Introduction
Acute myocardial infarction (AMI) is the leading
cause of morbidity and mortality worldwide (1). Reperfusion, the restoration of blood
flow, has been considered the most effective treatment of ischemic
heart disease, in particular AMI (2). However, reperfusion has further been
referred to as a double-edged sword, as reperfusion may cause
aggravation of myocardial injury, termed myocardial ischemia
reperfusion injury (MIRI) (3). With
the wide application of reperfusion therapy, including drug
thrombolysis, percutaneous coronary intervention and coronary
artery bypass grafting, the elucidation of the mechanism of MIRI
prevention has become imperative (4–7).
The mitochondrial permeability transition pore
(MPTP) is a non-specific channel located in the inner mitochondrial
membrane (8). MPTP remains closed
during ischemia, but rapidly opens following the commencement of
reperfusion (9). MPTP opening is
considered an important mechanism of MIRI (10). Additionally, the inhibition of MPTP
opening by cyclosporine A attenuates MIRI (11). Therefore, MPTP is considered an
important therapeutic target for the prevention of MIRI.
Hydroxytyrosol (HT), known as 3,4-dihydroxyphenyl
ethanol, is a phenolic compound extracted from Mediterranean virgin
olive oil (12). Biological effects
of HT include the suppression of oxidative stress, inflammation and
tumor formation, and protection of the cardiovascular system and
neurological function (13–17). HT serves a role in the protection
against liver ischemia/reperfusion injury in mice (18,19). Pei
et al (20) revealed that HT
affects MIRI via the phosphatidylinositol-4,5-bisphosphate 3 kinase
(PI3K)/protein kinase B (Akt) signaling pathway. However, the
effect of HT on MPTP in MIRI remains unknown. Therefore, the
present study suggested that HT attenuated MIRI by inhibiting MPTP
opening.
The aim of the present study was to investigate the
effects of HT on MIRI in an isolated rat heart model and to further
explore the role of MPTP in the cardioprotection of HT.
Materials and methods
Experimental animals and reagents
A total of 100 healthy male Wistar rats (age, 4
weeks; weight, 250±20 g) were purchased from Changsheng
Biotechnology Co., Ltd. (Beijing, China). Rats were housed in
environmentally controlled conditions (20–25°C, 5–65% relative
humidity, with a 12-h light/dark cycle) with a common 1 week
acclimatization period. All rats had access to fresh food and water
ad libitum. The procedures for handling and caring for animals
adhered to the guidelines in compliance with the Guide for the Care
and Use of Laboratory Animals (21).
The experimental protocol was approved by the Institutional Ethics
Committee of China Medical University (Shenyang, China).
HT was purchased from Dalian Meilun Biology
Technology Co., Ltd. (Dalian, China). Atractyloside (ATR) and
2,3,5-triphenyltetrazolium chloride (TTC) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Establishing MIRI in isolated rat
hearts
Pentobarbital sodium (100 mg/kg) was administered
intravenously to anesthetize the rats. Heparin (1,500 IU/kg) was
injected intravenously to prevent intracoronary clot formation.
Following opening of the thoracic cavity, hearts were swiftly
removed and immediately immersed in ice-cooled heparinized
Krebs-Henseleit (K-H) buffer (0.15 mol/l NaCl, 0.006 mol/l KCl,
0.002 mol/l CaCl2, 0.002 mol/l NaHCO3) and saturated with 100%
oxygen (22). Isolated hearts were
transferred to a Langendorff perfusion system (Taimeng Science and
Technology Ltd., Chengdu, China) to perform heart perfusion with
K-H solution, saturated with 95% O2 + 5% CO2
at 37°C. A water-filled latex balloon was inserted into the left
ventricle through the left atrium and connected to a pressure
transducer for pressure measurement. All isolated hearts were
continually perfused with K-H solution for 10 min stabilization,
prior to the commencement of ischemia. All isolated rat hearts were
subjected to 30 min global ischemia, followed by 120 min of
reperfusion to generate the MIRI model.
Experimental protocol
The experimental protocol consisted of two phases.
In the first phase, 40 rats were divided into the following 4
groups with 10/group: i) Ischemia reperfusion group (IR): As
described above; ii) 10 µM HT treatment group (HT 10 µM): Isolated
hearts were perfused with 10 µM HT for 10 min and K-H solution for
5 min prior to induction of ischemia; iii) 100 µM HT treatment
group (HT 100 µM): Isolated hearts were perfused with 100 µM HT for
10 min and K-H solution for 5 min prior to induction of ischemia;
iv) 1,000 µM HT treatment group (HT 1,000 µM): Isolated hearts were
perfused with 1,000 µM HT for 10 min and K-H solution for 5 min
prior to induction of ischemia.
According to the results of the first phase, 100 µM
HT was chosen for the second phase. A total of 60 rats were divided
into the following 3 groups with 20/group: i) IR group; ii) HT 100
µM group: The heart rate was stabilized and the heart was perfused
with HT for 10 min, then with K-H solution for 5 min,
ischemia/reperfusion was performed as in IR group. iii) 100 µM HT
in combination with ATR treatment group (HT 100 µM + ATR): 5 mg/kg
ATR was injected intraperitoneally 30 min prior to extraction of
the heart, the remainder of the procedure was as described for the
HT 100 µM group.
Cardiac function monitoring
Change in cardiac function was evaluated monitoring
heart rate (HR) and coronary flow (CF). HR and CF were measured
prior to ischemia, and 30 and 60 min post initiation of
reperfusion.
Hematoxylin and eosin (HE)
staining
Following reperfusion, hearts were harvested. Heart
tissues were fixed in 4% paraformaldehyde for 24–72 h at room
temperature and washed with flowing water for 4 h. Heart tissue
samples were subsequently dehydrated in an ascending ethanol series
(70% ethanol for 2 h, 80% ethanol overnight, 90% ethanol for 2 h,
100% ethanol I for 1 h and 100% ethanol II for 1 h) at room
temperature. The tissue samples were embedded in paraffin embedded
and the paraffin-embedded tissue samples were cut into 5-µm-thick
sections. The tissue sections were subsequently deparaffinized in
xylene I for 15 min and xylene II for 15 min at room temperature
and rehydrated in a descending ethanol series (100% ethanol I for 5
min, 100% ethanol II for 5 min, 95% ethanol for 2 min, 85% ethanol
for 2 min, 75% ethanol for 2 min) and distilled water for 2 min at
room temperature. Deparaffinized section were incubated with
hematoxylin solution for 5 min, 1% hydrochloric acid alcohol for 3
sec and eosin solution for 3 min at room temperature. Pathological
changes were observed under a light microscope (Olympus BX51;
Olympus Corporation, Tokyo, Japan; magnification, ×400).
Measurement of myocardial infarct
size
Following reperfusion, the hearts were removed,
frozen at −20°C for 1 h and sliced into 1–2 mm thick sections. The
sections were incubated in a 1% TTC solution for 20 min at 37°C.
Tissue sections were washed with 1X PBS and fixed in 4%
paraformaldehyde overnight at room temperature. Images of the
stained slices were captured using a digital camera and analyzed
using Image J2X analysis software (National Institutes of Health,
Bethesda, MD, USA). The severity of the myocardial infarction was
indicated by the ratio of the infarct area to the total area.
Apoptosis
Myocardial apoptosis was detected by terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay using the In Situ Cell Death Detection kit (cat. no.
11684817910; Roche Diagnostics, Indianapolis, IN, USA), as
previously described (23).
Apoptotic cells were observed under a light microscope
(magnification, ×400) in three randomly selected fields. Image-Pro
Plus (version 6.0; Media Cybernetics, Inc., Rockville, MD, USA) was
used for cell counting.
MPTP sensitivity to
Ca2+
Mitochondria were isolated from heart tissues using
the Tissue Mitochondria Isolation kit (Beyotime Institute of
Biotechnology, Shanghai, China), according to the manufacturer's
protocol. The sensitivity of MPTP to Ca2+ was determined
using the Purified Mitochondrial Membrane Pore Channel Colorimetric
Assay kit (cat. no. GMS10095; Shanghai Genmed Pharmaceutical
Technology Co., Ltd., Shanghai, China), according to the
manufacturer's protocol. The larger the min/max ratio, the lower
the sensitivity of MPTP opening to Ca2+, conversely, the
smaller the min/max ratio, the higher the sensitivity of MPTP
opening to Ca2+.
Western blot analysis
The left ventricle tissues were homogenized with
radioimmunoprecipitation buffer (Beyotime Institute of
Biotechnology) and protease inhibitor phenylmethanesulfonyl
fluoride (Beyotime Institute of Biotechnology) on ice for 20 min.
Proteins were extracted from lysates following centrifugation at
13,000 × g for 15 min at 4°C. Total protein was quantified using
the Enhanced BCA Protein Assay kit (Beyotime Institute of
Biotechnology). Subsequently, 50 µg protein was denatured at 100°C
for 10 min, and separated via SDS-PAGE on a 10% gel. The separated
proteins were transferred onto polyvinylidene difluoride membranes
and blocked with 5% skimmed milk for 1 h at room temperature. The
membranes were incubated with primary antibodies against B-cell
lymphoma 2 (Bcl-2)-associated X protein (Bax; 1:1,000; cat. no.
WL01637), Bcl-2 (1:1,000; cat. no. WL01556; both Shenyang Wan
Biotechnology Co., Ltd., Shenyang, China), cytochrome c (1:1,000;
cat. no. ab90529), apoptotic protease activating factor-1 (APAF-1;
1:1,000; cat. no. ab2001), cleaved caspase-3 (1:1,000; cat. no.
ab2302; all Abcam, Cambridge, UK), cleaved caspase-9 (1:1,000; cat.
no. 40503-1; Signalway Anitbody LLC, College Park, MD, USA) and
β-actin (1:1,000; cat. no. TA-09; OriGene Technologies, Inc.,
Beijing, China) overnight at 4°C. Following primary incubation, the
membranes were incubated with horseradish peroxidase-labeled goat
anti-rabbit immunoglobulin G (1:4,000; cat. no. E030120-01) or goat
anti-mouse immunoglobulin G (1:4,000; cat. no. E030110-01; both
EarthOx Life Sciences, Millbrae, CA, USA) at 37°C for 2 h. Protein
bands were visualized using BeyoECL Star (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. Levels of
phosphorylated proteins were normalized to the corresponding total
protein levels. Relative densitometry was calculated using ImageJ
(version 1.50e; National Institutes of Health).
Statistical analysis
Data are expressed as the mean ± standard deviation
from three independent experiments. All statistical analysis was
performed using SPSS 17.0 (SPSS, Inc., Chicago, IL, USA).
Differences between groups were evaluated using one-way analysis of
variance followed by Fisher's least significant difference tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of HT on HR and CF
As presented in Fig.
1, no statistical significance was detected for HR or CF in
rats of the various experimental groups at various times
(P>0.05).
Effects of HT on myocardial
injury
HE staining revealed that HT (100 and 1,000 µM)
pretreatment markedly alleviated pathological damage in the
ischemic myocardium; however, pretreatment with 10 µM HT did not
reveal any alleviation of pathological damage in the ischemic
myocardium (Fig. 2A). Results of the
TTC assay revealed the infarct size, which is presented as white
tissue sections compared with red normal tissue sections (Fig. 2B). The myocardial infarct size of the
100 and 1,000 µM HT groups was significantly decreased compared
with the IR group (30.9±2.9 and 32.3±3.2 vs. 54.3±3.0%; P<0.05;
Fig. 2C). However, pretreatment with
10 µM HT revealed no significant change in the myocardial infarct
size compared with the IR group (P>0.05; Fig. 2C). The protective effect on MIRI was
not improved when increasing HT from 100 to 1,000 µM. Therefore,
100 µM HT was selected for second-stage experiments.
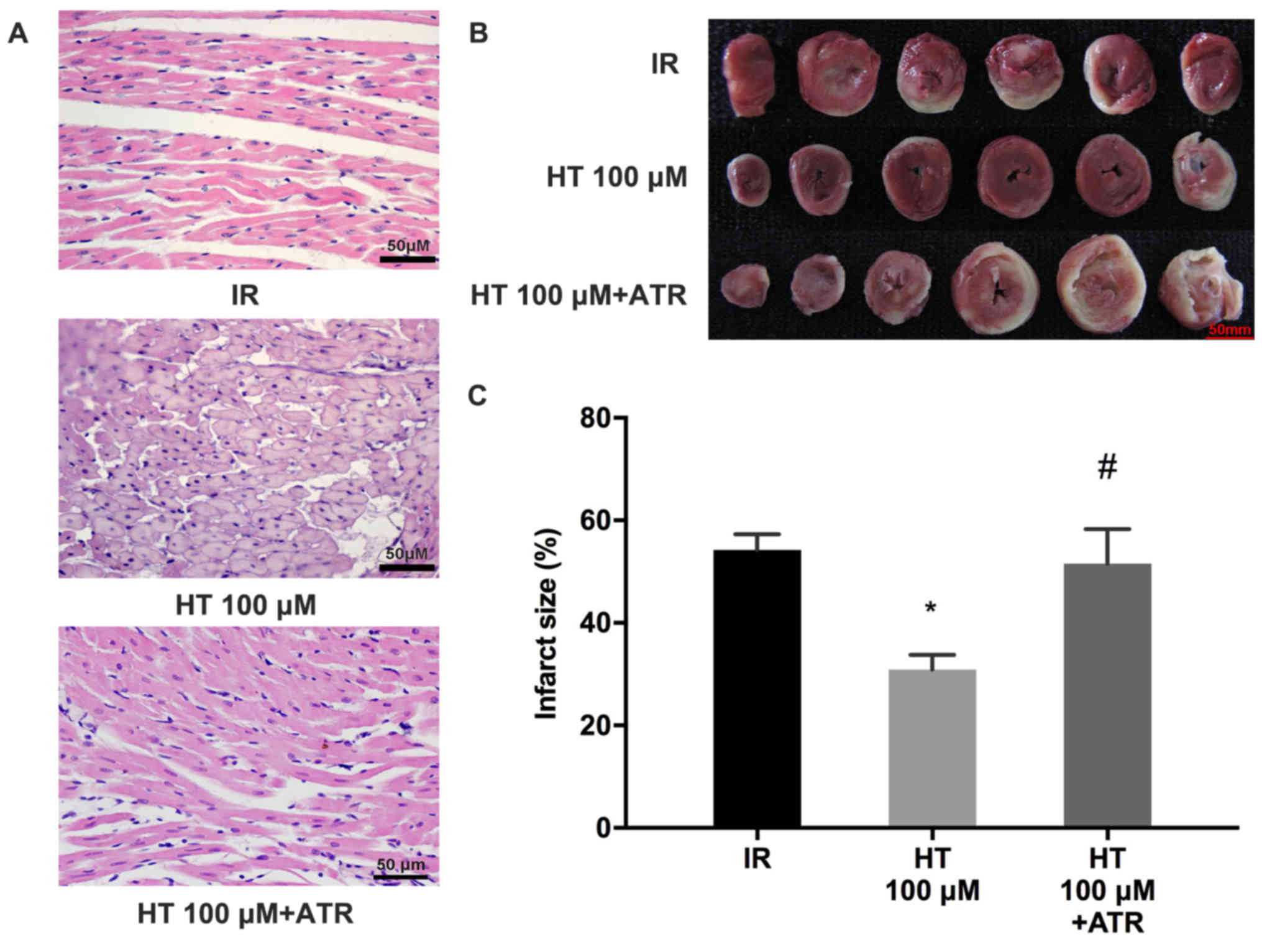
ATR reverses protective effects of
HT
To elucidate the role of HT in cardioprotection
during MIRI, the effects of ATR, a MPTP opener, were investigated.
The results revealed that myocardial injury was worsened in the HT
100 µM + ATR group compared with the HT 100 µM group (Fig. 3A). Myocardial infarct size was
significantly increased in the HT 100 µM + ATR group compared with
the HT 100 µM group (52.8±7.8 vs. 30.9±2.9%; P<0.05; Fig. 3B and C).
Effects of HT on MPTP opening
MPTP opening is generally induced by Ca2+
and the sensitivity of MPTP to Ca2+ is an indicator for
MPTP opening (24–26). The results indicated that changes in
Ca2+ induced MPTP opening. Isolated mitochondria from
the HT 100 µM group were significantly more resistant to
stimulation by Ca2+ compared with the IR group,
suggesting that HT inhibited MPTP opening (Fig. 4A and B). Additionally, the resistance
of the isolated mitochondria to Ca2+ was decreased in
the HT 100 µM + ATR group compared with the HT 100 µM group
(Fig. 4B).
Effects of HT on the mitochondrial
apoptotic pathway
MPTP opening leads to the release of cytochrome c
and activation of caspase-9 and −3, which results in apoptosis
(27). Results from the TUNEL assay
revealed that 100 µM HT significantly decreased the rate of
apoptosis (P<0.05; Fig. 5A and
B). In addition, compared with HT 100 µM group, the apoptosis
rate in the HT 100 µM + ATR group increased by 15.8% (Fig. 5). Similarly, western blot analysis
revealed that 100 µM HT significantly decreased cytochrome c,
cleaved caspase-9 and −3 protein levels compared with the IR group
(P<0.05; Fig. 6). In addition,
the cytochrome c, cleaved caspase-9 and −3 levels in the HT 100 µM
+ ATR group were significantly increased compared with the 100 µM
HT group (Fig. 6). The rate of
apoptosis and expression level of apoptosis-associated proteins was
not significantly different in the ATR-treated group compared with
the IR group, suggesting that ATR reversed cardioprotective effects
exerted by HT.
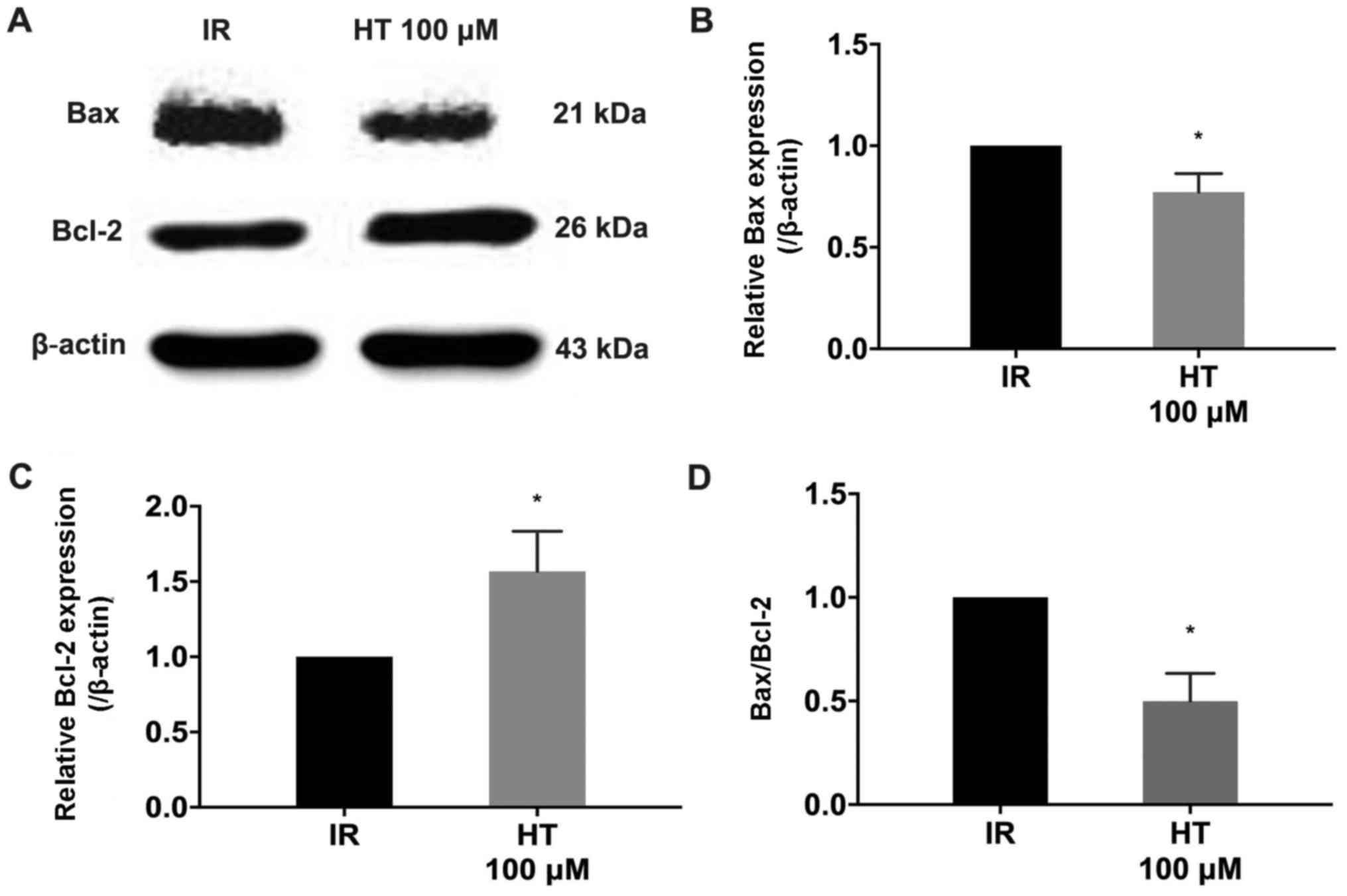
Effects of HT on Bax and Bcl-2 protein
expression
The Bcl-2 protein family is a key regulator in MPTP
opening (28). Western blot analysis
demonstrated that treatment with 100 µM HT significantly decreased
Bax expression and Bax/Bcl-2 compared with the IR group (P<0.05;
Fig. 7). Additionally, Bcl-2
expression was significantly increased compared with the IR group
(P<0.05; Fig. 7).
Discussion
In the present study, treatment with the
pharmacological agent HT at 100 or 1,000 µM was revealed to reduce
the myocardial infarction area and damage to the myocardium in rats
compared with the untreated animals, which suggested that HT may
protect against MIRI. However, there was no effect for 10 µM HT.
Additionally, there was no significant difference in the
cardioprotection exerted by 100 and 1,000 µM HT. Therefore, it was
suggested that a dose-associated effect of HT occurred at lower
doses (10–100 µM), which is consistent with a previous study by Pan
et al (18). Pei et al
(20) used SD rats to perform in
vivo cardiac ischemia for 30 min followed by reperfusion for 3
h. In their study, rats were intraperitoneally injected with HT at
a concentration of 20 mg/kg during ischemia. The results revealed
that HT attenuated MIRI via the PI3K/Akt signaling pathway, and
protected functional parameters of the heart. However, in the
present study, no difference in HR or CF of rats from various
groups was observed at various times. Reasons for this disparity
may include differences between in vivo and in vitro
models, differences in rat species, duration of procedures and
dosing methods.
To the best of our knowledge, this is the first
study to demonstrate the effect of HT on the inhibition of MPTP in
MIRI. The opening of MPTP causes irreversible damage to the heart
(29). According to previous
studies, core components of MPTP are voltage-dependent anion
channels (VDAC), adenine nucleotide transporter and cyclophilin-D
(30,31). The opening of MPTP is induced by
insufficient intracellular adenosine triphosphate synthesis,
reactive oxygen species-induced oxidative stress, and
Ca2+ and phosphate accumulation (32,33).
Several studies have revealed that MIRI is closely associated to
MPTP opening (34–36). Pretreatment with several
pharmacological agents, including irisin, melatonin and carnosic
acid, have been demonstrated to alleviate MIRI via inhibition of
MPTP opening (37–39). In the present study, it was
demonstrated that HT inhibited MPTP opening during MIRI.
Ca2+ treatment induces MPTP opening (40); compared with the IR group, it was
revealed that isolated mitochondria from rat hearts pretreated with
HT had a higher resistance to Ca2+ stimulation, which
indicated that HT inhibited MPTP opening. Additionally, it was
revealed that HT pretreatment reduced cytochrome c, cleaved
caspase-9 and −3 levels and decreased the rate of apoptosis. These
observations were similar to results reported by Soni et al
(41) studying rat brains. All
protective effects of HT were abolished with ATR treatment, which
strongly suggested that HT protected against MIRI by inhibiting
MPTP opening.
The Bcl-2 protein family is an important constituent
of the apoptotic pathway (42,43) and
serves an important regulatory role in MPTP opening (44). Members of the Bcl-2 family include
the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax
(45,46). Interactions of Bcl-2 and Bax with
VDAC regulate MPTP opening; Bax facilitates MPTP opening by binding
to VDAC, while Bcl-2 inhibits binding of Bax to VDAC (28). Bcl-2 and Bax co-express in tissue
cells and MPTP opening is closely associated with the ratio of Bax
to Bcl-2 (47). In the present
study, it was revealed that HT pretreatment enhanced Bcl-2
expression in MIRI and decreased Bax expression and Bax/Bcl-2
levels. This demonstrated that HT inhibited MPTP opening by
regulating Bcl-2 and Bax expression, which is consistent with a
previous study by Liu and Dong (39). Notably, a recent study suggested that
phosphorylated-Akt inhibits MPTP opening by regulating the Bcl-2
protein family (48). Furthermore,
Pei et al (20) demonstrated
that HT protects the rat myocardium from MIRI via direct activation
of the PI3K/Akt signaling pathway. It was therefore further
suggested that the PI3K/Akt/Bcl-2 signaling pathway may serve an
important role in the inhibition of MPTP opening by HT. This
hypothesis requires to be investigated in further studies for
confirmation.
There are limitations to the present study. The
isolated rat heart model used was deprived of neural and humoral
regulation and may not completely mimic pathophysiological changes
that occur during MIRI and in vivo cardioprotective effects
of HT require further validation. The present study solely
demonstrated that HT inhibited MPTP opening via Bcl-2; upstream
targets of the MPTP pathway, including PI3K/Akt, glycogen synthase
kinase 3β and Janus kinase/signal transducer and activator of
transcription pathways require further investigation.
In conclusion, the present study demonstrated for
the first time that HT protected against MIRI by inhibiting MPTP
opening and thereby providing a pharmacological basis for future
research and treatment of MIRI.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81670320 and 81800232) and
the Natural Science Foundation of Liaoning Province (grant no.
201602826).
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DJ and NW designed the experiments. JM, SL, ZH and
XL performed the experiments. JM, PJ and YG analyzed the data. JM
prepared the manuscript. NW revised the manuscript. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
All rats were treated in accordance with the Guide
for the Care and Use of Laboratory Animals. The experimental
protocol was approved by the Institutional Ethics Committee of
China Medical University (Shenyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moran AE, Forouzanfar MH, Roth GA, Mensah
GA, Ezzati M, Murray CJ and Naghavi M: Temporal trends in ischemic
heart disease mortality in 21 world regions, 1980 to 2010: The
Global Burden of Disease 2010 study. Circulation. 129:1483–1492.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bainey KR and Armstrong PW: Clinical
perspectives on reperfusion injury in acute myocardial infarction.
Am Heart J. 167:637–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu Y, Wu L, Chen A, Xu C and Feng Q:
Protective effects of olive leaf extract on acrolein-exacerbated
myocardial infarction via an endoplasmic reticulum stress pathway.
Int J Mol Sci. 19(pii): E4932018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bulluck H, Yellon DM and Hausenloy DJ:
Reducing myocardial infarct size: Challenges and future
opportunities. Heart. 102:341–348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng X, Xing X, Sun G, Xu X, Wu H, Li G
and Sun X: Guanxin danshen formulation protects against myocardial
ischemia reperfusion injury-induced left ventricular remodeling by
upregulating estrogen receptor β. Front Pharmacol. 8:7772017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Xiang Y, Zhang S, Wang Y, Yang J,
Liu W and Xue F: Intramyocardial injection of thioredoxin
2-expressing lentivirus alleviates myocardial ischemia-reperfusion
injury in rats. Am J Transl Res. 9:4428–4439. 2017.PubMed/NCBI
|
|
7
|
Liu H, Cala PM and Anderson SE: Na/H
exchange inhibition protects newborn heart from
ischemia/reperfusion injury by limiting Na+-dependent Ca2+
overload. J Cardiovasc Pharmacol. 55:227–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Griffiths EJ and Halestrap AP:
Mitochondrial non-specific pores remain closed during cardiac
ischaemia, but open upon reperfusion. Biochem J. 307:93–98. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Javadov S, Jang S, Parodi-Rullán R,
Khuchua Z and Kuznetsov AV: Mitochondrial permeability transition
in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable
target for cardioprotection? Cell Mol Life Sci. 74:2795–2813. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duan X, Ji B, Yu K, Liu J, Hei F and Long
C: Pharmacological postconditioning protects isolated rat hearts
against ischemia-reperfusion injury: The role of mitochondrial
permeability transition pore. ASAIO J. 57:197–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li X, Chen Z, Wu Y, Yan Y, Sun X and Yuan
Q: Establishing an artificial pathway for efficient biosynthesis of
hydroxytyrosol. ACS Synth Biol. 7:647–654. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Poudyal H, Lemonakis N, Efentakis P, Gikas
E, Halabalaki M, Andreadou I, Skaltsounis L and Brown L:
Hydroxytyrosol ameliorates metabolic, cardiovascular and liver
changes in a rat model of diet-induced metabolic syndrome:
Pharmacological and metabolism-based investigation. Pharmacol Res.
117:32–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Y, Zhou D and Shahidi F: Antioxidant
properties of tyrosol and hydroxytyrosol saturated fatty acid
esters. Food Chem. 245:1262–1268. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
González-Correa JA, Rodríguez-Pérez MD,
Márquez-Estrada L, López-Villodres JA, Reyes JJ,
Rodriguez-Gutierrez G, Fernández-Bolaños J and De La Cruz JP:
Neuroprotective effect of hydroxytyrosol in experimental diabetic
retinopathy: Relationship with cardiovascular biomarkers. J Agric
Food Chem. 66:637–644. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhi LQ, Yao SX, Liu HL, Li M, Duan N and
Ma JB: Hydroxytyrosol inhibits the inflammatory response of
osteoarthritis chondrocytes via SIRT6-mediated autophagy. Mol Med
Rep. 17:4035–4042. 2018.PubMed/NCBI
|
|
17
|
Zubair H, Bhardwaj A, Ahmad A, Srivastava
SK, Khan MA, Patel GK, Singh S and Singh AP: Hydroxytyrosol induces
apoptosis and cell cycle arrest and suppresses multiple oncogenic
signaling pathways in prostate cancer cells. Nutr Cancer.
69:932–942. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan S, Liu L, Pan H, Ma Y, Wang D, Kang K,
Wang J, Sun B, Sun X and Jiang H: Protective effects of
hydroxytyrosol on liver ischemia/reperfusion injury in mice. Mol
Nutr Food Res. 57:1218–1227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soto-Alarcon SA, Valenzuela R, Valenzuela
A and Videla LA: Liver protective effects of extra virgin olive
oil: Interaction between its chemical composition and the
cell-signaling pathways involved in protection. Endocr Metab Immune
Disord Drug Targets. 18:75–84. 2018.PubMed/NCBI
|
|
20
|
Pei YH, Chen J, Xie L, Cai XM, Yang RH,
Wang X and Gong JB: Hydroxytyrosol protects against myocardial
ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism.
Mediators Inflamm. 2016:12321032016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kastenmayer RJ, Moore RM, Bright AL,
Torres-Cruz R and Elkins WR: Select agent and toxin regulations:
Beyond the eighth edition of the guide for the care and use of
laboratory animals. J Am Assoc Lab Anim Sci. 51:333–338.
2012.PubMed/NCBI
|
|
22
|
Wu N, Zhang X, Guan Y, Shu W, Jia P and
Jia D: Hyper-cholesterolemia abrogates the cardioprotection of
ischemic postconditioning in isolated rat heart: Roles of glycogen
synthase kinase-3β and the mitochondrial permeability transition
pore. Cell Biochem Biophys. 69:123–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu N, Li W, Shu W and Jia D: Protective
effect of picroside II on myocardial ischemia reperfusion injury in
rats. Drug Des Devel Ther. 8:545–554. 2014.PubMed/NCBI
|
|
24
|
Endlicher R, Kriváková P, Lotkova H,
Milerová M, Drahota Z and Cervinková Z: Tissue specific sensitivity
of mitochondrial permeability transition pore to Ca2+ ions. Acta
Medica (Hradec Kralove). 52:69–72. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He F, Wu Q, Xu B, Wang X, Wu J, Huang L
and Cheng J: Suppression of Stim1 reduced intracellular calcium
concentration and attenuated hypoxia/reoxygenation induced
apoptosis in H9C2 cells. Biosci Rep. 37(pii): BSR201712492017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hurst S, Hoek J and Sheu SS: Mitochondrial
Ca2+ and regulation of the permeability transition pore.
J Bioenerg Biomembr. 49:27–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petit PX, Susin SA, Zamzami N, Mignotte B
and Kroemer G: Mitochondria and programmed cell death: Back to the
future. FEBS Lett. 396:7–13. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tong Z, Xie Y, He M, Ma W, Zhou Y, Lai S,
Meng Y and Liao Z: VDAC1 deacetylation is involved in the
protective effects of resveratrol against mitochondria-mediated
apoptosis in cardiomyocytes subjected to anoxia/reoxygenation
injury. Biomed Pharmacother. 95:77–83. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Penna C, Perrelli MG and Pagliaro P:
Mitochondrial pathways, permeability transition pore, and redox
signaling in cardioprotection: Therapeutic implications. Antioxid
Redox Signal. 18:556–599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Siemen D and Ziemer M: What is the nature
of the mitochondrial permeability transition pore and what is it
not? IUBMB Life. 65:255–262. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kwong JQ and Molkentin JD: Physiological
and pathological roles of the mitochondrial permeability transition
pore in the heart. Cell Metab. 21:206–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carraro M and Bernardi P: Calcium and
reactive oxygen species in regulation of the mitochondrial
permeability transition and of programmed cell death in yeast. Cell
Calcium. 60:102–107. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bernardi P, Rasola A, Forte M and Lippe G:
The mitochondrial permeability transition pore: Channel formation
by F-ATP synthase, integration in signal transduction, and role in
pathophysiology. Physiol Rev. 95:1111–1155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morciano G, Bonora M, Campo G, Aquila G,
Rizzo P, Giorgi C, Wieckowski MR and Pinton P: Mechanistic role of
mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 982:169–189.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bopassa JC, Michel P, Gateau-Roesch O,
Ovize M and Ferrera R: Low-pressure reperfusion alters
mitochondrial permeability transition. Am J Physiol Heart Circ
Physiol. 288:H2750–H2755. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fang R, Zhang LL, Zhang LZ, Li W, Li M and
Wen K: Sphingosine 1-phosphate postconditioning protects against
myocardial ischemia/reperfusion injury in rats via mitochondrial
signaling and Akt-Gsk3β phosphorylation. Arch Med Res. 48:147–155.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Zhao YT, Zhang S, Dubielecka PM,
Du J, Yano N, Chin YE, Zhuang S, Qin G and Zhao TC: Irisin plays a
pivotal role to protect the heart against ischemia and reperfusion
injury. J Cell Physiol. 232:3775–3785. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:2017. View Article : Google Scholar
|
|
39
|
Liu P and Dong J: Protective effects of
carnosic acid against mitochondria-mediated injury in H9c2
cardiomyocytes induced by hypoxia/reoxygenation. Exp Ther Med.
14:5629–5634. 2017.PubMed/NCBI
|
|
40
|
Marchi S and Pinton P: The mitochondrial
calcium uniporter complex: Molecular components, structure and
physiopathological implications. J Physiol. 592:829–839. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Soni M, Prakash C, Dabur R and Kumar V:
Protective effect of hydroxytyrosol against oxidative stress
mediated by arsenic-induced neurotoxicity in rats. Appl Biochem
Biotechnol. 186:27–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Siddiqui WA, Ahad A and Ahsan H: The
mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update.
Arch Toxicol. 89:289–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Narita M, Shimizu S, Ito T, Chittenden T,
Lutz RJ, Matsuda H and Tsujimoto Y: Bax interacts with the
permeability transition pore to induce permeability transition and
cytochrome c release in isolated mitochondria. Proc Natl Acad Sci
USA. 95:14681–14686. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ashkenazi A, Fairbrother WJ, Leverson JD
and Souers AJ: From basic apoptosis discoveries to advanced
selective BCL-2 family inhibitors. Nat Rev Drug Discov. 16:273–284.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li H, Sun JJ, Chen GY, Wang WW, Xie ZT,
Tang GF and Wei SD: Carnosic acid nanoparticles suppress liver
ischemia/reperfusion injury by inhibition of ROS, Caspases and
NF-κB signaling pathway in mice. Biomed Pharmacother. 82:237–246.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pastorino JG, Tafani M, Rothman RJ,
Marcinkeviciute A, Hoek JB and Farber JL: Functional consequences
of the sustained or transient activation by Bax of the
mitochondrial permeability transition pore. J Biol Chem.
274:31734–31739. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liao P, Sun G, Zhang C, Wang M, Sun Y,
Zhou Y, Sun X and Jian J: Bauhinia championii flavone attenuates
hypoxia-reoxygenation induced apoptosis in H9c2 cardiomyocytes by
improving mitochondrial dysfunction. Molecules. 21(pii): E14692016.
View Article : Google Scholar : PubMed/NCBI
|