Introduction
Cancer is one of the leading causes of mortality
worldwide and lung cancer is the highest cause of mortality among
all cancer types (1–5). Various strategies have been used to
combat lung cancer (6–8). For example, cisplatin-based compounds
have been widely used as therapeutics (9,10).
However, long-term exposure to cisplatin can induce drug
resistance, which compromises the therapeutic intervention.
Revealing and understanding the fundamental mechanisms of lung
cancer is essential for modifying current methods and improving
treatment. Therefore, there is a requirement to identify new
targets to improve the therapeutic index from current
therapeutics.
Multiple-factors mediate the tumor microenvironment
including inflammation, immune, hemostasis/coagulation and
metabolism, which serves a critical role in the generation and
progression of lung cancer (11,12). The
inflammatory response to various infections and injuries is a
critical cause of cancer through the initiation of molecular and
epigenetic regulatory mechanisms (13).
The nuclear factor-κB (NF-κB) family is a family of
transcription factors that serve a key role in the regulation of
inflammation (14). NF-κB regulates
cell growth, differentiation, proliferation and apoptosis (15). The NF-κB complex includes p65, RelB,
c-Rel, p50 and p52 (16,17). p65 is activated by kinase-mediated
phosphorylation and is translocated into the nucleus to mediate
gene expression levels (18,19).
Response gene to complement (RGC)-32 is a previously
identified molecule that regulates the cell cycle in response to
complement activation (20,21). RGC-32 may serve a critical role in
the control of the cell cycle, complement-mediated inflammation,
tumor metastasis, cell proliferation, migration and
tubulointerstitial fibrosis (22–25).
RGC-32 is widely expressed in various organs, including the liver,
skeletal muscle, heart, brain, kidney, pancreas, vessels and
placenta (26). RGC-32 gene
methylation is reported to be associated with the development of
lung cancer (27). However, the
mechanism on how RGC-32 mediates non-small-cell lung cancer (NSCLC)
cell proliferation remains unclear.
The present study demonstrated that RGC-32 is
involved in the progression of lung cancer by regulating the
NF-κB/p65-mediated inflammatory pathway. Therefore, RGC-32 is a
potential target for effective lung cancer therapy.
Materials and methods
Human samples (date range:
2016.1–2018.12)
A total of 17 male (age range, 48–68 years) and six
female (age range, 51–69 years) patients with NSCLC (adenocarcinoma
or squamous carcinoma) were recruited to the present study
(Table I). The study was performed
according to the International Ethical Guidelines for Biomedical
Research Involving Human Subjects published by The Council for
International Organizations of Medical Sciences and was approved by
the Institutional Review Board of Shiyan Taihe Hospital, Hubei
University of Medicine. Written informed consent was obtained from
all participating individuals. Lung tissues specimens were
collected during intraoperative and postoperative examinations with
informed consent from the patients or their family members.
 | Table I.Information of patients included in
the present study. |
Table I.
Information of patients included in
the present study.
| Patients, n | Sex | Age range,
years | Pathology | Stage |
|---|
| 17 | Male | 48–68 | Squamous carcinoma
Adenocarcinoma | IIB-IIIA |
| 6 | Female | 49–69 | Squamous carcinoma
Adenocarcinoma | IB-IIIA |
Immunohistochemistry
The pulmonary cancer tissue samples obtained from
patients with lung cancer were sliced for the immunohistochemistry
assay. The cancer tissue and normal adjacent tissue samples were
fixed in 4% paraformaldehyde for 24 h at room temperature and
embedded in paraffin. The tissue was then sliced to a thickness of
5-µm. Following rehydration and antigen retrieval, sections were
blocked with 5% goat serum (Sigma-Aldrich; Merck KGaA) for 1 h at
room temperature, permeabilized with 0.01% Triton X-100 in PBS and
incubated with RGC-32 antibody (cat. no. 4445000; Novus
Biologicals, LLC; 1:100) overnight at 4°C. Following incubation
with horseradish peroxidase-conjugated secondary antibody (cat. no.
111-055-003; Jackson ImmunoResearch Laboratories, Inc.; 1:200) for
30 min at room temperature, the sections were counterstained with
hematoxylin for 2 sec at room temperature. Cross-sectional images
were captured with a Nikon light microscope at room temperature
(magnification, ×200).
Construction of adenovirus (Ad)
expression vectors for RCG-32 and short hairpin (sh)RCG-32
Construction of RGC32 overexpression or short
hairpin RNA (shRNA) adenoviral vector were prepared as previously
described (22). shRGC-32 was
designed by OriGene Technologies, Inc. The shRNA sequences were as
follows: 5′-CTGAATTCTCCAACAGACT-3′. A vector expressing RGC-32 was
constructed as described previously (22) and cells were transfected with
Ad-RGC32 or Ad-shRGC32 for 48 h [10−10 PFU/ml; 100
multiplicity of infection (MOI)], according to the manufacturer's
protocol. Under a fluorescent microscope, it was determined that
>95% of the cells were transfected with different MOIs (Ad-RGC32
or Ad-shRGC32) to determine the optimal MOI. Ad-RGC32 was purified
by gradient density ultracentrifugation with cesium chloride.
Cells proliferation assay
A549 cells (cat. no. CCL-185; American Type Culture
Collection) and H460 cells (kindly provided by Stem Cell Bank,
Chinese Academy of Sciences) were cultured in DMEM (Gibco;
Invitrogen; Thermo Fisher Scientific, Inc.) containing 10% FBS
(Thermo Fisher Scientific, Inc.). Cells were transfected with
either Ad-RGC32 or Ad-shRGC32 for overexpression or knockdown of
RGC32. NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) was used
to evaluate the effect of RGC32 overexpression. The transfection of
adenoviral vector (Null) was used as a control. Briefly, the cells
were seeded into a 96-well plate at a density of 2×103
cells/well and incubated for 24 h at 37°C in 5% CO2
incubator. After transfection, cells were cultured for 24, 48 or 72
h for a time-dependent study. Subsequently, the culture medium was
discarded and the wells were washed twice with PBS, followed by the
addition of 10 µl Cell Counting Kit-8 reagent (cat. no. CK04;
Dojindo Molecular Technologies, Inc.) or 10 µl MTT dye (0.5 mg/ml;
Beijing Solarbio Science & Technology, Co., Ltd.) to each well,
respectively. The cells were incubated for a further 2 h at 37°C.
The samples were then detected at 450 nm using a Microplate reader
(Bio-Rad Laboratories, Inc.). Each treatment was replicated and
each experiment was repeated 3–5 times.
Immunofluorescence assay
To observe NF-κB p65 nuclear translocation, A549
cells transfected with Ad-Null, Ad-RGC32 or Ad-shRGC32 were rinsed
with PBS and fixed with 4% paraformaldehyde for 20 min at room
temperature, followed by blocking with 5% goat serum in 0.01%
Triton X-100 in PBS for 1 h at room temperature The cells were then
incubated with anti-NF-κB p65 antibody (cat. no. sc-109; Santa Cruz
Biotechnology, Inc.; 1:100) overnight at 4°C, followed by
tetramethylrhodamine-conjugated secondary antibody (cat. no.
711-585-152; Jackson ImmunoResearch Laboratories, Inc.; 1:200) for
1 h at room temperature. The cellular nucleus was then
counterstained with DAPI (Sigma-Aldrich; Merck KGaA) for 10 min at
room temperature. Cells were subsequently imaged using fluorescence
microscopy (magnification, ×400).
Nuclear protein extraction
CHEMICON® Nuclear Extraction kit (EMD
Millipore) was used. Briefly, A549 cells (4×104
cells/well) were washed with cold PBS buffer, followed by
incubation with cytoplasmic lysis buffer. The whole lysis solution
was centrifuged at 4°C at a speed of 1,800 × g for 5 min and the
cellular pellet was further incubated with nuclear extraction
buffer. The supernatant was then used for western blot
analysis.
Western blotting analysis
The lung tissues were ground in liquid nitrogen and
homogenized on ice with RIPA buffer containing protease inhibitors
(Beyotime Institute of Biotechnology, China). Protein concentration
was measured using a bicinchoninic acid protein assay kit.
Subsequently, 20 µg protein from tissues and whole cell lysates was
subjected to 12% SDS-PAGE and transferred to PVDF membranes
(Bio-Rad Laboratories, Inc.). The blots were blocked with 5%
fat-free milk at room temperature for 1 h by shaking and then
incubated with rabbit anti-RGC32 (cat. no. 4445000; Novus
Biologicals, LLC; 1:500) and rabbit p65 (sc-109; Santa Cruz
Biotechnology, Inc.; 1:1,000) at 4°C overnight, followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies (cat. no. 111-035-003; Jackson ImmunoResearch
Laboratories, Inc.; 1:200) at room temperature for 1 h by shaking.
The immunized blots were then visualized and analyzed with
chemiluminescence reagent (Novex™ ECL Chemiluminescent Substrate
Reagent kit; cat. no. WP20005) and Image J 1.51 software (National
Institutes of Health).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Roche Diagnostics GmbH). RNA (1 µg) was reverse
transcribed with a RevertAid RT Reverse Transcription kit (cat. no.
K1691; Thermo Fisher Scientific, Inc.) to amplify the genes using
the following temperature protocol: 25°C for 5 min, 42°C for 60 min
and 70°C for 5 min. The primers used for qPCR were as follows:
Vascular cell adhesion protein 1 (VCAM1) forward,
5′-CCTGAGCCCTGTGAGTTTTG-3′ and reverse, 5′-GGGTACACGCTAGGAACCTT-3′;
interleukin-6 (IL-6) forward, 5′-AGTCCTGATCCAGTTCCTGC-3′ and
reverse, 5′-CTACATTTGCCGAAGAGCCC-3′; vascular endothelial growth
factor A (VEGFA) forward, 5′-GAACTTTCTGCTGTCTTGGG-3′ and reverse,
5′-CTTCGTGATGATTCTGCCCT-3′; cyclin dependent kinase inhibitor 2C
(CDKN2C) forward, 5′-CCCGATTTGAAAGACCGAAC-3′ and reverse,
5′-TCACCAGGAACTCCACCACC-3′; and TES forward,
5′-AAGAAGATGGGCTTAGGTC-3′ and reverse, 5′-TTTTGCAATCAGAGTGGTA-3′.
All qPCR reactions (cat. no. 04913850001; Roche Diagnostics GmbH)
were performed in triplicate with an ABI-7000 PCR detection machine
(28). Denaturation was performed at
95°C for 10 min, followed by 40 cycles at 95°C for 30 sec and 30
sec annealing at 60°C.
Statistical analysis
The results are expressed as the mean ± standard
error of the mean. A Student's t-test was used for comparisons
between two groups and one-way analysis of variance was used for
comparisons of more than two groups. Significance was confirmed by
post-hoc analysis using Fisher's least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference. Each experiment was repeated three times.
Results
NSCLC tissue exhibits a high
expression level of RGC-32
Immunohistochemistry staining of tissues derived
from patients and healthy controls demonstrated that RGC-32 was
specifically expressed in the NSCLC tissue (Fig. 1A). Semi-quantification of the image
revealed that the expression of RGC-32 was 5.2-fold higher in NSCLC
tissue compared with normal tissue (Fig.
1B). Western blot analysis demonstrated that the protein
expression level of RGC-32 was significantly increased in NSCLC
tissues compared with normal tissues (P<0.001; Fig. 1C and D). These data suggest that
RGC-32 is highly expressed in NSCLC.
RGC-32 promotes lung cancer cell
proliferation
To determine the role of RGC-32 in human NSCLC
cells, RGC-32 was either overexpressed or knocked down in A549
cells and H460 cells. The results revealed that RGC-32-knockdown in
both cell lines significantly inhibited proliferation compared with
the control cells (P<0.05; Fig.
2). In addition, the effect of RGC-32 overexpression on the
proliferation of A549 and H460 cells was examined by CCK-8 assay,
which revealed that overexpression of RGC-32 significantly
increased A549 and H460 cell proliferation (P<0.05; Fig. 2B and E). An MTT assay was used to
evaluate the effect of RGC-32 on the proliferation of NSCLC cells
and similar results were obtained (Fig.
2C and F). These data indicate that RGC-32 is a novel inducer
of NSCLC cell proliferation.
RGC-32 promotes the proliferation of
lung cancer cells via NF-κB
NF-κB promotes tumor growth and activation of NF-κB
is often identified in cancer, including human hepatocellular
carcinoma (29). To determine
whether the NF-κB signaling pathway mediates RGC-32 function in
A549 cell proliferation, the NF-κB inhibitor PDTC was used to treat
A549 cells expressing RGC-32. As presented in Fig. 3, overexpression of RGC-32 increased
A549 cell proliferation in a time-dependent manner. A significant
increase in cell proliferation was observed after incubation for 48
h with Ad-RGC32 (P<0.05). However, the NF-κB inhibitor PDTC
markedly inhibited the effect of Ad-RGC-32 on A549 cell
proliferation and significant differences were identified at the
48-h time point compared with the untreated RGC32-overexpressing
control cells (P<0.05). These results suggest that NF-κB is
critical in regulating RGC-32-associated A549 cell
proliferation.
RGC-32 activates NF-κB by promoting
the nuclear translocation of p65
Due to the involvement of NF-κB in RGC-32-induced
cell proliferation, the present study further investigated the role
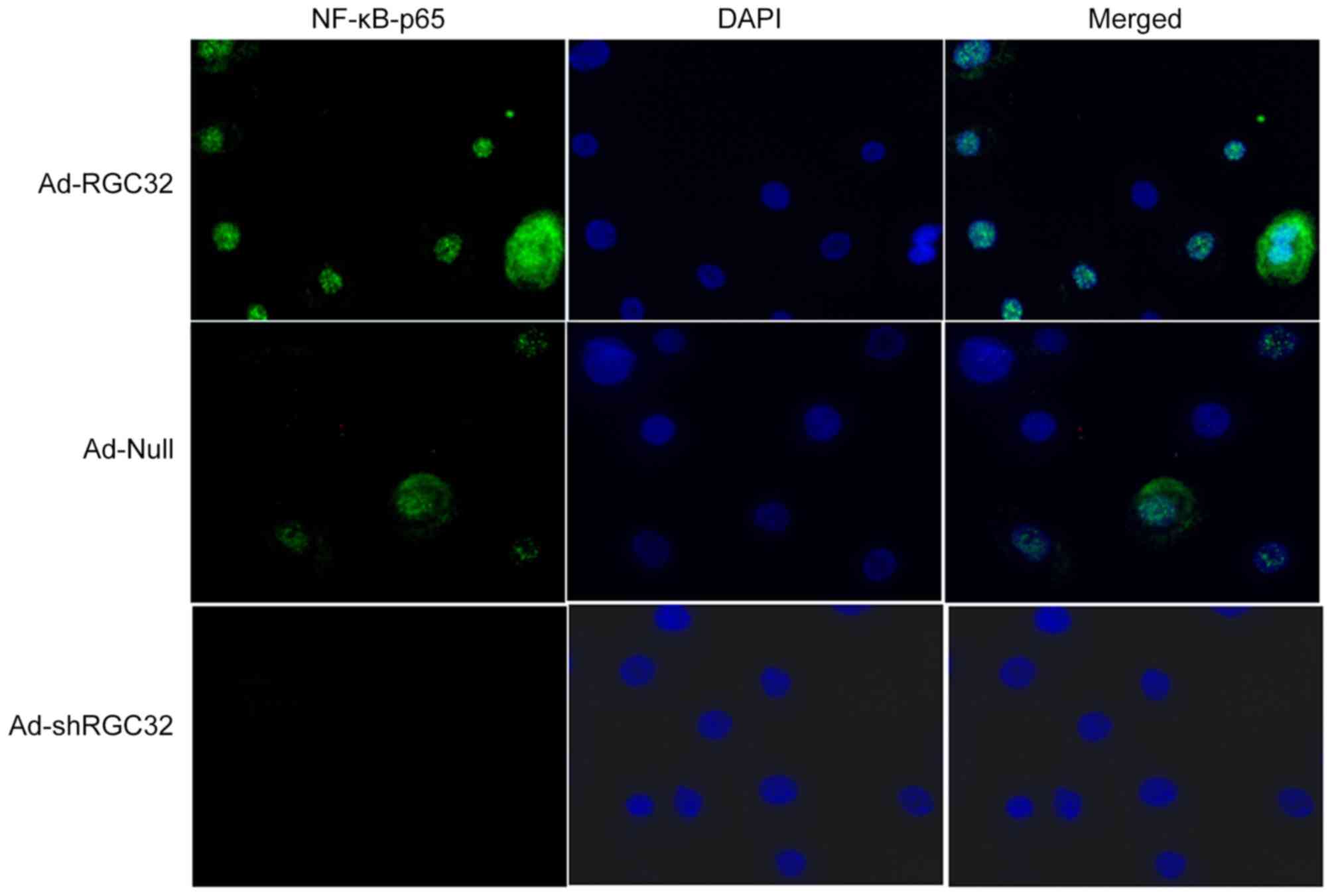
of RGC-32 in NF-κB expression. Translocation of NF-κB from the
cytoplasm to the nucleus is critical for the activation of
downstream target genes (30). Thus,
the NF-κB translocation behavior was examined. Immunofluorescence
staining demonstrated that RGC-32 overexpression increased p65
nucleus translocation compared with the control group and RGC32
knockdown reduced p65 nuclear translocation (Fig. 4). Furthermore, western blot analysis
revealed that the nuclear expression level of p65 protein in
RGC-32-overexpressing cells was significantly increased compared
with the control cells (P<0.05) and the increased level of RGC32
in the Ad-RGC32 group was also confirmed (Fig. 5). Knockdown of RGC32 by shRNA
significantly decreased the nuclear p65 level (P<0.05; Fig. 5). These results indicate that RGC-32
could regulate A549 cell proliferation by activating p65.
RGC-32 regulates gene expression
levels downstream of NF-κB p65
Active NF-κB is able to control the transcription of
various target genes, which mediates the expression of inflammatory
cytokines, chemokines and cell adhesion molecules (31). The present study examined target
genes of NF-κB, including IL-6, VCAM1, VEGFA, CDKN2C and TES. As
presented in Fig. 6, VCAM1, VEGFA
and IL-6 mRNA levels were significantly increased in the Ad-RGC32
group (P<0.05). By contrast, knockdown of RGC32 by shRNA
decreased the expression levels. Meanwhile, the expression levels
of CDKN2C and TES, a cell cycle inhibitor (32–34),
were decreased in Ad-RGC32 cells expressing RGC32 compared with the
untreated control cells. Knockdown of RGC32 by shRNA increased
CDKN2C and TES mRNA levels. Thus, the findings suggest that
RGC-32-driven tumor growth is associated with genes downstream of
NF-κB p65.
 | Figure 6.RGC-32 regulates the downstream genes
of NF-κB p65. The expression levels of NF-κB downstream genes (A)
VCAM1, (B) VEGFA, (C) IL-6, (D) CDKN2C and (E) TES in A549 cells
following treatment with Ad-RGC32 or Ad-shRGC32, as determined by
reverse transcription-quantitative polymerase chain reaction. n=3;
*P<0.01 vs. Ad-Null group. RGC32, response gene to
complement-32; VCAM1, vascular cell adhesion protein 1; VEGFA,
vascular endothelial growth factor A; CDKN2C, cyclin dependent
kinase inhibitor 2C; IL-6, interleukin-6; sh, short hairpin; NF,
nuclear factor; Ad, adenovirus; sh, short hairpin. |
Discussion
RGC-32 is a previously identified molecule that acts
in response to complement activation (20). RGC-32 is a cell-cycle modulator and
RGC-32 overexpression has been reported in breast and colon cancers
(35,36). Highly expressed RGC-32 activates the
cell cycle by regulating CDC2 (35).
The epigenetic modulation of RGC-32 (promoter methylation) is
reported to be significantly associated with the prognosis of
patients with lung cancer (27). The
present study identified that RGC-32 was overexpressed in human
lung cancer tissue compared with normal controls and knockdown of
RGC-32 inhibited the activity of lung cancer cells, which is
consistent with the previous study by Xu et al (37). Thus, RGC-32 could be used as an
important marker for lung cancer.
Inflammation targeting endothelial cells in the
pulmonary tract could act as the most common cause of various lung
diseases, including lung cancer (13). NF-κB activation following an
inflammatory response contributes to abnormalities of the pulmonary
tract (38). NF-κB is a major factor
of inflammation and serves a critical role in the progression of
lung cancer (39). Therefore, it is
important to identify the upstream factors associated with the
NF-κB pathway that regulate lung cancer development. RGC-32 has
been reported to stimulate epithelial-mesenchymal transition in
lung cancer cells via the NF-κB signaling pathway (39). Similarly, the present study
demonstrated that RGC-32 accelerated the translocation of p65 into
the nucleus. Hence, activation of NF-κB/p65 by RGC-32 could play an
important role in lung cancer development.
In addition, NF-κB/p65 can regulate downstream genes
associated with cancer development, including VCAM1, IL-6, CDKN2C,
TES and VEGFA (40–42). In the present study, VCAM1, VEGFA and
IL-6 mRNA levels were increased in RGC32-overexpressed A549 cells.
By contrast, knockdown of RGC32 by shRNA reduced the expression
levels. CDKN2C and TES, cell cycle inhibitors (32–34),
exhibited decreased mRNA levels in Ad-RGC32-treated A549 cells, and
knockdown of RGC32 by shRNA increased CDKN2C and TES mRNA levels.
Therefore, RGC-32 could regulate lung cancer growth by regulating
the expression of genes downstream of NF-κB p65. The physical
interaction of RGC32 with NF-κB has been confirmed by a
co-immunoprecipitation assay (unpublished data). These results
suggest that RGC32 may be associated with NF-κB both functionally
and physically.
In conclusion, RGC-32 may be a novel and specific
marker of NSCLC. RGC-32-mediated NSCLC development has been
demonstrated to involve p65 activation. Therefore, RGC-32 may be a
new target for preventative and immuno-pharmacological treatments
of NSCLC.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from Hubei
Provincial Department of Education Youth Project (grant no.
Q20102104) and the Taihe Hospital Scientific Research Project
(grant no. 2017042).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ carried out main human sample collection and
detection, cell experiments and drafted the manuscript. JL carried
out qPCR, cell proliferation assays and data evaluation. LY
participated in the immunostaining and protein assays. RW
participated in the design of the study and manuscript writing. JY
conceived of the study, participated in the experimental design and
helped to draft the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Institutional
Review Board of Shiyan Taihe Hospital, Hubei University of
Medicine. Written informed consent was obtained from all
participating individuals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu WJ, Zhu KL, Xu J, Wang JL and Zhu H:
Enediyne-activated, EGFR-targeted human β-defensin 1 has
therapeutic efficacy against non-small cell lung carcinoma. Lab
Invest. 98:1538–1545. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu J, Du Y, Liu XJ, Zhu BY, Zhang SH, Li
L, Li Y, Wang XF, Shan CK, Wang RQ and Zhen YS: Recombinant
EGFR/MMP-2 bi-targeted fusion protein markedly binding to
non-small-cell lung carcinoma and exerting potent therapeutic
efficacy. Pharmacol Res. 126:66–76. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun YW, Xu J, Zhou J and Liu WJ: Targeted
drugs for systemic therapy of lung cancer with brain metastases.
Oncotarget. 9:5459–5472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wen R, Umeano AC, Chen P and Farooqi AA:
Polymer-based drug delivery systems for cancer. Crit Rev Ther Drug
Carrier Syst. 35:521–553. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Basu U, Banik B, Wen R, Pathak RK and Dhar
S: The platin-X series: Activation, targeting, and delivery. Dalton
Trans. 45:12992–13004. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pathak RK, Wen R, Kolishetti N and Dhar S:
A prodrug of two approved drugs, cisplatin and chlorambucil, for
chemo war against cancer. Mol Cancer Ther. 16:625–636. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wysoczynski M and Ratajczak MZ: Lung
cancer secreted microvesicles: Underappreciated modulators of
microenvironment in expanding tumors. Int J Cancer. 125:1595–1603.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blanco D, Vicent S, Fraga MF,
Fernandez-Garcia I, Freire J, Lujambio A, Esteller M,
Ortiz-de-Solorzano C, Pio R, Lecanda FJ and Montuenga LM: Molecular
analysis of a multistep lung cancer model induced by chronic
inflammation reveals epigenetic regulation of p16 and activation of
the DNA damage response pathway. Neoplasia. 9:840–852. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JI and Burckart GJ: Nuclear factor
kappa B: Important transcription factor and therapeutic target. J
Clin Pharmacol. 38:981–993. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tak PP and Firestein GS: NF-κB: A key role
in inflammatory diseases. J Clin Invest. 107:7–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dixit V and Mak TW: NF-kappaB signaling:
Many roads lead to Madrid. Cell. 111:615–619. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghosh S and Karin M: Missing pieces in the
NF-kappaB puzzle. Cell. 109 (Suppl):S81–S96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naumann M and Scheidereit C: Activation of
NF-kappa B in vivo is regulated by multiple phosphorylations. EMBO
J. 13:4597–4607. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sakurai H, Chiba H, Miyoshi H, Sugita T
and Toriumi W: IkappaB kinases phosphorylate NF-kappaB p65 subunit
on serine 536 in the transactivation domain. J Biol Chem.
274:30353–30356. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Badea T, Niculescu F, Soane L, Fosbrink M,
Sorana H, Rus V, Shin ML and Rus H: RGC-32 increases p34CDC2 kinase
activity and entry of aortic smooth muscle cells into S-phase. J
Biol Chem. 277:502–508. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schlick SN, Wood CD, Gunnell A, Webb HM,
Khasnis S, Schepers A and West MJ: Upregulation of the cell-cycle
regulator RGC-32 in Epstein-Barr virus-immortalized cells. PLoS
One. 6:e286382011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang JN, Shi N, Xie WB, Guo X and Chen SY:
Response gene to complement 32 promotes vascular lesion formation
through stimulation of smooth muscle cell proliferation and
migration. Arterioscler Thromb Vasc Biol. 31:e19–e26. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo X, Jose PA and Chen SY: Response gene
to complement 32 interacts with Smad3 to promote
epithelial-mesenchymal transition of human renal tubular cells. Am
J Physiol Cell Physiol. 300:C1415–C1421. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu L, Qin H, Li PY, Xu SN, Pang HF, Zhao
HZ, Li DM and Zhao Q: Response gene to complement-32 enhances
metastatic phenotype by mediating transforming growth factor
β-induced epithelial-mesenchymal transition in human pancreatic
cancer cell line BxPC-3. J Exp Clin Cancer Res. 31:292012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang JM, Shi N, Dong K, Brown SA, Coleman
AE, Boegehold MA and Chen SY: Response gene to complement 32
maintains blood pressure homeostasis by regulating α-adrenergic
receptor expression. Circ Res. 123:1080–1090. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Badea TC, Niculescu FI, Soane L, Shin ML
and Rus H: Molecular cloning and characterization of RGC-32, a
novel gene induced by complement activation in oligodendrocytes. J
Biol Chem. 273:26977–26981. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim DS, Lee JY, Lee SM, Choi JE, Cho S and
Park JY: Promoter methylation of the RGC32 gene in nonsmall cell
lung cancer. Cancer. 117:590–596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and
Ben-Neriah Y: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Njikan S, Manning AJ, Ovechkina Y, Awasthi
D and Parish T: High content, high-throughput screening for small
molecule inducers of NF-κB translocation. PLoS One.
13:e01999662018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jalili A, Wagner C, Pashenkov M, Pathria
G, Mertz KD, Widlund HR, Lupien M, Brunet JP, Golub TR, Stingl G,
et al: Dual suppression of the cyclin-dependent kinase inhibitors
CDKN2C and CDKN1A in human melanoma. J Natl Cancer Inst.
104:1673–1679. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu J, Li X, Kong X, Moran MS, Su P,
Haffty BG and Yang Q: Testin is a tumor suppressor and prognostic
marker in breast cancer. Cancer Sci. 103:2092–2101. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kravtsova-Ivantsiv Y, Shomer I,
Cohen-Kaplan V, Snijder B, Superti-Furga G, Gonen H, Sommer T, Ziv
T, Admon A, Naroditsky I, et al: KPC1-mediated ubiquitination and
proteasomal processing of NF-κB1 p105 to p50 restricts tumor
growth. Cell. 161:333–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fosbrink M, Cudrici C, Niculescu F, Badea
TC, David S, Shamsuddin A, Shin ML and Rus H: Overexpression of
RGC-32 in colon cancer and other tumors. Exp Mol Pathol.
78:116–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu Y and Hu XB: C5a stimulates the
proliferation of breast cancer cells via Akt-dependent RGC-32 gene
activation. Oncol Rep. 32:2817–2823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu R, Shang C, Zhao J, Han Y, Liu J, Chen
K and Shi W: Knockdown of response gene to complement 32 (RGC32)
induces apoptosis and inhibits cell growth, migration, and invasion
in human lung cancer cells. Mol Cell Biochem. 394:109–118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jo H, Zhang R, Zhang H, McKinsey TA, Shao
J, Beauchamp RD, Ballard DW and Liang P: NF-kappaB is required for
H-ras oncogene induced abnormal cell proliferation and
tumorigenesis. Oncogene. 19:841–849. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen W, Li Z, Bai L and Lin Y: NF-kappaB
in lung cancer, a carcinogenesis mediator and a prevention and
therapy target. Front Biosci (Landmark Ed). 16:1172–1185. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tichet M, Prod'Homme V, Fenouille N,
Ambrosetti D, Mallavialle A, Cerezo M, Ohanna M, Audebert S, Rocchi
S, Giacchero D, et al: Tumour-derived SPARC drives vascular
permeability and extravasation through endothelial VCAM1 signalling
to promote metastasis. Nat Commun. 6:69932015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen PH, Chang H, Chang JT and Lin P: Aryl
hydrocarbon receptor in association with RelA modulates IL-6
expression in non-smoking lung cancer. Oncogene. 31:2555–2565.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Naik NA, Bhat IA, Afroze D, Rasool R, Mir
H, Andrabi SI, Shah S, Siddiqi MA and Shah ZA: Vascular endothelial
growth factor A gene (VEGFA) polymorphisms and expression of VEGFA
gene in lung cancer patients of Kashmir Valley (India). Tumour
Biol. 33:833–839. 2012. View Article : Google Scholar : PubMed/NCBI
|