Introduction
Atrial fibrillation (AF), a highly prevalent heart
disorder with a significant genetic component (1), is regarded as the most common sustained
arrhythmia in clinical practice worldwide, which can result in
heart failure and represents an important risk factor for ischemic
stroke, thereby leading to significant morbidity and mortality
(2–4). A higher rate of adverse outcomes for
the elderly are connected with AF. According to literature,
arrhythmia has been demonstrated to arise because of the
interaction between genetic and acquired risk factors (5). However, the molecular pathogenesis of
AF was not well elucidated, leading to the need of exploring the
precise mechanisms of this disease as well as developing novel
treatments. Extraction of novel biomarkers for AF is critically
important to the understanding and future prevention of this
disease.
Metabolites have been regarded as the eventual
response of biological systems to the changes of inheritance or
environment, whose level directly reflect the physiological status
of the body (6). Consequently,
selecting disease-associated metabolites is very important for
enhancing clinical diagnosis (7,8). Of
note, functionally associated metabolites and genes are tended to
infer to phenotypically similar disorders. Notably, ‘omics’ data,
for example, metabolic, phenomic and genomic, will offer valuable
information for identification of disease risk candidate
metabolites. From a biological perspective, a biological system is
expressed as a multi-omics network. It is a natural method to
combine gene, metabolite and phenotype data to establish a
composite network for detecting disease-related metabolites, and
this combination approach can offer accurate information (9,10).
Nevertheless, few attempts have been made to explore the possible
involvement of cardiac metabolism in AF and the underlying pathways
remain poorly understood.
In the present study, with the aim of further
clarifying metabolic processes in the progression of AF and to
confirm whether metabolic derangements may exert important
functions in contributing to the arrhythmia occurrence, human
atrial tissues obtained from the matched cohorts in sinus rhythm
(SR) were analyzed to identify and prioritize candidate metabolites
by constructing a composite network that combined metabolic,
phenomic, and genomic data. Specifically, microarray data selection
(E-GEOD-79768) and differentially expressed genes (DEGs)
identification were first conducted. Then, a multi-omics composite
network by combining six data sets was established. Next, we mapped
the AF-related seed genes and known metabolites to the composite
network, following by the prioritization of candidate metabolite
related to AF according to the global functional relations of the
composite network using random walk with restart (RWR). Via using
the interaction score among metabolites, we extracted the top 50
metabolites and identified the top 100 co-expressed genes
interacted with the top 50 metabolites.
Materials and methods
Acquisition of data set and
pretreatment of the original data
The genechip data on AF (E-GEOD-79768) (11) were downloaded from the EMBL-EBI
database according to the platform of A-AFFY-44 - Affymetrix
GeneChip Human Genome U133 Plus 2.0 [HG-U133_Plus_2]. E-GEOD-79768
included 26 samples including data from patients with AF (n=14),
and SR samples as control (n=12).
Subsequently, the original data were pre-processed
based on a series of steps including background correction,
quantile normalization, perfectmatch/mismatch correction, and
mediapolish summarization, and transformation of probe IDs into
human gene symbols.
Identification of DEGs
As reported, the change of gene expression reflects
the propensity of a given disease. In the present study, detecting
DEGs between two groups was performed using Student's t-test. Then,
we utilized the multiple test to correct the original P-values
based on Benjamini-Hochberg (12)
using false discovery rate (FDR). DEGs were screened out when the
FDR was <0.05. These DEGs were used to establish a gene
network.
Constructing multi-omics composite
network
A composite network was constructed by combing six
data, which could be denoted by six networks, namely gene,
phenotype, metabolite, phenotype-gene association, gene-metabolite
association network, as well as phenotype-metabolite association
network.
Gene network construction
In the present study, all protein-protein
interactions (PPIs) of human having combine scores (1,048,576
interactions) were downloaded from the STRING to build the original
PPI network. Following removal of the duplicated PPIs, and
transformation of proteins into gene symbols, 1,515,370 highly
correlated gene-gene interactions among 16,785 genes were
identified to construct the background PPI network (combine-score
of edges <0.8). Subsequently, the intersection between the
16,785 genes in the background PPI network and DEGs was extracted
to construct the informative gene-gene network.
Metabolite network construction
First, a total of 4,994 human metabolites were
collected from KEGG, HMDB, Reactome, MSEA (13) and SMPDB (14). Subsequently, metabolite-metabolite
associations of human and their corresponding confidence scores
were extracted from STITCH (15),
where the the 4,994 human metabolites were covered. Finally, a
total of 3,764 human metabolites and 74,667 human
metabolite-metabolite interactions were obtained.
Phenotype network construction
In the phenotype-phenotype similarity associations
(16), there are 5,080 phenotypes
and similarity scores across them. The majority of recorded human
phenotypes were included in these phenotypes. Based on the
phenotype-phenotype similarity associations, we constructed a
phenotype network.
Gene-metabolite association network
construction
With the goal of extracting human gene-metabolite
interactions, we downloaded the chemical and gene associations of
human and the corresponding confidence scores from STITCH. After
discarding the metabolites that were not included in the metabolite
network mentioned above and removing the genes that were not
involved in the above gene network, overall 192,763 gene-metabolite
interactions were obtained among 12,342 genes as well as 3,278
metabolites.
Phenotype-gene association network
construction
We got the phenotype-gene interactions according to
the OMIM database. Following removing the phenotypes not appearing
in the phenotype network and the genes not covered in the gene
network, 2,603 gene-phenotype associations were found between 1,715
genes and 1,886 phenotypes. The weighted score was determined as 1
for every phenotype-gene interaction.
Phenotype-metabolite association
network construction
We obtained the phenotype-metabolite associations
from the HMDB. Analogously, a total of 664 associations between 388
metabolites and 149 phenotypes were reserved after filtration.
Moreover, the weighted score was defined as 1 for every
phenotype-metabolite interaction.
Establishment of a multi-omics
composite network
To identify and prioritize the potential metabolite,
the six networks mentioned above were merged into one weighted
composite network. Specific steps were as described in Yao et
al (17).
Prioritization of candidate metabolite
relying on the multi-omics composite network
The corresponding seed genes of AF were ABCC9,
GJA5, KCNA5, KCNE2, KCNJ2, KCNQ1, NPPA, NUP155, SCN1B, SCN2B,
and SCN5A, which were deposited in the database of OMIM.
Next, we mapped these seed genes and known disease metabolites to
the multi-omics composite network.
In an attempt to obtain the candidate metabolite
prioritization from the weighted composite network, the RWR method
was expanded to the multi-omics composite network (18). Then, we scored the candidate
metabolites and ranked these candidate metabolites based on
distance proximity. Based on the interaction score, we identified
the top 50 metabolites which were defined as the AF-prioritized
metabolites.
Then, based on the top 50 metabolites, co-expressed
genes interacted with the top 50 metabolites were identified, and
then, we analyzed these co-expressed genes. According to the score
distribution, the top 100 co-expressed genes were extracted.
The subnetwork on the top 50 metabolites obtained
from the composite network, and the co-expressed network were
constructed. Furthermore, degree analyses were performed for these
two networks to further identify several important AF-related
metabolites.
Results
DEG identification and construction of
multi-omics composite network
In the present study, we used DEGs to comprise the
gene nodes of the composite network, thus, we first identified the
DEGs between the two groups. Based on the FDR <0.05, a total of
622 genes were extracted as DEGs. The top 20 DEGs are shown in
Table I. This method identified and
prioritized the disease-related metabolites via combining
multi-omics information. In the present study, we first established
a multi-omics composite network by merging information derived from
the genome, phenome, and metabolome. There were three kinds of
nodes (metabolite, gene, and phenotype) and six kinds of
interactions (gene-gene, phenotype-phenotype,
metabolite-metabolite, phenotype-gene, gene-metabolite, and
phenotype-metabolite) in the multi-omics composite network. In this
network, there were 9,415 nodes and 10,227,292 edges (Table II).
 | Table I.The top 20 differentially expressed
genes (DEGs). |
Table I.
The top 20 differentially expressed
genes (DEGs).
| Gene symbols | FDR | Gene symbols | FDR |
|---|
| LBH | 9.23E-06 | IDH3A | 1.73E-04 |
| PPP4C | 7.52E-05 | RNF141 | 1.85E-04 |
| NOL6 | 7.48E-05 | TOMM22 | 2.14E-04 |
| AGK | 1.25E-04 | TSPAN12 | 2.78E-04 |
| DEDD | 1.37E-04 | PDK1 | 2.92E-04 |
| THOC6 | 1.55E-04 | SAFB | 3.20E-04 |
| COL21A1 | 1.61E-04 | LRIF1 | 3.21E-04 |
| CAPZA2 | 1.65E-04 | INPP4A | 3.26E-04 |
| FLI1 | 1.69E-04 | ZBTB43 | 3.32E-04 |
| KLHL12 | 1.71E-04 | SETD5 | 3.39E-04 |
| Table II.The composite network. |
Table II.
The composite network.
| Statistics of the
composite network | Nodes | Edges |
|---|
| Gene-gene
network | 571 | 4,254 |
| Metabolite-metabolite
network | 3764 | 74,667 |
| Phenotype-phenotype
network | 5080 | 10,140,046 |
| Gene-metabolite
association network | 571 | 3,763 |
| Phenotype-gene
association network | 5080 | 2,600 |
|
Phenotype-metabolite association
network | 537 | 664 |
| Sum | 9415 | 10,227,292 |
Prioritization of the risk metabolites
of AF
There are 11 disease-related genes related to AF in
OMIM, including ABCC9, GJA5, KCNA5, KCNE2, KCNJ2, KCNQ1, NPPA,
NUP155, SCN1B, SCN2B, and SCN5A, which were extracted
and defined as seed genes. Nevertheless, no known disease
metabolite data on AF were deposited in HMDB. In our work, the
whole metabolome as candidates, the phenotype of AF and the 11
disease genes were utilized as seeds. With the goal of illustrating
the intrinsic mode of this method, we sorted the metabolites of the
composite network in descending order based on the interaction
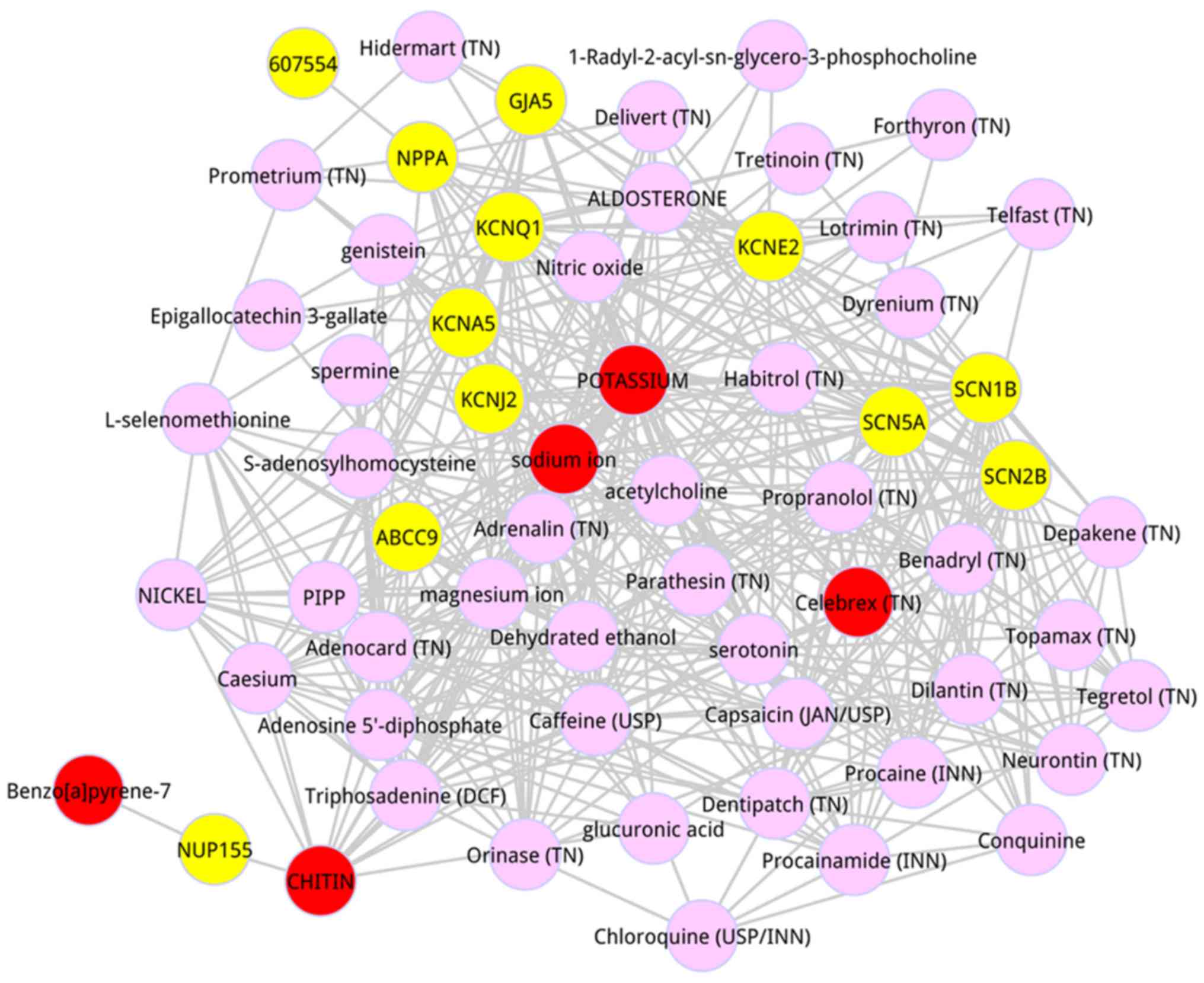
scores. The top 50 metabolites were dissected, as shown in Table III. The top 5 metabolites were
respectively potassium (score = 0.004268), sodium ion (score =
0.002251), Chitin (score = 0.001765),
Benzo[a]pyrene-7,8-dihydrodiol-9,10-oxide (score = 0.001509), and
Celebrex (TN) (score = 0.001131). A subnetwork was detected from
the whole composite network in which the top 50 metabolites were
included (Fig. 1). Based on the
degree analysis for the entire composite network, we found that
potassium had the highest degree (degree = 42), and sodium ion also
possessed the higher degree (degree = 39).
| Table III.The top 50 metabolites. |
Table III.
The top 50 metabolites.
| Metabolite ID | Metabolite
name | Score |
|---|
| 813 | Potassium | 0.004268 |
| 923 | Sodium ion | 0.002251 |
| 24139 | Chitin | 0.001765 |
| 41322 |
Benzo[a]pyrene-7,8-dihydrodiol-9,10-oxide | 0.001509 |
| 2662 | Celebrex (TN) | 0.001131 |
| 5957 | Triphosadenine
(DCF) | 0.001094 |
| 3676 | Dentipatch
(TN) | 0.001053 |
| 935 | Nickel | 0.001025 |
| 187 | Acetylcholine | 0.000897 |
| 1775 | Dilantin (TN) | 0.000829 |
| 2554 | Tegretol (TN) | 0.000805 |
| 5816 | Adrenalin (TN) | 0.000798 |
| 1103 | Spermine | 0.000772 |
| 105024 |
L-selenomethionine | 0.000758 |
| 945 | Nitric oxide | 0.000753 |
| 3348 | Telfast (TN) | 0.000735 |
| 888 | Magnesium ion | 0.000725 |
| 24755493 | PIPP | 0.000711 |
| 4913 | Procainamide
(INN) | 0.000709 |
| 3446 | Neurontin (TN) |
0.00064 |
| 5505 | Orinase (TN) |
0.00063 |
| 5284627 | Topamax (TN) | 0.000603 |
| 2337 | Parathesin
(TN) | 0.000546 |
| 4946 | Propranolol
(TN) | 0.000536 |
| 5202 | Serotonin | 0.000535 |
| 439155 |
S-adenosylhomocysteine | 0.000521 |
| 2812 | Lotrimin (TN) | 0.000515 |
| 89594 | Habitrol (TN) | 0.000484 |
| 4914 | Procaine (INN) | 0.000483 |
| 24798682 |
1-Radyl-2-acyl-sn-glycero-3-phosphocholine |
0.00048 |
| 5839 | Aldosterone |
0.00047 |
| 5354618 | Caesium | 0.000439 |
| 5280961 | Genistein | 0.000429 |
| 10114 | Hidermart (TN) | 0.000426 |
| 441074 | Conquinine | 0.000409 |
| 6022 | Adenosine
5-diphosphate | 0.000395 |
| 444795 | Tretinoin (TN) |
0.00038 |
| 1548943 | Capsaicin
(JAN/USP) | 0.000367 |
| 3121 | Depakene (TN) | 0.000364 |
| 702 | Dehydrated
ethanol | 0.000361 |
| 5819 | Forthyron (TN) | 0.000342 |
| 172198 | Delivert (TN) | 0.000327 |
| 60961 | Adenocard (TN) | 0.000327 |
| 94715 | Glucuronic
acid | 0.000317 |
| 5994 | Prometrium
(TN) |
0.0003 |
| 2519 | Caffeine (USP) | 0.000293 |
| 65064 | Epigallocatechin
3-gallate | 0.000273 |
| 8980 | Benadryl (TN) | 0.000271 |
| 5546 | Dyrenium (TN) | 0.000267 |
| 2719 | Chloroquine
(USP/INN) | 0.000264 |
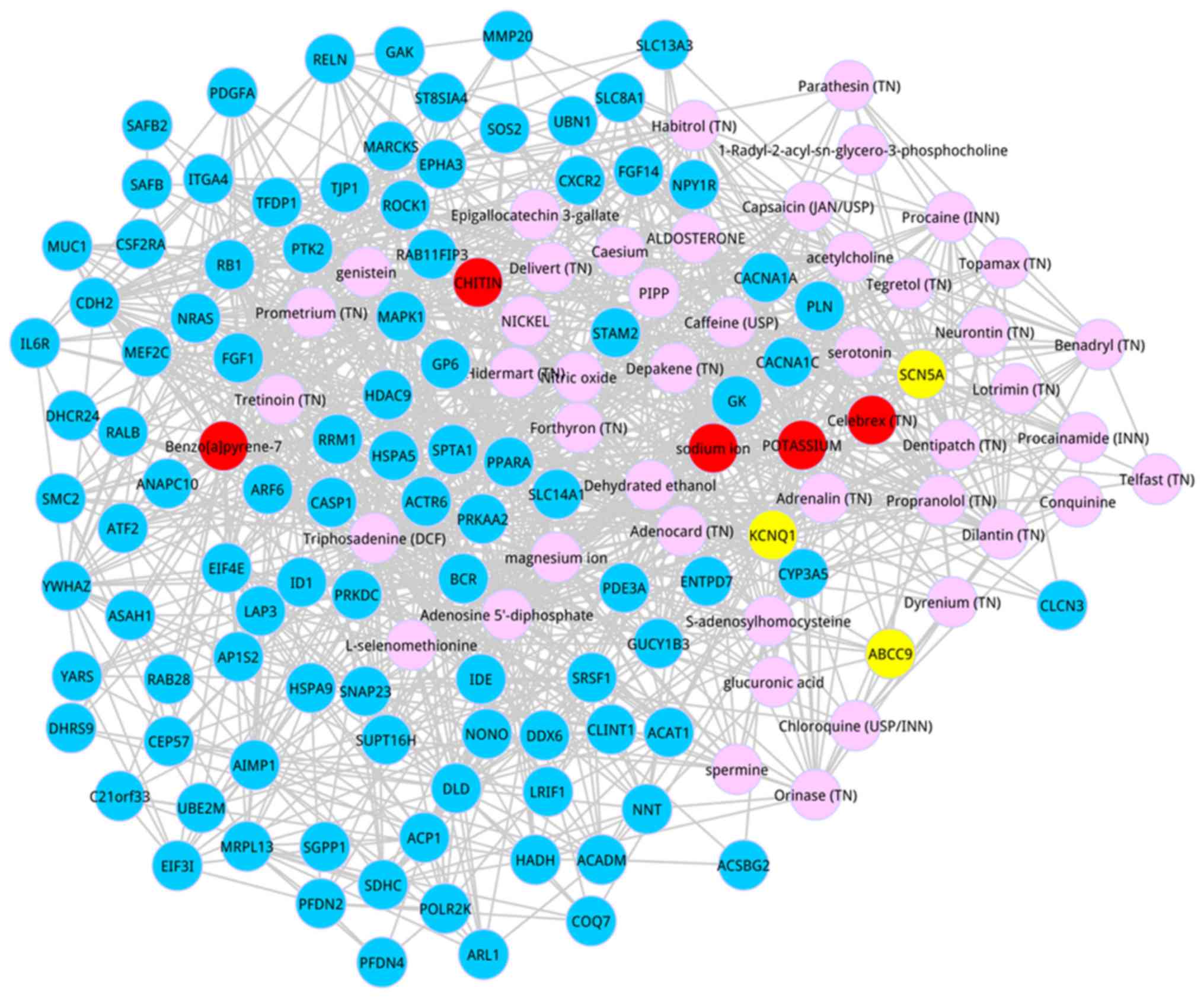
Subsequently, we identified the co-expressed genes
which interacted with the top 50 metabolites based on the score
ranking. Based on the defined condition, we identified the top 100
genes, and the co-expressed network of the top 100 genes and the
top 50 metabolites are shown in Fig.
2. After degree analysis for the co-expressed network, 4
metabolites possessed the degree >50, including adenosine
5-diphosphate (degree = 58), magnesium ion (degree = 57), potassium
(degree = 52), and sodium ion (degree = 52).
Discussion
AF is the most common cardiac arrhythmia, but the
molecular metabolites remain undefined. To the best of our
knowledge, the present study was the first to compare the
expression of metabolites between both atria in AF and SR. Based on
the results, we found that the top metabolites of potassium and
sodium ion had the highest degrees in the composite network and
co-expressed network.
Potassium is reported to be connected with a higher
risk of cardiovascular disease, for example, ventricular
arrhythmias and cardiac arrest (19). Many studies have suggested that
increased inward-rectifier K+ current (IK1) is a prominent feature
of atrial electrical remodeling (20–22).
Moreover, AF is a final common endpoint of atrial remodeling
(23). Olesen et al (24) demonstrated that enhanced potassium
current increases AF susceptibility and Linz et al (25) demonstrated that blocking the
activated atrial potassium currents inhibits AF in a pig model of
obstructive apnea. Accordingly, we infer that the changes of
potassium during AF might contribute to the self-perpetuating
nature of the arrhythmia.
Another metabolite of sodium ion also had the higher
degree in our study.
Sodium current is responsible for the early fast
depolarization upstroke of the cardiac action potential (26). Reduced sodium current has been
demonstrated to shorten the refractory period, and then to create a
substrate for re-entry, thereby contributing to AF susceptibility
(27,28). Importantly, decreased sodium current
may slow the electric conduction, and electrical remodeling plays
important roles in the development of AF (29). Moreover, AF-related mutations have
been found in sodium ion channel subunits, for example, SCN5A,
SCN1B, and SCN2B (30).
Thus, we speculate that sodium ion is highly associated with the
progression of AF.
Our work is the first to implement the analysis on
AF by identifying the metabolites through systematically
integrating multi-data including metabolic, phenomic, and genomic
information. This is the main strong advantage of our work. We
successfully detected several significant metabolites according to
this computational method. Nevertheless, in the process of our
study, we carried out an in-depth analysis using extremely
well-matched but small size samples. The evaluation of the
influence of metabolism underlying AF progression will need further
exploration based on larger independent data sets. Moreover, our
study merely fixed on analysis based on the bioinformatics, but the
findings were not proven by the experiments. Further, this work is
based on the gene expression in transcriptional level, not protein
level. Consequently, further validation studies and functional
experiments are required to confirm the significance of these
initial discoveries. Despite these limitations, we believe that the
prioritized metabolites can provide researchers valuable
information for focusing research efforts to explore the molecular
mechanisms of disease, and extracting potential bio-signatures for
diagnosis and treatment of AF.
In a nutshell, metabolites such as potassium and
sodium ion might be potential biomarkers for AF. Moreover, these
metabolites might provide worthy clues for early diagnostic and
therapeutic targets for AF.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the
National Natural Science Foundation of China: No. 81460055.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZTY and JMH designed the study, revised the figures
and drafted the manuscript. WLL, JWL and KZ acquired and analyzed
the data, and prepared the figures. All authors read and approved
the final manuscript.
Ethics' approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mahida S: Transcription factors and atrial
fibrillation. Cardiovasc Res. 101:194–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Santulli G and Totary-Jain H: Tailoring
mTOR-based therapy: Molecular evidence and clinical challenges.
Pharmacogenomics. 14:1517–1526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thomas IC and Sorrentino MJ: Bleeding risk
prediction models in atrial fibrillation. Curr Cardiol Rep.
16:4322014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garg A and Akoum N: Atrial fibrillation
and heart failure: Beyond the heart rate. Curr Opin Cardiol.
28:332–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Santulli G, Iaccarino G, De Luca N,
Trimarco B and Condorelli G: Atrial fibrillation and microRNAs.
Front Physiol. 5:152014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fiehn O: Metabolomics - the link between
genotypes and phenotypes. Plant Mol Biol. 48:155–171. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nicholson JK and Wilson ID: Opinion:
Understanding ‘global’ systems biology: Metabonomics and the
continuum of metabolism. Nat Rev Drug Discov. 2:668–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Holmes E, Wilson ID and Nicholson JK:
Metabolic phenotyping in health and disease. Cell. 134:714–717.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ritchie MD, Holzinger ER, Li R,
Pendergrass SA and Kim D: Methods of integrating data to uncover
genotype-phenotype interactions. Nat Rev Genet. 16:85–97. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blekherman G, Laubenbacher R, Cortes DF,
Mendes P, Torti FM, Akman S, Torti SV and Shulaev V: Bioinformatics
tools for cancer metabolomics. Metabolomics. 7:329–343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tsai FC, Lin YC, Chang SH, Chang GJ, Hsu
YJ, Lin YM, Lee YS, Wang CL and Yeh YH: Differential left-to-right
atria gene expression ratio in human sinus rhythm and atrial
fibrillation: Implications for arrhythmogenesis and thrombogenesis.
Int J Cardiol. 222:104–112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benjamini Y, Drai D, Elmer G, Kafkafi N
and Golani I: Controlling the false discovery rate in behavior
genetics research. Behav Brain Res. 125:279–284. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia J and Wishart DS: MSEA: A web-based
tool to identify biologically meaningful patterns in quantitative
metabolomic data. Nucleic Acids Res 38 (Web Server). W71–7. 2010.
View Article : Google Scholar
|
|
14
|
Jewison T, Su Y, Disfany FM, Liang Y, Knox
C, Maciejewski A, Poelzer J, Huynh J, Zhou Y, Arndt D, et al: SMPDB
2.0: Big improvements to the Small Molecule Pathway Database.
Nucleic Acids Res. 42:478–484. 2014. View Article : Google Scholar
|
|
15
|
Kuhn M, Szklarczyk D, Franceschini A,
Campillos M, von Mering C, Jensen LJ, Beyer A and Bork P: STITCH 2:
An interaction network database for small molecules and proteins.
Nucleic Acids Res. 38:552–556. 2010. View Article : Google Scholar
|
|
16
|
van Driel MA, Bruggeman J, Vriend G,
Brunner HG and Leunissen JA: A text-mining analysis of the human
phenome. Eur J Hum Genet. 14:535–542. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yao Q, Xu Y, Yang H, Shang D, Zhang C,
Zhang Y, Sun Z, Shi X, Feng L, Han J, et al: Global prioritization
of disease candidate metabolites based on a multi-omics composite
network. Sci Rep. 5:172012015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Jiang R, Zhang MQ and Li S:
Network-based global inference of human disease genes. Mol Syst
Biol. 4:1892008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Macdonald JE and Struthers AD: What is the
optimal serum potassium level in cardiovascular patients? J Am Coll
Cardiol. 43:155–161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Atienza F, Almendral J, Moreno J,
Vaidyanathan R, Talkachou A, Kalifa J, Arenal A, Villacastín JP,
Torrecilla EG, Sánchez A, et al: Activation of inward rectifier
potassium channels accelerates atrial fibrillation in humans:
Evidence for a reentrant mechanism. Circulation. 114:2434–2442.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gaborit N, Steenman M, Lamirault G, Le
Meur N, Le Bouter S, Lande G, Léger J, Charpentier F, Christ T,
Dobrev D, et al: Human atrial ion channel and transporter subunit
gene-expression remodeling associated with valvular heart disease
and atrial fibrillation. Circulation. 112:471–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nattel S, Maguy A, Le Bouter S and Yeh YH:
Arrhythmogenic ion-channel remodeling in the heart: Heart failure,
myocardial infarction, and atrial fibrillation. Physiol Rev.
87:425–456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wakili R, Voigt N, Kääb S, Dobrev D and
Nattel S: Recent advances in the molecular pathophysiology of
atrial fibrillation. J Clin Invest. 121:2955–2968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Olesen MS, Bentzen BH, Nielsen JB,
Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen
JH and Schmitt N: Mutations in the potassium channel subunit KCNE1
are associated with early-onset familial atrial fibrillation. BMC
Med Genet. 13:242012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Linz D, Schotten U, Neuberger H, Böhm M
and Wirth K: Combined blockade of early and late activated atrial
potassium currents suppresses atrial fibrillation in a pig model of
obstructive apnea. Heart rhythm. 8:1933–1999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Darbar D, Kannankeril PJ, Donahue BS,
Kucera G, Stubblefield T, Haines JL, George AL Jr and Roden DM:
Cardiac sodium channel (SCN5A) variants associated with atrial
fibrillation. Circulation. 117:1927–1935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nattel S: New ideas about atrial
fibrillation 50 years on. Nature. 415:219–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Terrenoire C, Simhaee D and Kass RS: Role
of sodium channels in propagation in heart muscle: How subtle
genetic alterations result in major arrhythmic disorders. J
Cardiovasc Electrophysiol. 18:900–905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Opacic D, van Bragt KA, Nasrallah HM,
Schotten U and Verheule S: Atrial metabolism and tissue perfusion
as determinants of electrical and structural remodelling in atrial
fibrillation. Cardiovasc Res. 109:527–541. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Watanabe H, Darbar D, Kaiser DW,
Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ and Roden
DM: Mutations in sodium channel β1- and β2-subunits associated with
atrial fibrillation. Circ Arrhythm Electrophysiol. 2:268–275. 2009.
View Article : Google Scholar : PubMed/NCBI
|