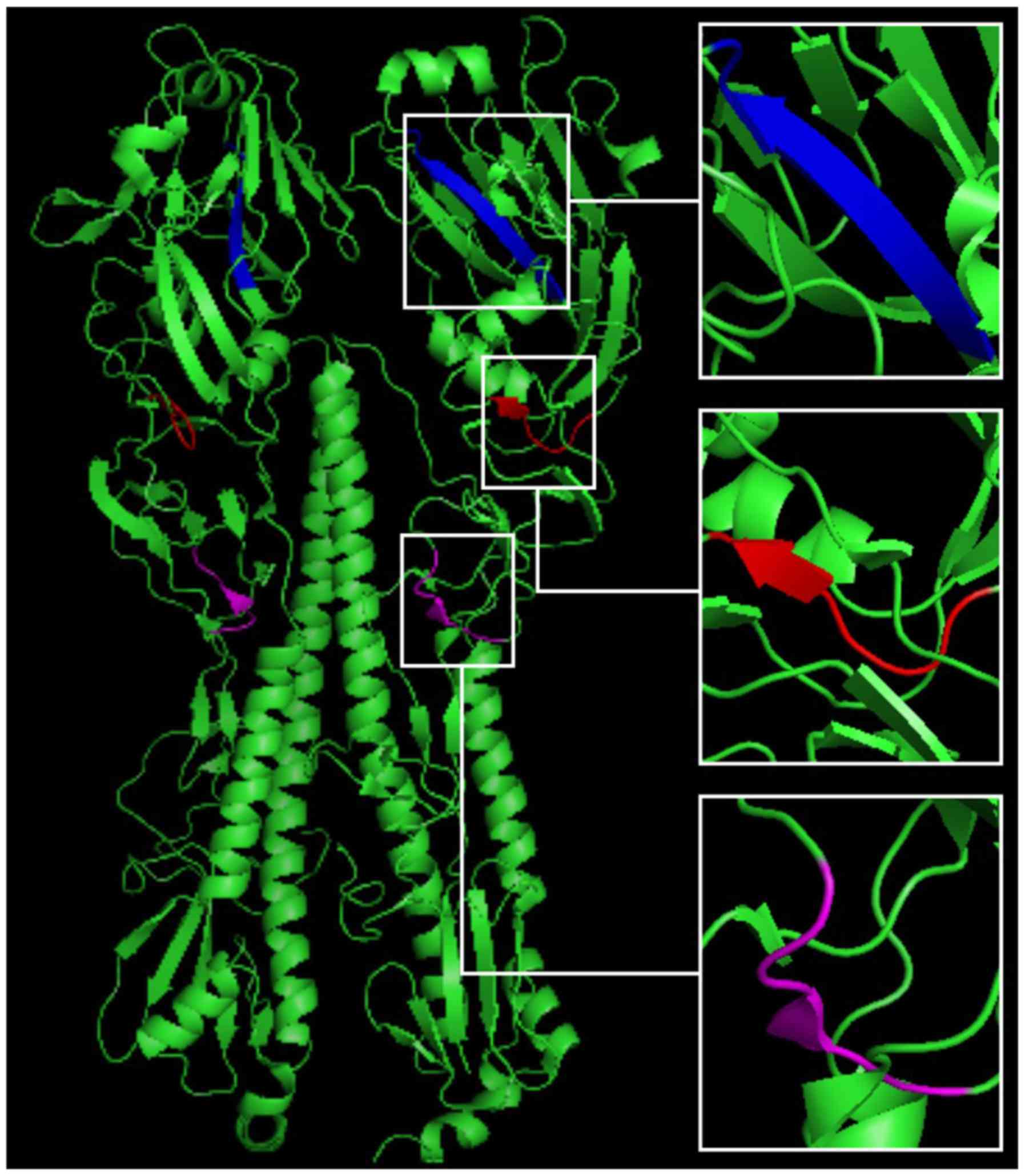
After the predicted epitopes were located, three
peptides (including 93-WSYIVE-98, 191-LVLWGIHHP-199 and
307-LPFQNI-312), located in the continuous conserved amino acid
sequences in all five HA antigens, were chosen for distribution
analysis. PyMOL software analysis identified the three peptides
sequences in the HA crystal structure, and predicted their location
in the 3D structure of HA (Fig.
4).
Discussion
Variability in the HA proteins of the influenza
virus impacts on the suitability and efficacy of existing vaccines.
Developing universal vaccines effective against various subtypes of
influenza is the primary approach for controlling the spread of
infection (22). As epitopes are a
key feature of viruses, several strategies have been successfully
applied in the design and development of ‘epitope-focused’ vaccines
(23,24), which demonstrate advantages such as
high specificity, fewer side effects, simple preparation, and easy
storage and transportation (25,26).
These rapid and accurate strategies have become the foundation for
the development of influenza virus vaccines, as well as supporting
clinical diagnosis and treatment.
In the present study, we predicted the epitopes of
multiple subtypes of the influenza virus HA protein using 40
previously developed mAbs, and extracted the common continuous
amino acid sequences as linear epitopes. Following this, we
determined the localization and distribution with candidate peptide
analysis, to verify and confirm 9 linear epitopes of the HA
protein. For five common subtypes of the influenza virus, 3
epitopes (peptides 1, 2 and 9) showed a strong association with
multiple influenza viruses. Additionally, three epitopes are known
to overlap with three neutralizing epitopes, HA183 ~195, HA127 ~133
and HA92 ~105, of the H3 subtype influenza virus HA protein, as
reported by Li et al (27).
It is also suspected that the three peptides 1-LVLWGIHHP, 2-LPFQNI
and 9-WSYIVE may stimulate organisms to produce neutralizing
antibodies and promote immunogenicity, which may benefit the
development of universal influenza vaccines.
In the second group, which included the 2009
influenza A virus H1N1, and the seasonal influenza A1 and A3 virus,
four linear epitopes were identified. The epitopes in the conserved
sequences of this group were immunodominant epitopes, capable of
stimulating organisms to produce a high volume of antibodies in
response. Therefore, there were more chances of mixed infection of
three of them presently (28). In
the third group, two linear epitopes were predicted, which are the
main markers used to distinguish between the HA proteins of H1 and
other subtypes. Our results suggested that only 15% (6/40) of the
antibodies are produced by organisms when stimulated by epitopes in
group 3, due to there being fewer common epitopes between these two
subtypes of the influenza virus. To an extent, this observation may
also explain the significant difference between the H1N1 influenza
virus subtypes in terms of the infection frequency, pathogenicity
and infection scale, among other variables (29).
As identified in ELISA experiments, 13/40
anti-influenza virus HA antigens were positioned at 9 epitopes. In
group 1, we synthesized 9 peptides after analyzing the common
sequences of the human and avian influenza viruses using DNAMAN
software and 20 mAbs against epitopes common to both viruses; we
positioned 5 antigens to 3 epitopes. Li et al (27), used an E. coli model to
demonstrate that rabbits and mice are immune to recombinant
multi-epitope peptides specific to three neutralizing epitopes,
HA183~195, HA127~133 and HA92~105, from the H3 subtype of influenza
virus HA, and neutralizing antibodies with high titer were
produced. This indicated that, in the first group, among peptides
able to identify multiple subtypes of the influenza virus,
1-LVLWGIHHP and 9-WSYIVE could potentially stimulate organisms to
produce neutralizing antibodies, which would benefit the
development of universal influenza vaccines. In the second group,
we designed 7 peptides complementary to the antigen-conserved areas
of 14 mAbs. We positioned 4 epitopes to 6 mAbs. Epitopes in the
conserved areas of the 2009 influenza A virus H1N1, and the
seasonal influenza A1 and A3 viruses were immunodominant, and
stimulated organisms to produce an abundance of antibodies in
response. In the third group, 2 antigens were positioned to 2
epitopes. Epitopes corresponding with these antibodies are the
primary markers used to distinguish between the HA proteins of H1
and other subtypes.
Influenza virus HA proteins include 562–566 amino
acids and consist of a HA1 spherical head (319-328aa) and a HA2
bacilliform stalk (221-222aa). HA1 includes 8 anti-parallel
β-laminated structures, including a receptor-binding domain (RBD)
and 5 antigenic determinants: A, B, C, D and E (30). The RBD domain is composed of a helix
at site 190, and of rings at sites 130 and 220; one of the
predicted epitopes, verified by peptide 191-LVLWGIHHP-199, was
located near this domain (Fig. 4),
indicating that the current method was effective and reliable, and
could be used to investigate the mechanisms underlying the spread
of influenza, its genetic variation, and in the development of
epitope-specific vaccines.
To predict the epitopes of influenza HA proteins, we
used the antigen-antibody reaction method. Multiple reactivity
modes were observed, including the one-to-one mode, the one-to-many
mode (H1-74 reacted with peptides 1 and 2; H1-51 reacted with
peptides 10 and 11; H1-58 reacted with peptides 17 and 27), and the
unresponsive mode. Two findings were notable: First, two peptides
that react with the same antibody were close to the 3D structure of
HA, and formed a conformational epitope, although they were
separated by a long sequence in the primary structure; second, 40
mAbs were obtained using the split influenza virus vaccine, and
these immunogens can induce organisms to produce the same
antibodies as those induced by natural pathogens. Synthesized
peptides, for which the design and utilization were based on the
primary sequence of the protein, were used for linear epitope
prediction and identification.
These short peptides can be also used as good
immunogens to research different subtypes of influenza virus
epitope vaccines. Li et al (31) applied short-peptide immunization to
the mice directly, and screened the prepared mAbs. In order to
enhance immunogenicity, connection of polypeptides and
macromolecular protein can also be used. Gong et al
(32) coupled the short peptides
P1~P6 of the chemically synthesized influenza virus H3N2 sequence
with the Keyhole Limpet Hemocyanin (KLH) carrier protein in order
to increase the immunogenicity of the polypeptide, and induced a
strong humoral immune response. We have previously linked 9
different polypeptides with KLH one by one, obtaining high titer
and high affinity polyclonal antibodies after immunizing mice.
Polyclonal antibodies were then tested for their neutralizing
activity and cross-reactivity with human tissues. These experiments
are underway.
In conclusion, the present study identified 9 linear
epitopes of the influenza HA protein via traditional mAb and
antigen interaction analysis, and verified these using ELISA and 3D
structure location analyses with synthesized peptides. The results
provide a novel, effective and reliable method for investigating
the mechanisms underlying the spread and variation of influenza
virus and other pathogenic microorganisms, in addition to improving
the development of epitope-focused vaccines.
Acknowledgements
Not applicable.
Funding
This work was supported by Natural Science Basic
Research Program of Shaanxi Province (grant no. 2016JM8065); The
National Key Research and Development Program of China (grant no.
2016YFD0500700); The Natural Science Foundation of China (grant no.
81202373).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CG, HZ, XX and JH conceived and designed the
experiments; CG, HL, PY, HH, JS, YL, QF, XZ, DL and ZW performed
the experiments; CG, HZ and JH analyzed the data; CG, HZ, YL, PY
and LS made data interpretation and critical manuscript revisions;
CG and HZ wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
mAbs
|
monoclonal antibodies
|
|
HA
|
hemagglutinin
|
|
IR
|
inhibition rate
|
|
PDB
|
protein database
|
References
|
1
|
Cao L, Zhu F and Zeng CL: To explore the
clinical value of the Hepatitis B virus mutation detection by the
gene chip technology testing. Chin J Lab Diagn. 19:1301–1303.
2015.
|
|
2
|
Tedbury PR, Mercredi PY, Gaines CR,
Summers MF and Freed EO: Elucidating the mechanism by which
compensatory mutations rescue an HIV-1 matrix mutant defective for
gag membrane targeting and envelope glycoprotein incorporation. J
Mol Biol. 427:1413–1427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nishioka R, Satomura A, Yamada J, Kuroda K
and Ueda M: Rapid preparation of mutated influenza Hemagglutinins
for Influenza virus pandemic prevention. AMB Express. 6:82016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang J and Honda W: CED: A conformational
epitope database. BMC Immunol. 7:72016. View Article : Google Scholar
|
|
5
|
Huang X, Lu D, Ji G, Sun Y, Ma L, Chen Z,
Zhang L, Huang J and Yu L: Hepatitis B virus (HBV) vaccine-induced
escape mutants of HBV S gene among children from Qidong area,
China. Virus Res. 99:63–68. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zerbe K, Moehle K and Robinson JA: Protein
epitope mimetics: From new antibiotics to supramolecular synthetic
vaccines. Acc Chem Res. 50:1323–1331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khairy WOA, Wang L, Tian X, Ye J, Qian K,
Shao H and Qin A: Identification of a novel linear B-cell epitope
in the p27 of Avian leukosis virus. Virus Res. 238:253–257. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nezafat N, Eslami M, Negahdaripour M,
Rahbar MR and Ghasemi Y: Designing an efficient multi-epitope oral
vaccine against Helicobacter pylori using immunoinformatics and
structural vaccinology approaches. Mol Biosyst. 13:699–713. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Liu R, Zhang W, Sun L, Ning Z, Ji
F, Cui J and Zhang G: Identification of epitopes on nonstructural
protein 7 of porcine reproductive and respiratory syndrome viru
tecohnlogy. s recognized by monoclonal antibodies using
phage-display. Virus Genes. 53:623–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Groot AS, Sbai H, Aubin CS, McMurry J
and Martin W: Immuno-informatics: Mining Genomes for vaccine
components. Immunol Cell Biol. 80:255–269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
El-Manzalawy Y and Honavar V: Recent
advances in B-cell epitope prediction methods. Immunome Res. 6
Suppl 2:S22010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang L, Huang P, Wen M, Ni H, Tan S,
Zhang Y and Chen Q: Epitope peptides of influenza H3N2 virus
neuraminidase gene designed by immunoinformatics. Acta Biochim
Biophys Sin (Shanghai). 44:113–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Igarashi M, Ito K, Yoshida R, Tomabechi D,
Kida H and Takada A: Predicting the antigenic structure of the
pandemic (H1N1) 2009 influenza virus hemagglutinin. PLos One.
5:e85532010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pan W, Chen DS, Lu YJ, Sun FF, Xu HW,
Zhang YW, Yan C, Fu LL, Zheng KY and Tang RX: Bioinformatic
prediction of the epitopes of Echinococcus granulosus antigen 5.
Biomed Rep. 6:181–187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen W, Zhong Y, Qin Y, Sun S and Li Z:
The evolutionary pattern of glycosylation sites in influenza virus
(H5N1) hemagglutinin and neuraminidase. PLoS One. 7:e492242012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang YX, Bao YL and Li YX: Advances in
immunological information methods for prediction of antigenic
epitopes. Chin J Immunol. 24:857–860. 2008.
|
|
17
|
Xiao C, Liu Y, Jiang Y, Magoffin DE, Guo
H, Xuan H, Wang G, Wang LF and Tu C: Monoclonal antibodies against
the nucleocapsid proteins of henipaviruses: Production, epitope
mapping and application in immunohistochemistry. Arch Virol.
153:273–281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O'Brien CM, Chy HS, Zhou Q, Blumenfeld S,
Lambshead JW, Liu X, Kie J, Capaldo BD, Chung TL, Adams TE, et al:
New monoclonal antibodies to defined cell surface proteins on human
pluripotent stem cells. Stem Cells. 35:626–640. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia XY, Yu JT, Hu SY, Li JN, Wang M, Wang
C, Chen M, Cui Z and Zhao MH: Antibodies against linear epitopes on
Goodpasture autoantigen in patients with anti-neutrophil
cytoplasmic antibody-associated vasculitis. Clin Rheumatol.
26:2017.
|
|
20
|
Jones ML, Legge FS, Lebani K, Mahler SM,
Young PR, Watterson D, Treutlein HR and Zeng J: Computational
identification of antibody epitopes on the dengue virus NS1
protein. Molecules. 22:E6072017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo CY, Tang YG, Qi ZL, Liu Y, Zhao XR,
Huo XP, Li Y, Feng Q, Zhao PH, Wang X, et al: Development and
characterization of a panel of cross-reactive monoclonal antibodies
generated using H1N1 influenza virus. Immunobiology. 8:941–946.
2015. View Article : Google Scholar
|
|
22
|
Jegaskanda S, Vanderven HA, Wheatley AK
and Kent SJ: Fc or not Fc; that is the question: Antibody
Fc-receptor interactions are key to universal influenza vaccine
design. Hum Vaccin Immunother. 13:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Correia BE, Bates JT, Loomis RJ, Baneyx G,
Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal
V, et al: Proof of principle for epitope-focused vaccine design.
Nature. 507:201–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McBurney SP, Sunshine JE, Gabriel S, Huynh
JP, Sutton WF, Fuller DH, Haigwood NL and Messer WB: Evaluation of
protection induced by a dengue virus serotype 2 envelope domain III
protein scaffold/DNA vaccine in non-human primates. Vaccine.
34:3500–3507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao Y, Li D, Fu Y, Bai Q, Chen Y, Bai X,
Jing Z, Sun P, Bao H, Li P, et al: Rational design and efficacy of
a multi-epitope recombinant protein vaccine against foot-and-mouth
disease virus serotype A in pigs. Antiviral Res. 140:133–141. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baratelli M, Pedersen LE, Trebbien R,
Larsen LE, Jungersen G, Blanco E, Nielsen J and Montoya M:
Identification of cross-reacting T-cell epitopes in structural and
non-structural proteins of swine and pandemic H1N1 influenza A
virus strains in pigs. J Gen Virol. 98:895–899. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Ding J and Chen YH: Recombinant
protein comprising multi-neutralizing epitopes induced high titer
of antibodies against influenza A virus. Immunobiology.
207:305–313. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Myers CA, Kasper MR, Yasuda CY, Savuth C,
Spiro DJ, Halpin R, Faix DJ, Coon R, Putnam SD, Wierzba TF and
Blair PJ: Dual infection of novel influenza viruses A/H1N1 and
A/H3N2 in a cluster of Cambodian patients. Am J Trop Med Hyg.
85:961–963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kilbourne ED: Influenza pandemics of the
20th century. Emerg Infect Dis. 12:9–14. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han T and Marasco WA: Structural basis of
influenza virus neutralization. Ann N Y Acad Sci. 1217:178–190.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Hu HY, Qi ZL, Sun LJ, Li Y, Feng Q,
Guo CY, Wang HF, Zhao PH, Liu Y, et al: Identification and
characterization of epitopes from influenza A virus hemagglutinin
that induce broadly cross-reactive antibodies. Int J Mol Med.
3:1673–1682. 2018.
|
|
32
|
Gong X, Yin H, Shi YH, Guan SS, He XQ,
Yang L, Yu YJ, Kuai ZY, Jiang CL, Kong W, et al: Conserved stem
fragment from H3 influenza hemagglutinin elicits cross-clade
neutralizing antibodies through stalk-targeted blocking of
conformational change during membrane fusion. Immunol Lett.
172:11–20. 2016. View Article : Google Scholar : PubMed/NCBI
|