Introduction
Idiopathic ventricular fibrillation occurs in
patients with structural normal heart and causes unexpected cardiac
death (1,2). The primary electrical disorders
resulting from ion-channel mutations are believed to play a crucial
role. In recent years, early repolarization (ER) or J wave have
been reported to be associated with idiopathic VF (3-5),
‘Gain-of-function’ mutations in KCNJ8 (6,7) and
‘loss-of-function’ mutations in L-type calcium channel genes,
including CACNA1C, CACNB2B, and CACNA2D1(8), have been identified in patients with
early repolarization syndrome (ERS). Several missense mutations in
SCN5A gene caused ERS have also been reported (9-11).
Although there are clinical studies that reveal the
relationship between ER and higher risk of VF (12,13),
the mechanism responsible for the J wave (or ER) and its
arrhythmogenesis remains controversial. Coronary-perfused wedge
preparation shows that the transient outward current (Ito) mediated
transmural voltage gradient resulted in the inscription of J wave
(14). Conversely, several
clinical non-invasive electrophysiological studies support the
hypothesis that J waves may be more strongly associated with a
‘depolarization-dependent’ abnormality (15,16).
The present study presented the electrophysiological
characteristics of an novel frame-shift mutation of C280Sfs*61 in
the SCN5A gene in a male proband with IVF and transient appearance
of J wave in the inferior leads and prolonged S-wave upstroke in
precordial leads.
Methods and materials
Study subject
The present study was approved by the Ethics
Committee of Fuwai Hospital (approval no. 080133) and all
experiments conformed to the principles outlined in the Declaration
of Helsinki. Blood samples were obtained after the patient
volunteered to participate in this study and provided written
informed consent for publication.
Mutation screening
The genomic DNA was isolated from leukocytes with
TIANamp Blood DNA isolation kit (Tiangen Biotech Co., Ltd.)
according to the manufacturer's instructions. The known arrhythmia
syndrome suspected genes, such as KCNQ1, KCNH2, KCNE1, KCNE2,
KCNE3, KCNJ8, CACNA1C, CACNB2, GPD1L, SCN5A, SCN1B and SCN3B,
underwent comprehensive open-reading frame/splice site mutational
screening using denaturing high-performance liquid chromatography
(DHPLC) and verified by direct DNA sequencing as previously
described (9,17,18).
From March 2015 to September 2015, a total of 200 unrelated healthy
Chinese Han individuals (male 56%, female 44%; aged 18-30 years
old) consisted of the control group in Fuwai Hospital (Beijing,
China).
Site-directed mutagenesis and
heterologous expression
The full-length wild-type (WT) human SCN5A cDNA
(GenBank ID: NM198056) was subcloned into pcDNA3.1 vector for
mammalian expression in the Pathophysiology Laboratory of Fuwai
Hospital (Invitrogen; Thermo Fisher Scientific, Inc.). The mutation
(C280Sfs*61) was constructed using a QuikChange site-directed
mutagenesis kit (Stratagene; Agilent Sumitomo Dainippon Pharma Co.,
Ltd.) on the WT-SCN5A background, and verified by direct
sequencing. 293 cells were transfected with 0.6 µg cDNA of WT or
mutant channels using the Effectene method (Qiagen GmbH) (19,20).
293 cells were cultured in DMEM containing l00 ml/l fetal bovine
serum. Cells were spread in 6-well plates 24 h before transfection
and the density of cells was 70-80% at the time of transfection.
The cells were transfected into pEGFP-SCN5A, pEGFPI.100lQSCN5A and
pEGFP-SCN5A/pEGFP-L100lQ SCN5A (1:1) plasmids respectively
according to the instructions of I. ipofectaIIIi- neTM2000 and then
incubated at 37˚C with 50 ml/l CO2 for 48 h. Then,
subsequent experiments were performed. In the coexpression
experiments, WT and mutant were transfected at a 1:1 molar ratio.
Enhanced green fluorescent protein gene (0.2 µg) was co-transfected
and served as an indicator. All experiments were performed 24-48 h
after transfection. Over three independent experiments were
conducted to confirm the reproducibility of the results.
Patch-clamp recordings
Sodium current was measured using whole-cell patch
clamp techniques with Axonpatch 700B amplifiers (Molecular Devices,
LLC.) at a room temperature of 22-24˚C (21,22).
Pipette resistance ranged from 1.5-2.5 MΩ when filled with
recording solution. The bath solution contained (in mmol/l), NaCl
140, KCl 4, CaCl2 1.8, MgCl2 0.75 and HEPES 5 (pH7.4 set with
NaOH). The pipette medium contained (in mmol/l), CsF 120, CsCl 20,
EGTA 2.0, MgCl2 1.0 and HEPES 5 (pH 7.4 set with CsOH). The
standard voltage clamp protocols are presented with the data and as
described in detail previously (10).
Immunocytochemistry
The immunocytochemical experiments were performed in
293 cells transfected with WT or mutant SCN5A plasmid as previously
described. Briefly, cells were fixed in 4% paraformaldehyde and
permeabilized with 0.1% Triton X-100. Nonspecific binding was
blocked with 5% bovine serum albumin (BSA; Beyotime Institute of
Biotechnology) in PBS. Then cells were incubated with anti-Nav1.5
N-terminal monoclonal antibody (1:50; Abcam) and anti-Nav1.5
C-terminal polyclonal antibody (1:200; Alomone Labs) overnight at
4˚C (23). Then the cells were
incubated with Cy3-conjugated goat anti-rabbit (1:1,000; Jackson
ImmunoResearch Laboratories, Inc.) and FITC- conjugated goat
anti-mouse (1:500; Jackson ImmunoResearch Laboratories, Inc.) for 1
h at room temperature in the dark. Confocal images were obtained
using a confocal laser scanning microscope (FV1000; Olympus
Corporation).
Statistical analysis
INa data were analyzed using Clampfit
10.0 (Molecular Devices, LLC.) and non-linear curve fitting was
performed with OriginPro 8.5 software (OriginLab Corporation). Data
were presented as the means ± standard error of the mean (SEM).
Student's t-test analysis was used for the comparison of two means.
All statistical analyses were performed with SPSS, version 17.0.
(SPSS Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical evaluation
The proband, a 55-year-old otherwise healthy man
suffered from agonal respiration during sleep. After being woken by
his family, his respiration became smooth. However, the next day he
suddenly lost consciousness when talking with his colleagues at
work and was admitted to the local emergency room in Weixian
People's Hospital. There was no prior syncope episode and no family
history of sudden cardiac death (SCD). In the hospital, six
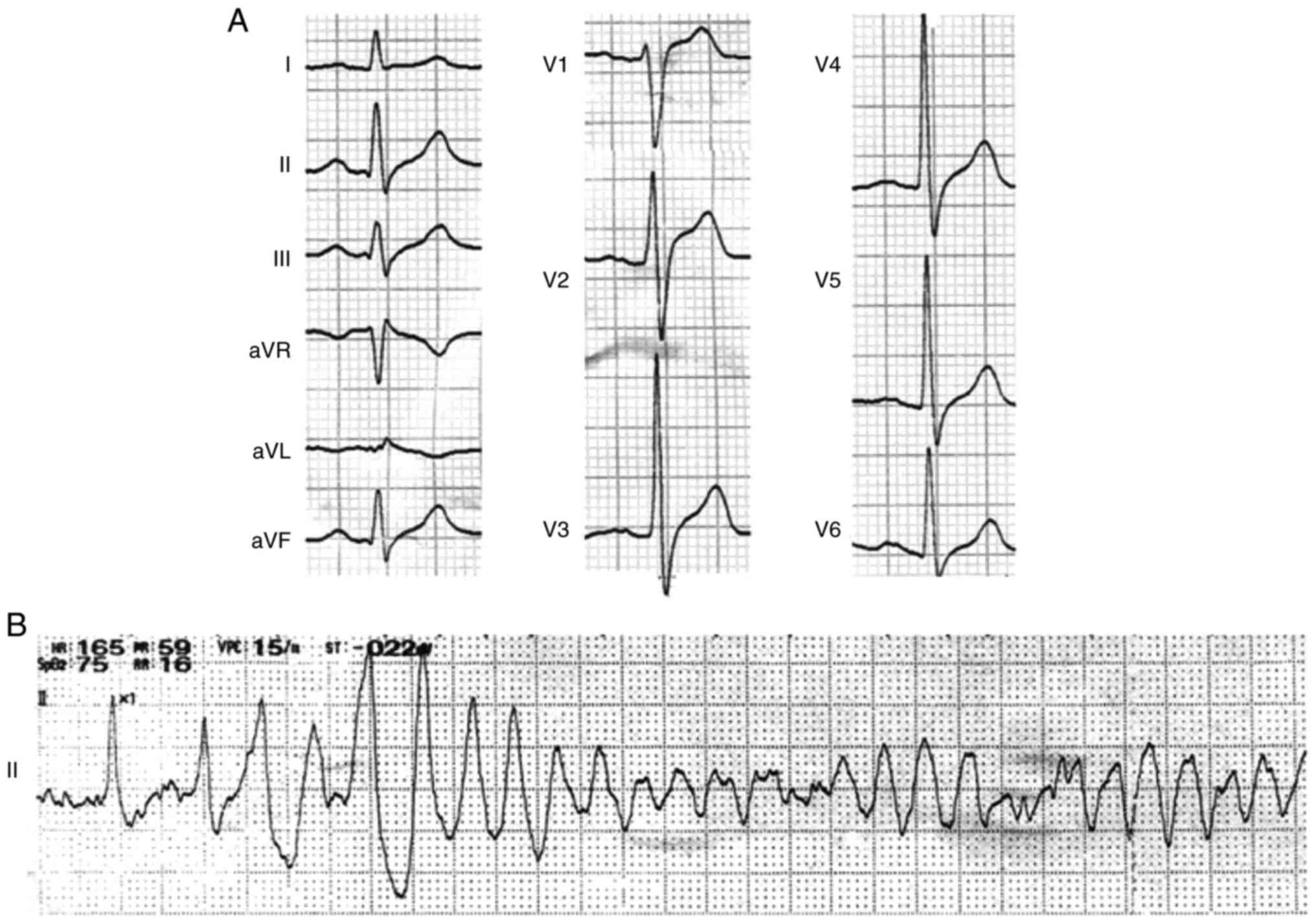
episodes of spontaneous VF (Fig.
1) were recorded during daytime and aborted by immediate
external defibrillation. However, the diagnosis is not clear, so
the patient sent to Fuwai Hospital with information on previous
tests. In Fuwai Hospital, laboratory test revealed normal levels of
serum electrolytes and cardiac enzymes. The subsequent
echocardiogram and coronary angiography showed structurally normal
heart (left ventricular ejection fraction was 61%) and normal
coronary arteries.
During the hospitalization, the repeated baseline
ECG with sinus rhythm exhibited wide and notched P wave (138 msec)
and prolongation of QRS wave (140 msec) without signs of bundle
branch block. There were also no signs of QT prolongation (QTc
interval, 389 msec). The transient J waves, however, were recorded
in the inferior leads (II, III and aVF), and prolonged S-wave
upstroke appeared in the precordial leads of V1-V3 in the same
time-frame (Fig. 2). Typical type
I Brugada ECG was not shown in the repeated recordings. In
addition, 24-h Holter recording did not reveal bradycardia,
atrioventricular block or other arrhythmias. The metoprolol and
potassium magnesium aspartata were administered after discharge.
Then, three months later, the patient came in for further
consultation. Since the sodium channel blockers, such as ajmaline,
flecainide and procainamide, were not available in China, drug
challenge was not performed. Although strongly suggested, the
patient still refused to receive an implantation of implantable
cardiac defibrillator (ICD). Regular one year and a half follow-up
showed no subsequent arrhythmic event.
Genetic analysis
A novel heterozygous 1 base (G) deletion at position
839 (c.839delG) locating in exon2 of SCN5A gene was revealed in the
proband (Fig. 3). This deletion
produced a frame-shift annotated as C280Sfs*61, which indicated the
cysteine (C) at position 280 was replaced by serine (S) resulting
in 61 amino acids frame-shift before a premature stop codon. Thus,
the mutation produced a severe truncated protein which ended at the
extracellular domain between segment 5 and 6 in domain I. No other
disease-causing mutation was identified in the proband and the
mutation was not found in 200 control subjects. Neither his
daughter or son inherited this mutation.
Electrophysiological
characterization
The transfected 293 cells transiently expressing the
Nav1.5-WT or Nav1.5-C280Sfs*61 were voltage clamped after 24-48 h
incubation. Given the nature and severity of the truncation
mutation, the C280Sfs*61 mutant produced no detectable sodium
current as expected, which conformed that the truncated protein was
nonfunctional. Fig. 4 showed the
representative current traces.
 | Figure 4Electrophysiological characterization
of Nav1.5 channels. (A) Representative sodium current
(INa) recordings of WT, C280Sfs*61 and WT + C280Sfs*61.
(B) Current-voltage relationships for WT, C280Sfs*61 and WT +
C280Sfs*61 channels. (C) Voltage-dependence of activation for WT
(n=8) and WT + C280Sfs*61 (n=10). The line represents a fit to the
Boltzmann function GNa=[1+exp(V1/2-V)/κ]-1, where V1/2
and κ are the midpoint and the slope factor, respectively, and
GNa=INa(norm)/(V-Vrev), where Vrev is the
reversal potential and V is the membrane potential. The results
showed the mutant channel had no effect on the steady state
activation. (D) Steady-stage inactivation relationships for WT
(n=10) and WT + C280Sfs*61 (n=10) fitted with Boltzmann function:
Ina=INa-max[1+exp(Vc-V1/2)/κ]-1, where the V1/2 and κ are the
midpoint and the slope factor, respectively, and Vc is the membrane
potential. and there was no significant difference between WT and
WT + C280Sfs*61 channels. All data points are shown as the mean
value, and the bars represent the standard error of the mean. WT,
wild-type. |
To mimic the heterozygous state of the proband, the
INa was also recorded in cells co-transfected with
NaV1.5-WT and NaV1.5-C280Sfs*61 at a 1:1 ratio. Comparing with WT
channels, the peak current density of heterozygous state showed a
significant reduction by ~55%, but the kinetics of steady-state
activation and inactivation were not altered (Fig. 4). Similarly, no difference in
time-dependent recovery from inactivation was found (data not
shown). These results suggested that the haploinsufficiency rather
than dominant negative effect of the C280Sfs*61 mutation of the
sodium channel was the probable cause of clinical phenotype.
Confocal imaging
To investigate the cellular localization of the WT
and mutant channels, immunocytochemical experiments were performed.
293 cells transfected with the WT or C280Sfs*61 channels were
double-stained by anti-Nav1.5 N-terminal and anti-Nav1.5 C-terminal
antibodies. As expected, cells expressing the mutant channels
showed no fluorescence staining identified using the
anti-C-terminal antibody, indicating that the truncated peptide of
mutant channel was located in the plasma and not functioning
(Fig. 5).
Discussion
Main findings
In the present study, a novel SCN5A frame-shift
mutation of C280Sfs*61 was identified in a patient suffered from
spontaneous VF episodes. The patch clamp studies revealed that
C280S*fs61 mutant channel failed to produce any sodium current. The
ECGs of the proband revealed the distinguishing
electrocardiographic anomalies of transient J wave in inferior
leads along with prolonged S-wave upstroke in precordial leads. In
the absence of type I Brugada ECG and other well-defined
electrophysiological disorders, this patient was diagnosed as ERS
according to the expert consensus statement (24).
‘Loss-of-function’ SCN5A mutation
SCN5A encodes the α-subunit of cardiac sodium
channel, which drives the impulse conduction in the heart.
‘Loss-of-function’ in SCN5A is associated with a wide range of
inherited arrhythmia syndromes, such as Brugada syndrome,
progressive cardiac conduction disease and sick sinus syndrome
(25-27).
In Brugada syndrome, SCN5A mutations are responsible for 15-20% of
the cases (28-30).
Among these mutations, frame-shift mutations are less commonly seen
as other mutations. In accordance with prior reported early
truncated mutations (30-32),
the C280S*fs61 mutant channel identified in the present study was
not functional. In addition, in the heterozygous state, the current
density was reduced by half without affecting the biophysical
gating characteristics of WT channel. Accordingly, the truncated
C280S*fs61 channel may not be able to reach to cell membrane, but
be degraded by nonsense-mediated mRNA decay (NMD) mechanism
(9,18).
Depolarization disorder underlying J
wave
As a risk factor of SCD, J wave or early
repolarization is attracting more attention. The association
between J-waves and idiopathic VF has been described in
case-control studies (33,34). In the patient of the present study,
the transient appearance of J wave in the inferior leads were
recorded, meanwhile, a simultaneous remarkable prolongation of the
S-wave upstroke in leads V1-V3 was noticed.
Since Miyazaki et al described the
association between ER and idiopathic VF in the year of
2008(3), studies have demonstrated
that inferior/inferior-lateral location of the J wave is associated
with higher risk of ventricular arrhythmias (33-36).
However, the mechanisms of J wave formation remains to be
elucidated. There are two hypotheses regarding the formation of the
J wave: The repolarization hypothesis and the depolarization
hypothesis. J-wave can be the phenotypic expression of either
delayed depolarization or early repolarization. The mechanism of
repolarization suggests that J-waves can result from different
distributions and functions of transient outward currents as a
consequence of transmembrane repolarization gradients. A
late-depolarization J-wave is more likely associated with gene
variants in the Na channel, connexins and structural proteins,
while mutations in the ion channels carrying Ito, IK-ATP or ICa are
more associated with early repolarization (37). With the wedge preparation of canine
ventricle, Yan and Antzelevitch (14,38)
proposed that, on the basis of uneven level of transient outward
current (Ito) in epi- and endocardia wall, a larger inward notch in
action potential phase 1 in epicardium was responsible for J-point
elevation in surface ECG. However, there are still questions
regarding the ‘repolarization-disorder theory’ (39). Wellens (40) states that ‘abnormality at the end
of QRS could be interpreted as early repolarization or as delayed
activation of depolarization’. Clinicians report that J wave could
be depolarization-dependent' (41,42).
For example, Abe et al (16) shows no association between
repolarization parameters (TWA and QTD) and J-wave. However, the
late potentials reflecting the abnormal depolarization were
commonly observed in ERS patients. In addition, Kamakura et
al (43) divided the inferior
lateral ERS into two groups: ER group A, the inferiolateral ER plus
non-type 1 anterior ER (anterior ER consisting of notching or
saddleback ST-segment elevation in any of the right precordial
leads) and ER group B, the pure inferolateral ER group. They found
in ER group A patients that the J waves were augmented by sodium
channel blockers and showed saddleback ST-segment elevation in the
anterior lead, clinical profiles similar to Brugada syndrome. The
positive J-wave responses to sodium channel blockers accompanied
with frequent VF episodes in the parasympathomimetic status is
considered to indicate a repolarization abnormality. By contrast,
in ER group B patients, most J waves were attenuated or disappeared
along with QRS prolongation under sodium channel blockers and the
VF episodes occurred in an awaken state, which does not seem to
indicate the presence of significant transmural dispersion of
repolarization in the ventricle. The J waves in this group seem to
be an expression of a depolarization abnormality in some
ventricular areas, although subsequent ST-segment elevation is
explainable by transmural dispersion of repolarization (44).
The prolongation of S-wave upstroke in leads V1-V3
in our patient, which could be a minor diagnostic criteria of
arrthymogenic right ventricular cardiomyopathy (ARVC) (45), represented the activation delay or
abnormal depolarization of the right ventricle. The co-existence of
J wave and prolonged S-wave, to some extent, may support the
hypothesis that J wave is associated with abnormal depolarization
rather than repolarization.
In the present study, the C280Sfs*61 mutation
causing haploinsufficiency of sodium channel has been identified.
In a study of animal model of SCN5A+/- haploinsufficiency mice, the
increased fibrosis was more severe in the right ventricle, which may
cause conduction delay and prolonged S-wave in right precordal
leads (46). Similarly, concealed
disarrangement of cardiomyocytes and interstitial fibrosis was also
reported in a young man with SCN5A mutation-related ERS (11). Considering the age of our patient,
it may also be hypothesized that there concealed abnormalities
existed in the right ventricle that may have led to conduction
delay and provide the substrate for ventricular fibrillation
(46). How loss-of-function of
sodium channels explains the manifestation of Brugada or ER
syndromes is still not fully understand at the molecular level.
Other genetic or environmental factors may be involved in modifying
the clinical phenotype (9).
Moreover, Papadotos et al (47) reported that slow conduction and
significant impairment of impulse propagation were identified in
SCN5a+/- mice within the atrium, which is in accordance with the
present study, in that the ECG of the proband showed wide and
notched P wave with normal atrium size.
The present study identified a novel frame-shift
mutation of C280Sfs*61 in SCN5A gene in a patient manifested
transient J wave in the inferior leads and prolonged S-wave
upstroke in precordial leads follow by ventricular fibrillation.
Functional study revealed that the mutation caused
haploinsufficiency effect of the sodium channel. The results
suggested that the depolarization delay may underly the mechanism
of the J wave in the inferior leads and prolonged S-wave upstroke
in the precordial leads.
Acknowledgements
The authors also warmly thank technicians Mr Jian
Huang and Ms Yinhui Zhang, both technicians at Fuwai Hospital,
Chinese Academy of Medical Science & Peking Union Medical
College (Beijing, China), for carefully preparing each experiment
and helping to collect data.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant nos. 81470460 and 81770323) and
Top-level Clinical Discipline Project of Shanghai Pudong District
Grant/Award (grant no. PWYgf2021-01).
Availability of data and materials
All data generated and/or analyzed during the
present study are included in this published article.
Authors' contributions
JP conceived and designed present study. YC was
responsible for administrative and financial support. LR and JP
were responsible for the provision of study materials and the
patient. LR, JH and YZ were responsible for collection and assembly
of data. LR, XZ, YC and JP were responsible for data analysis
and/or interpretation. XZ was responsible for writing the
manuscript. LR and XZ confirm the authenticity of all the raw data.
All authors read and approved the final manuscript. The authors are
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Fuwai Hospital (approval no. 080133) and all experiments conformed
to the principles outlined in the Declaration of Helsinki. The
patient volunteered to participate in this study and provided
written informed consent for publication
Patient consent for publication
The patient volunteered to participate in this study
and provided written informed consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Conte G, Giudicessi JR and Ackerman MJ:
Idiopathic ventricular fibrillation: The ongoing quest for
diagnostic refinement. Europace. 23:4–10. 2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Conte G, Belhassen B, Lambiase P, Ciconte
G, de Asmundis C, Arbelo E, Schaer B, Frontera A, Burri H, Calo' L,
et al: Out-of-hospital cardiac arrest due to idiopathic ventricular
fibrillation in patients with normal electrocardiograms: Results
from a multicentre long-term registry. Europace. 21:1670–1677.
2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Miyazaki S, Shah AJ and Haïssaguerre M:
Early repolarization syndrome-a new electrical disorder associated
with sudden cardiac death-. Circ J. 74:2039–2044. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Siebermair J, Sinner MF, Beckmann BM,
Laubender RP, Martens E, Sattler S, Fichtner S, Estner HL, Kääb S
and Wakili R: Early repolarization pattern is the strongest
predictor of arrhythmia recurrence in patients with idiopathic
ventricular fibrillation: Results from a single centre long-term
follow-up over 20 years. Europace. 18:718–725. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nakagawa K, Nagase S, Morita H and Ito H:
Left ventricular epicardial electrogram recordings in idiopathic
ventricular fibrillation with inferior and lateral early
repolarization. Heart Rhythm. 11:314–317. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Medeiros-Domingo A, Tan BH, Crotti L,
Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q and
Kopp D: Gain-of-function mutation S422L in the KCNJ8-encoded
cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave
syndromes. Heart Rhythm. 7:1466–1471. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Barajas-Martínez H, Hu D, Ferrer T, Onetti
CG, Wu Y, Burashnikov E, Boyle M, Surman T, Urrutia J, Veltmann C,
et al: Molecular genetic and functional association of Brugada and
early repolarization syndromes with S422L missense mutation in
KCNJ8. Heart Rhythm. 9:548–555. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Watanabe H, Nogami A, Ohkubo K, Kawata H,
Hayashi Y, Ishikawa T, Makiyama T, Nagao S, Yagihara N, Takehara N,
et al: Electrocardiographic characteristics and SCN5A mutations in
idiopathic ventricular fibrillation associated with early
repolarization. Circ Arrhythm Electrophysiol. 4:874–881.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li N, Wang R, Hou C, Zhang Y, Teng S and
Pu J: A heterozygous missense SCN5A mutation associated with early
repolarization syndrome. Int J Mol Med. 32:661–667. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Watanabe H, Ohkubo K, Watanabe I,
Matsuyama TA, Ishibashi-Ueda H, Yagihara N, Shimizu W, Horie M,
Minamino T and Makita N: SCN5A mutation associated with ventricular
fibrillation, early repolarization, and concealed myocardial
abnormalities. Int J Cardiol. 165:e21–e23. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Roten L, Derval N, Maury P, Mahida S,
Pascale P, Leenhardt A, Jesel L, Deisenhofer I, Kautzner J, Probst
V, et al: Benign vs. malignant inferolateral early repolarization:
Focus on the T wave. Heart Rhythm. 13:894–902. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Iwakami N, Aiba T, Kamakura S, Takaki H,
Furukawa TA, Sato T, Sun W, Shishido T, Nishimura K, Yamada-Inoue
Y, et al: Identification of malignant early repolarization pattern
by late QRS activity in high-resolution magnetocardiography. Ann
Noninvasive Electrocardiol. 25(e12741)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yan GX and Antzelevitch C: Cellular basis
for the electrocardiographic J wave. Circulation. 93:372–379.
1996.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Peeters HA, Sippensgroenewegen A, Wever
EF, Potse M, Daniëls MC, Grimbergen CA, Hauer RN and Robles de
Medina EO: Electrocardiographic identification of abnormal
ventricular depolarization and repolarization in patients with
idiopathic ventricular fibrillation. J Am Coll Cardiol.
31:1406–1413. 1998.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Abe A, Ikeda T, Tsukada T, Ishiguro H,
Miwa Y, Miyakoshi M, Mera H, Yusu S and Yoshino H: Circadian
variation of late potentials in idiopathic ventricular fibrillation
associated with J waves: Insights into alternative pathophysiology
and risk stratification. Heart Rhythm. 7:675–682. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Medeiros-Domingo A, Tan BH,
Iturralde-Torres P, Tester DJ, Tusié-Luna T, Makielski JC and
Ackerman MJ: Unique mixed phenotype and unexpected functional
effect revealed by novel compound heterozygosity mutations
involving SCN5A. Heart Rhythm. 6:1170–1175. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Calloe K, Refaat MM, Grubb S, Wojciak J,
Campagna J, Thomsen NM, Nussbaum RL, Scheinman MM and Schmitt N:
Characterization and mechanisms of action of novel NaV1.5 channel
mutations associated with Brugada syndrome. Circ Arrhythm
Electrophysiol. 6:177–184. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Teng S, Huang J, Gao Z, Hao J, Yang Y,
Zhang S, Pu J, Hui R, Wu Y and Fan Z: Readthrough of SCN5A nonsense
mutations. p.R1623X and p.S1812X questions gene-therapy in Brugada
syndrome. Curr Gene Ther. 17:50–58. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guo Q, Ren L, Chen X, Hou C, Chu J, Pu J
and Zhang S: A novel mutation in the SCN5A gene contributes to
arrhythmogenic characteristics of early repolarization syndrome.
Int J Mol Med. 37:727–733. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang JT, Huang J, Teng SY, Wang RR, Zhang
YH, Pu JL, Hui RT and Zhang S: Readthrough of nonsense mutation
W822X in the SCN5A gene can effectively restore expression of
cardiac Na+ channels W822X. Zhonghua Xin Xue Guan Bing Za Zhi.
39:238–241. 2011.PubMed/NCBI(In Chinese).
|
|
22
|
Wang RR, Li N, Zhang YH, Ran YQ and Pu JL:
The effects of paeoniflorin monomer of a Chinese herb on cardiac
ion channels. Chin Med J (Engl). 124:3105–3111. 2011.PubMed/NCBI
|
|
23
|
Chatelier A, Mercier A, Tremblier B,
Thériault O, Moubarak M, Benamer N, Corbi P, Bois P, Chahine M and
Faivre JF: A distinct de novo expression of Nav1.5 sodium channels
in human atrial fibroblasts differentiated into myofibroblasts. J
Physiol. 590:4307–4319. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Macfarlane PW, Antzelevitch C,
Haissaguerre M, Huikuri HV, Potse M, Rosso R, Sacher F, Tikkanen
JT, Wellens H and Yan GX: The early repolarization pattern: A
consensus paper. J Am Coll Cardiol. 66:470–477. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li W, Stauske M, Luo X, Wagner S, Vollrath
M, Mehnert CS, Schubert M, Cyganek L, Chen S, Hasheminasab SM, et
al: Disease phenotypes and mechanisms of iPSC-derived
cardiomyocytes from Brugada syndrome patients with a
loss-of-function SCN5A mutation. Front Cell Dev Biol.
8(592893)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Benson DW, Wang DW, Dyment M, Knilans TK,
Fish FA, Strieper MJ, Rhodes TH and George AL Jr: Congenital sick
sinus syndrome caused by recessive mutations in the cardiac sodium
channel gene (SCN5A). J Clin Invest. 112:1019–1028. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Zhang ZH, Barajas-Martínez H, Xia H, Li B,
Capra JA, Clatot J, Chen GX, Chen X, Yang B, Jiang H, et al:
Distinct features of probands with early repolarization and Brugada
syndromes carrying SCN5A pathogenic variants. J Am Coll Cardiol.
78:1603–1617. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wilde AAM and Amin AS: Clinical spectrum
of SCN5A mutations: long QT syndrome, Brugada syndrome, and
cardiomyopathy. JACC Clin Electrophysiol. 4:569–579.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kapplinger JD, Tester DJ, Alders M, Benito
B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A,
Harris-Kerr C, et al: An international compendium of mutations in
the SCN5A-encoded cardiac sodium channel in patients referred for
Brugada syndrome genetic testing. Heart Rhythm. 7:33–46.
2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tfelt-Hansen J, Jespersen T, Hofman-Bang
J, Rasmussen HB, Cedergreen P, Skovby F, Abriel H, Svendsen JH,
Olesen SP, Christiansen M and Haunso S: Ventricular tachycardia in
a Brugada syndrome patient caused by a novel deletion in SCN5A. Can
J Cardiol. 25:156–160. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Holst AG, Liang B, Jespersen T, Bundgaard
H, Haunso S, Svendsen JH and Tfelt-Hansen J: Sick sinus syndrome,
progressive cardiac conduction disease, atrial flutter and
ventricular tachycardia caused by a novel SCN5A mutation.
Cardiology. 115:311–316. 2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ziyadeh-Isleem A, Clatot J, Duchatelet S,
Gandjbakhch E, Denjoy I, Hidden-Lucet F, Hatem S, Deschênes I,
Coulombe A, Neyroud N and Guicheney P: A truncating SCN5A mutation
combined with genetic variability causes sick sinus syndrome and
early atrial fibrillation. Heart Rhythm. 11:1015–1023.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Antzelevitch C and Yan GX: J-wave
syndromes: Brugada and early repolarization syndromes. Heart
Rhythm. 12:1852–1866. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cheng YJ, Li ZY, Yao FJ, Xu XJ, Ji CC,
Chen XM, Liu LJ, Lin XX, Yao H and Wu SH: Early repolarization is
associated with a significantly increased risk of ventricular
arrhythmias and sudden cardiac death in patients with structural
heart diseases. Heart Rhythm. 14:1157–1164. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Demidova MM, Martín-Yebra A, van der Pals
J, Koul S, Erlinge D, Laguna P, Martínez JP and Platonov PG:
Transient and rapid QRS-widening associated with a J-wave 375
pattern predicts impending ventricular fibrillation in experimental
myocardial infarction. Heart Rhythm. 11:1195–1201. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsuda T, Hayashi K, Konno T, Sakata K,
Fujita T, Hodatsu A, Nagata Y, Teramoto R, Nomura A, Tanaka Y, et
al: J waves for predicting cardiac events in hypertrophic
cardiomyopathy. JACC Clin Electrophysiol. 3:1136–1142.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Haïssaguerre M, Nademanee K, Hocini M,
Cheniti G, Duchateau J, Frontera A, Sacher F, Derval N, Denis A,
Pambrun T, et al: Depolarization versus repolarization abnormality
underlying inferolateral J-wave syndromes: New concepts in sudden
cardiac death with apparently normal hearts. Heart Rhythm.
16:781–790. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yan GX and Antzelevitch C: Cellular basis
for the Brugada syndrome and other mechanisms of arrhythmogenesis
associated with ST-segment elevation. Circulation. 100:1660–1666.
1999.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Mizusawa Y and Bezzina CR: Early
repolarization pattern: Its ECG characteristics, arrhythmogeneity
and heritability. J Interv Card Electrophysiol. 39:185–192.
2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wellens HJ: Early repolarization
revisited. N Engl J Med. 358:2063–2065. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Badri M, Patel A and Yan GX: Cellular and
ionic basis of J-wave syndromes. Trends Cardiovasc Med. 25:12–21.
2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Aizawa Y, Sato M, Kitazawa H, Aizawa Y,
Takatsuki S, Oda E, Okabe M and Fukuda K: Tachycardia-dependent
augmentation of ‘notched J waves’ in a general patient population
without ventricular fibrillation or cardiac arrest: not a
repolarization but a depolarization abnormality? Heart Rhythm.
12:376–383. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kamakura T, Kawata H, Nakajima I, Yamada
Y, Miyamoto K, Okamura H, Noda T, Satomi K, Aiba T, Takaki H, et
al: Significance of non-type 1 anterior early repolarization in
patients with inferolateral early repolarization syndrome. J Am
Coll Cardiol. 62:1610–1618. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bourier F, Denis A, Cheniti G, Lam A,
Vlachos K, Takigawa M, Kitamura T, Frontera A, Duchateau J, Pambrun
T, et al: Early repolarization syndrome: Diagnostic and therapeutic
approach. Front Cardiovasc Med. 5(169)2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Nasir K, Bomma C, Tandri H, Roguin A,
Dalal D, Prakasa K, Tichnell C, James C, Spevak PJ, Marcus F and
Calkins H: Electrocardiographic features of arrhythmogenic right
ventricular dysplasia/cardiomyopathy according to disease severity:
A need to broaden diagnostic criteria. Circulation. 110:1527–1534.
2004.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang Y, Guzadhur L, Jeevaratnam K,
Salvage SC, Matthews GD, Lammers WJ, Lei M, Huang CL and Fraser JA:
Arrhythmic substrate, slowed propagation and increased dispersion
in conduction direction in the right ventricular outflow tract of
murine Scn5a+/- hearts. Acta Physiol (Oxf). 211:559–573.
2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Papadatos GA, Wallerstein PMR, Head CEG,
Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AEO, Huang
CLH, Vandenberg JI, et al: Slowed conduction and ventricular
tachycardia after targeted disruption of the cardiac sodium channel
gene Scn5a. Proc Natl Acad Sci USA. 99:6210–6215. 2002.PubMed/NCBI View Article : Google Scholar
|